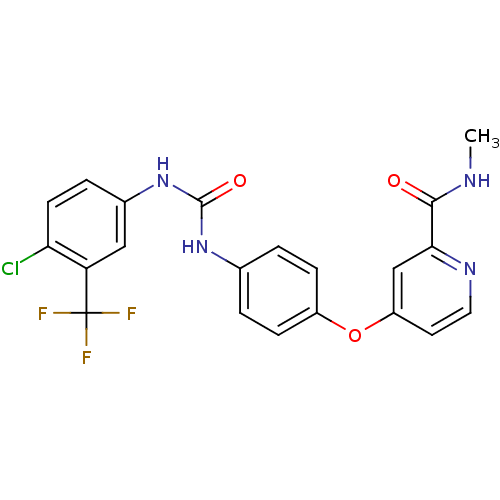
 US10183928, Sorafenib US9902709, Comparative example 1 US10980809, Example Sorafenib US11505527, Compound Sorafenib BAY 439006 US10227329, Compound Sorafenib US10774070, Compound Sorafenib Xarelto CHEMBL1336 BAY439006 Sorafenib, 4 BDBM16673 Hit compound, 8 US10584114, Compound Sorafenib US11279688, Compound Sorafenib 4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenoxy]-N-methyl-picolinamide;tosylic acid US11912663, Compound Sorafenib US9469639, Sorafenib US10202365, Compound Sorafenib 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)phenoxy]-N-methylpyridine-2-carboxamide US20250129067, Compound Sorafenib BAY 43-9006 Nexavar Sorafenib cid_216239 US9029401, Sorafenib
US10183928, Sorafenib US9902709, Comparative example 1 US10980809, Example Sorafenib US11505527, Compound Sorafenib BAY 439006 US10227329, Compound Sorafenib US10774070, Compound Sorafenib Xarelto CHEMBL1336 BAY439006 Sorafenib, 4 BDBM16673 Hit compound, 8 US10584114, Compound Sorafenib US11279688, Compound Sorafenib 4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenoxy]-N-methyl-picolinamide;tosylic acid US11912663, Compound Sorafenib US9469639, Sorafenib US10202365, Compound Sorafenib 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)phenoxy]-N-methylpyridine-2-carboxamide US20250129067, Compound Sorafenib BAY 43-9006 Nexavar Sorafenib cid_216239 US9029401, Sorafenib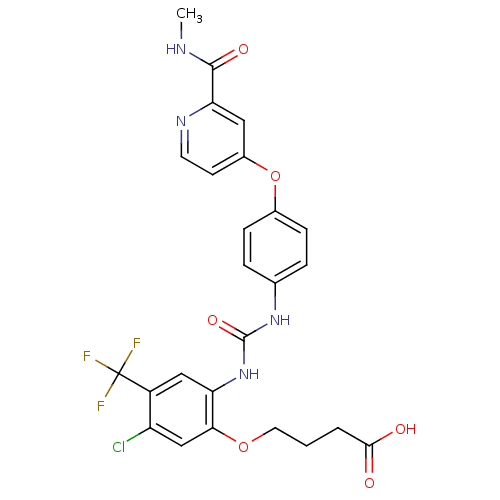 BDBM92353 Sorafenib derivative, 16
BDBM92353 Sorafenib derivative, 16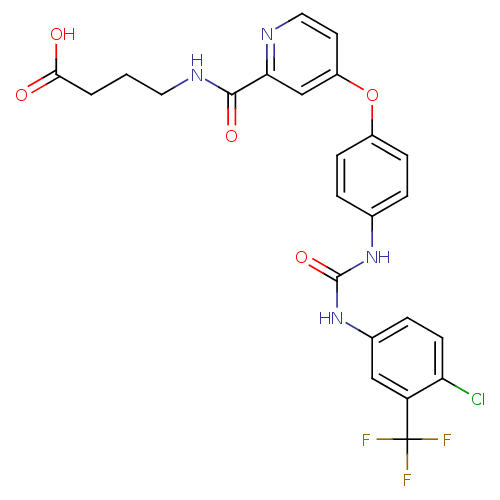 Sorafenib derivative, 17 BDBM92354
Sorafenib derivative, 17 BDBM92354
- Hammock, BD; Hwang, SH; Wecksler, AT; Morisseau, C Sorafenib derivatives as sEH inhibitors US Patent US9029401 (2015)
- Chen, F; Fang, Y; Zhao, R; Le, J; Zhang, B; Huang, R; Chen, Z; Shao, J Evolution in medicinal chemistry of sorafenib derivatives for hepatocellular carcinoma. Eur J Med Chem 179: 916-935 (2019)
- Wang, W; Wu, C; Wang, J; Luo, R; Wang, C; Liu, X; Li, J; Zhu, W; Zheng, P Synthesis, activity and docking studies of phenylpyrimidine-carboxamide Sorafenib derivatives. Bioorg Med Chem 24: 6166-6173 (2016)
- Wang, M; Xu, S; Wu, C; Liu, X; Tao, H; Huang, Y; Liu, Y; Zheng, P; Zhu, W Design, synthesis and activity of novel sorafenib analogues bearing chalcone unit. Bioorg Med Chem Lett 26: 5450-5454 (2016)
- Hwang, SH; Wecksler, AT; Zhang, G; Morisseau, C; Nguyen, LV; Fu, SH; Hammock, BD Synthesis and biological evaluation of sorafenib- and regorafenib-like sEH inhibitors. Bioorg Med Chem Lett 23: 3732-7 (2013)
- Wang, M; Xu, S; Lei, H; Wang, C; Xiao, Z; Jia, S; Zhi, J; Zheng, P; Zhu, W Design, synthesis and antitumor activity of Novel Sorafenib derivatives bearing pyrazole scaffold. Bioorg Med Chem 25: 5754-5763 (2017)
- Xie, Y; Fan, S; Ni, D; Wan, W; Xu, P; Ding, Y; Zhang, R; Lu, J; Zhang, N; Zhang, Y; Xiao, W; Zhao, K; Luo, C An ATG4B inhibitor blocks autophagy and sensitizes Sorafenib inhibition activities in HCC tumor cells. Bioorg Med Chem 84: (2023)
- Lin, X; Huang, XP; Chen, G; Whaley, R; Peng, S; Wang, Y; Zhang, G; Wang, SX; Wang, S; Roth, BL; Huang, N Life beyond kinases: structure-based discovery of sorafenib as nanomolar antagonist of 5-HT receptors. J Med Chem 55: 5749-59 (2012)
- Bozdag, M; Ferraroni, M; Ward, C; Carta, F; Bua, S; Angeli, A; Langdon, SP; Kunkler, IH; Al-Tamimi, AS; Supuran, CT Carbonic anhydrase inhibitors based on sorafenib scaffold: Design, synthesis, crystallographic investigation and effects on primary breast cancer cells. Eur J Med Chem 182: (2019)
- Sun, S; He, Z; Huang, M; Wang, N; He, Z; Kong, X; Yao, J Design and discovery of thioether and nicotinamide containing sorafenib analogues as multikinase inhibitors targeting B-Raf, B-Raf Bioorg Med Chem 26: 2381-2391 (2018)
- Jiao, Y; Xin, BT; Zhang, Y; Wu, J; Lu, X; Zheng, Y; Tang, W; Zhou, X Design, synthesis and evaluation of novel 2-(1H-imidazol-2-yl) pyridine Sorafenib derivatives as potential BRAF inhibitors and anti-tumor agents. Eur J Med Chem 90: 170-83 (2015)
- Sbenati, RM; Zaraei, SO; El-Gamal, MI; Anbar, HS; Tarazi, H; Zoghbor, MM; Mohamood, NA; Khakpour, MM; Zaher, DM; Omar, HA; Alach, NN; Shehata, MK; El-Gamal, R Design, synthesis, biological evaluation, and modeling studies of novel conformationally-restricted analogues of sorafenib as selective kinase-inhibitory antiproliferative agents against hepatocellular carcinoma cells. Eur J Med Chem 210: (2021)
- ChEMBL_2263475 Activation of PKM2 (unknown origin) in presence of sorafenib
- ChEMBL_1838867 (CHEMBL4339000) Inhibition of human CYP3A4 using sorafenib as substrate by LC/MS/MS analysis
- ChEMBL_2069798 (CHEMBL4725051) Inhibition of human liver microsome CYP3A4 using sorafenib as substrate incubated for 20 mins by LC-MS/MS with HPLC analysis
- ChEMBL_2248038 (CHEMBL5162248) Inhibition of CYP3A4 in human liver microsomes using sorafenib as cocktail probe substrate preincubated for 5 mins followed by NADPH addition and measured after 30 mins by HPLC-MS/MS analysis
- Enzyme-Linked Immunosorbent Assay Experimental Procedures(1) Kinase reaction substrate Poly(Glu, Tyr, 4:1) was diluted to 20 g/ml with potassium ion-free PBS, and enzyme labeled plate was coated by the substrate, reacted at 37° C. for 12-16 h, and then the liquid in holes was removed.(2) Enzyme labeled plate was washed with T-PBS for three times, 10 min each time.(3) Enzyme labeled plate was dried at 37° C. in a dryer.(4) The tested samples were added into holes of the coated enzyme labeled plate:The tested samples were firstly formulated to be a 10 2 M stock solution with DMSO, and then diluted to required concentration with reaction buffer before use, added into the experiment holes to reach corresponding final concentration in a 100 uL reaction system. Meanwhile, positive control holes were created, and compound Sorafenib was added. The rest of the stock solutions was storaged at 20° C. after subpackage.
- Test of Small Molecule Compounds for Inhibiting the Activity of C-RAF and B-RAF Kinases 1. Preparation of test compounds: according to the molecular weight of the compounds, an appropriate volume of DMSO was directly added to dissolve the test compounds. For storing the compound, the concentration of DMSO is 100%, and the final concentration of DMSO in the experimental system is 1%. The compounds were 3-fold serially diluted with DMSO to obtain a total of 8 dilutions, with a maximum concentration of 1000 nM and a minimum concentration of 0.46 nM.2. Preparation of the sorafenib positive control: sorafenib, a selective inhibitor of BRAF and RAF1, was used as the positive control of this experiment, and the dilution method thereof was the same as that of the above test compounds.3. Test Conditions:Enzyme: B-RAF: 0.1 ng/l (the final concentration in the reaction system); C-RAF: 0.1 ng/μl (the final concentration in the reaction system)Substrate and ATP: inactive MEKI: 2 ng/μl (the final concentration in the reaction system); ATP: 35 μM (the final concentration in the reaction system)HPE: the reaction without enzyme (1% DMSO)ZPE: the reaction with enzyme but without compound (1% DMSO)4. Test procedure:a) 1 ul of 10-fold diluted compound or 10% DMSO was added to a 384-well assay plate,b) 4 ul enzyme solution or assay buffer was added to the wells of the assay plate,c) the plate was centrifuged at 1000 rpm for 1 minute to homogeneous,d) 5 ul of ATP-substrate mixture was added to the wells of the assay plate,e) the plate was shaked for mixing for 2 minutes,f) the plate was incubated at 30° C. for 1 hour,g) 10 ul of ADP-Glo reagent was added to the wells of the assay plate, and the plate was incubated for 40 minutes at 27° C.,h) 20 ul of kinase assay solution was added to the wells of the plate, and the plate was incubated for 30 minutes at 27° C.,i) the chemiluminescent signal was read with Envision.5. Analysis of the results: calculation of the compound inhibition rate:Inhibition rate (%)=(control measurement without compound−sample measurement)/(control measurement without compound−control measurement without enzyme)*100%The IC50 values of the positive control compound and the test compounds were calculated using the Prism software according to the variable slope of the curve.
- LanthaScreen Eu Kinase Binding Assay Binding of an Alexa Fluor™ conjugate or “tracer” to a kinase is detected by addition of an Eu-labeled anti-tag antibody. Binding of the tracer and antibody to a kinase result in a high degree of FRET, whereas displacement of the tracer with a kinase inhibitor results in a loss of FRET. This assay is carried out by mixing the compound tested with the reagents and reading, no development step is required. Life Technologies' Kinase Tracers are based on ATP-competitive kinase inhibitors, making them suitable for detection of any compounds that bind to the ATP site. Inhibitors that bind the ATP site include both Type I kinase inhibitors, which bind solely to the ATP site, and Type II inhibitors (e.g., Gleevec /Imatinib, Sorafenib, BIRB-796), which bind to both the ATP site and a second site often referred to as the allosteric site. The following protocol is used to carry out this assay: The Test Compounds are screened in 1% DMSO (final) in the well. For 10-point titrations, 3-fold serial dilutions are conducted from the starting concentration. All Kinase/Antibody Mixtures are diluted to a 2× working concentration in the specified kinase buffer. The 4× AlexaFluor labeled Tracer is prepared in Kinase Buffer. Assay Protocol Bar-coded, low volume, white 384-well plate (Greiner Cat. #784207) 1. 160 nL100× Test Compound in 100% DMSO 2. 3.84 μL—Kinase Buffer 3. 8.0 μL2× Kinase/Antibody Mixture 4. 4.0 μL—4× Tracer 5. 30-second plate shake 6. 60-minute incubation at room temperature7. Read on fluorescence plate reader and analyze the data. The following controls are made for each individual kinase and are located on the same plate as the kinase:0% Displacement Control: the maximum Emission Ratio is established by the 0% Displacement Control wells, which do not contain known inhibitor in the reaction and therefore exhibits no displacement of the tracer. 100% Displacement Control: the minimum Emission Ratio is established by the 100% Displacement Control wells, which contain the highest concentration of the known inhibitor used in that assay. Known Inhibitor Control Protocol: a known inhibitor control standard curve, 10-point titration, is run for each individual kinase on the same plate as the kinase to ensure the inhibitor is displaced within an expected IC50 range previously determined.
- Biochemical Assay for mTOR TR-FRET assays for protein kinases uses a long-lifetime lanthanide Terbium or Europium chelates as the donor species which overcome interference by compound autofluorescence or light scatter from precipitated compounds, by introducing a delay after excitation by a flashlamp excitation source. Results are often expressed as a ratio of the intensities of the acceptor and donor fluorophores. The ratiometric nature of such a value corrects for differences in assay volumes between wells, as well as corrects for quenching effects due to colored compounds.Binding Assays are based on the binding and displacement of an Alexa Fluor 647-labeled, ATP-competitive kinase inhibitors to the kinase of interest. Invitrogen's “Kinase Tracers” have been developed to address a wide range of kinase targets and are based on ATP-competitive kinase inhibitors, making them suitable for detection of any compounds that bind to the ATP site or to an allosteric site altering the conformation of the ATP site. Inhibitors that bind the ATP site include both Type I kinase inhibitors, which bind solely to the ATP site, and Type II inhibitors (e.g., Gleevec/Imatinib, Sorafenib, BIRB-796), which bind to both the ATP site and a hydrophobic site exposed in the DFG-out (non-active) conformation. Type III inhibitors are compounds that do not compete with ATP are loosely referred to as allosteric inhibitors. A study of 15 diverse Type III inhibitors demonstrated that all but one compound was detected in the binding assay with equivalent potency to activity assays. The sole exception was a substrate-competitive compound, and thus not a true allosteric inhibitor.In contrast to most fluorescence-based kinase activity assays, LanthaScreen Eu3+ Kinase Binding Assays can be read continuously, which facilitates evaluation of compounds with slow binding kinetics. Also, unlike most activity assays, binding assays can be performed using either active or non-activated kinase preparations, which enables characterization of compounds that bind preferentially to non-activated kinases, such as Gleevec /imatinib and some allosteric inhibitors.In the Lanthascreen™ kinase binding assay, the donor (Eu3+-anti-GST antibody) is excited at 340 nm and will transfer its energy to the acceptor (Alexa Fluor 647-labeled ATP-competitive kinase inhibitor=Tracer-314). The emission from the Tracer-314 (Alexa Fluor 647 inhibitor) can be monitored with a filter centered at 665 nm because it is located between the emission peaks of the donor, which is measured at 615/620 nm. The binding of both, the Tracer-314 and Eu3+-anti-GST antibody, to the kinase results in a high degree of FRET from the Eu3+-donor fluorophore to the Alexa-Fluor 647-acceptor fluorophore on the Tracer-314. Binding of an inhibitor to the kinase competes for binding with the tracer, resulting in a loss of FRET.50 nL of compound dilutions were dispensed onto white 384-well small volume polystyrene plate as described in section 2.2. Then 5 μL of GST-mTOR and Europium-anti-GST antibody followed by 5 μL of tracer-314 (final assay volume 10 μL) are incubated at RT. The standard reaction buffer for the Lanthascreen™ kinase binding assay contained 50 mM HEPES pH 7.5, 5 mM MgCl2, 1 mM EGTA, 0.01% Pluronic F-127. Plates are read 60 mins later in a Synergy2 reader using an integration time of 0.2 microseconds and a delay of 0.1 microseconds.To calculate the emission ratio, the signal emitted at 665 nm from the acceptor (Alexa Fluor 647-labeled Tracer-314) is divided by the signal emitted at 620 nm from the donor (Eu3+ anti-GST antibody)Control for the 0% inhibition was given by the solvent vehicle of the compounds (90% DMSO in H2O). Control for the relative 100% inhibition was performed by adding 10 μM in the mix containing GST-mTOR and Europium anti-GST antibody. An additional control for the absolute 0% inhibition is given by Eu3+ anti-GST antibody without GST-mTOR.