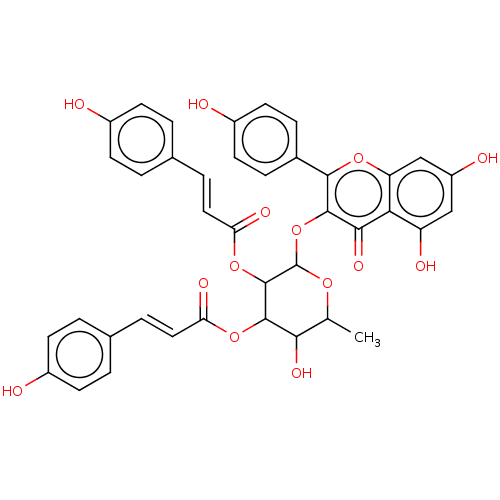
 BDBM241950 Neolitsea aciculata extract, 5
BDBM241950 Neolitsea aciculata extract, 5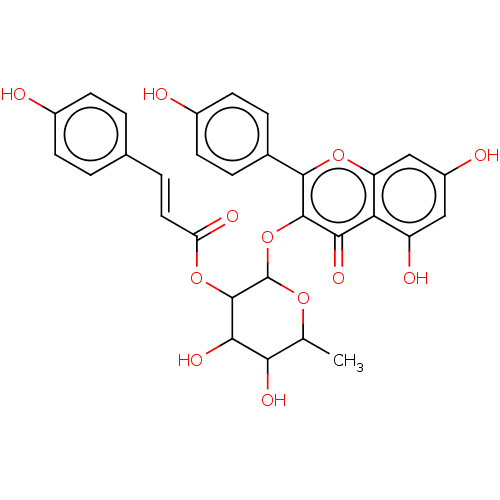 BDBM241951 Neolitsea aciculata extract, 6
BDBM241951 Neolitsea aciculata extract, 6
- Balestrieri, E; Pizzimenti, F; Ferlazzo, A; Giofrè, SV; Iannazzo, D; Piperno, A; Romeo, R; Chiacchio, MA; Mastino, A; Macchi, B Antiviral activity of seed extract from Citrus bergamia towards human retroviruses. Bioorg Med Chem 19: 2084-9 (2011)
- Rainer, B; Revoltella, S; Mayr, F; Moesslacher, J; Scalfari, V; Kohl, R; Waltenberger, B; Pagitz, K; Siewert, B; Schwaiger, S; Stuppner, H From bench to counter: Discovery and validation of a peony extract as tyrosinase inhibiting cosmeceutical. Eur J Med Chem 184: (2019)
- Rho, HS; Ahn, SM; Lee, BC; Kim, MK; Ghimeray, AK; Jin, CW; Cho, DH Changes in flavonoid content and tyrosinase inhibitory activity in kenaf leaf extract after far-infrared treatment. Bioorg Med Chem Lett 20: 7534-6 (2010)
- Rempel, V; Fuchs, A; Hinz, S; Karcz, T; Lehr, M; Koetter, U; Müller, CE Magnolia Extract, Magnolol, and Metabolites: Activation of Cannabinoid CB2 Receptors and Blockade of the Related GPR55. ACS Med Chem Lett 4: 41-5 (2013)
- Cuéllar, MJ; Giner, RM; Recio, MC; Just, MJ; Máñez, S; Cerdá, M; Hostettmann, K; Ríos, JL Zanhasaponins A and B, antiphospholipase A2 saponins from an antiinflammatory extract of Zanha africana root bark. J Nat Prod 60: 1158-60 (1998)
- Park, MH; Kim, IS; Kim, SA; Na, CS; Hong, CY; Dong, MS; Yoo, HH Inhibitory effect of Rhus verniciflua Stokes extract on human aromatase activity; butin is its major bioactive component. Bioorg Med Chem Lett 24: 1730-3 (2014)
- Kasangana, PB; Haddad, PS; Eid, HM; Nachar, A; Stevanovic, T Bioactive Pentacyclic Triterpenes from the Root Bark Extract of Myrianthus arboreus, a Species Used Traditionally to Treat Type-2 Diabetes. J Nat Prod 81: 2169-2176 (2018)
- Wu, T; Jiang, C; Wang, L; Morris-Natschke, SL; Miao, H; Gu, L; Xu, J; Lee, KH; Gu, Q 3,5-Diarylpyrazole Derivatives Obtained by Ammonolysis of the Total Flavonoids from Chrysanthemum indicum Extract Show Potential for the Treatment of Alzheimer's Disease. J Nat Prod 78: 1593-9 (2015)
- Hüsch, J; Gerbeth, K; Fricker, G; Setzer, C; Zirkel, J; Rebmann, H; Schubert-Zsilavecz, M; Abdel-Tawab, M Effect of phospholipid-based formulations of Boswellia serrata extract on the solubility, permeability, and absorption of the individual boswellic acid constituents present. J Nat Prod 75: 1675-82 (2012)
- Sharma, R; Gatchie, L; Williams, IS; Jain, SK; Vishwakarma, RA; Chaudhuri, B; Bharate, SB Glycyrrhiza glabra extract and quercetin reverses cisplatin resistance in triple-negative MDA-MB-468 breast cancer cells via inhibition of cytochrome P450 1B1 enzyme. Bioorg Med Chem Lett 27: 5400-5403 (2017)
- ChEMBL_2346015 Inhibition of HDAC in human HeLa nuclear extract
- ChEMBL_2346044 Inhibition of HDAC1 in human HeLa nuclear extract
- ChEMBL_2346045 Inhibition of HDAC2 in human HeLa nuclear extract
- ChEMBL_472921 (CHEMBL921164) Inhibition of HDAC1 in rat liver extract
- ChEMBL_1552208 (CHEMBL3761210) Inhibition of HDAC in human HeLa nuclear extract
- ChEMBL_1875110 (CHEMBL4376399) Inhibition of HDAC in human K562 nuclear extract
- ChEMBL_1919932 (CHEMBL4422777) Inhibition of HDAC in human HeLa nuclear extract
- ChEMBL_2439282 Inhibition of HDAC in human HeLa cells nuclear extract
- ChEMBL_1919890 (CHEMBL4422735) Inhibition of HDAC1/HDAC2 in human HeLa nuclear extract
- ChEMBL_852177 (CHEMBL2157598) Inhibition of aCDase expressed in human HL60 cell extract
- ChEBML_28421 Inhibition of AICAR formyltransferase from extract of Manca human lymphoma cells
- ChEMBL_432005 (CHEMBL918789) Inhibition of telomerase in JR8 cell extract by TRAP assay
- ChEMBL_472923 (CHEMBL921166) Inhibition of HDAC1 in rat liver extract by trypsin assay
- ChEMBL_873193 (CHEMBL2183424) Inhibition of DNA-PK isolated from human HeLa cell extract
- ChEMBL_69929 (CHEMBL678809) Inhibition of GAR formyltransferase from extract of Manca human lymphoma cells
- ChEMBL_87390 (CHEMBL691505) Tested for Histone deacetylase enzyme inhibition assay using Eimeria tenella extract
- ChEMBL_87718 (CHEMBL697246) Inhibition of histone deacetylase (HDAC) activity in HeLa cell nuclear extract
- ChEMBL_2105981 (CHEMBL4814656) Inhibition of telomerase in human SGC-7901 cell extract by TRAP assay
- ChEMBL_2201302 (CHEMBL5114010) Inhibition of telomerase derived from human A2780 cell extract by TRAP assay
- ChEMBL_2275541 Inhibition of human HDAC in human HeLa cell nuclear extract by fluorescence assay
- ChEMBL_457624 (CHEMBL923825) Inhibition of HDAC activity in HeLa cell nuclear extract by fluorescent assay
- ChEMBL_54532 (CHEMBL664849) Affinity for DNA-dependent protein kinase(DNA-PK) from HeLa cell extract
- ChEMBL_583224 (CHEMBL1055023) Inhibition of ALR2 from Sprague-Dawley albino rat lens extract by spectrophotometrically
- ChEMBL_610879 (CHEMBL1065034) Inhibition of HDAC in human HeLa cell nuclear extract by fluorescence assay
- ChEMBL_956022 (CHEMBL2380117) Inhibition of telomerase in human HL60 cell extract by TRAP-LIG assay
- ChEBML_54534 Inhibition of DNA-dependent protein kinase (DNA-PK) of HeLa cell nuclear cell extract
- ChEMBL_209485 (CHEMBL810077) Inhibition of pure human thymidylate synthase from extract of Manca human lymphoma cells
- ChEMBL_34783 (CHEMBL646224) In vitro inhibition of Angiotensin I converting enzyme isolated from rabbit lung extract.
- ChEMBL_653705 (CHEMBL1227029) Inhibition of HDAC in human HeLa cell extract by fluorescence plate reader assay
- ChEMBL_87532 (CHEMBL694903) In vitro inhibitory activity against histone deacetylase (HDAC) isolated from HeLa nuclear extract
- ChEMBL_87542 (CHEMBL695140) In vitro inhibitory activity against human histone deacetylase (HDAC) using HeLa nuclear extract
- ChEMBL_1434455 (CHEMBL3388272) Inhibition of HDAC in human HeLa cell extract after 15 mins by fluorescence assay
- ChEMBL_1580211 (CHEMBL3811519) Inhibition of HDAC1 in human Jurkat cells extract after 30 mins by immunoprecipitation assay
- ChEMBL_1580212 (CHEMBL3811520) Inhibition of HDAC3 in human Jurkat cells extract after 30 mins by immunoprecipitation assay
- ChEMBL_1990603 (CHEMBL4624338) Inhibition of NF-kappaB p65 in human HeLa cells nuclear extract by chemiluminescent assay
- ChEMBL_28279 (CHEMBL645820) Inhibition of AGT activity to 50% of control rate in HT-29 cell extract
- ChEMBL_557374 (CHEMBL955704) Inhibition of PSMA in human LNCaP cell extract using [3H]NAAG by radiometric assay
- ChEMBL_756042 (CHEMBL1804152) Inhibition of NFkappa p65 isolated from nuclear extract of human HeLa cells by ELISA
- ChEMBL_1663210 (CHEMBL4012891) Binding affinity to TNKS1 in human HeLa cell extract after 2 hrs by HTS assay
- ChEMBL_2201829 (CHEMBL5114537) Inhibition of recombinant human Top1 derived from human MCF7 cell extract incubated for 30 mins
- ChEMBL_2213599 (CHEMBL5126731) Inhibition of HDAC1 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate
- ChEMBL_2213600 (CHEMBL5126732) Inhibition of HDAC2 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate
- ChEMBL_2213601 (CHEMBL5126733) Inhibition of HDAC8 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate
- ChEMBL_2272806 Inhibition of HDAC1 in human HeLa nuclear extract incubated for 30 mins by fluorescence based analysis
- ChEMBL_2339423 Inhibition of HDAC in human HeLa nuclear extract measured after 30 mins by fluorescence based assay
- ChEMBL_2488043 Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate
- ChEMBL_422105 (CHEMBL907102) In vitro inhibition of histone deacetylase activity using HeLa cell nuclear extract as enzyme source
- ChEMBL_557375 (CHEMBL955705) Inhibition of PSMA in human LNCaP cell extract using [3H]NAAG by fluorescence-based assay
- ChEMBL_776071 (CHEMBL1912767) Inhibition of HDAC in human HeLa cell extract assessed as fluorophore release by fluorescence spectrophotometry
- ChEMBL_1663203 (CHEMBL4012884) Binding affinity to PARP1 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663204 (CHEMBL4012885) Binding affinity to PARP2 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663205 (CHEMBL4012886) Binding affinity to PARP4 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663206 (CHEMBL4012887) Binding affinity to PARP11 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663208 (CHEMBL4012889) Binding affinity to PARP14 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663209 (CHEMBL4012890) Binding affinity to PARP16 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663213 (CHEMBL4012894) Binding affinity to PARP10 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1700237 (CHEMBL4051219) Inhibition of HDAC 10 in human HeLa nuclear extract measured after 60 mins by fluorometric analysis
- ChEMBL_2201312 (CHEMBL5114020) Inhibition of telomerase derived from human K562 cell extract incubated for 30 mins by TRAP assay
- ChEMBL_223549 (CHEMBL845855) concentration required to reduce AGT activity to 50% of control rate in HT-29 cell extract.
- ChEMBL_2236190 (CHEMBL5150086) Inhibition of HDAC in human HeLa nuclear extract measured after 30 mins by fluorescence based assay
- ChEMBL_2239302 (CHEMBL5153198) Inhibition of HDAC in human NALM-6 nuclear extract incubated for 48 hrs by fluorometric analysis
- ChEMBL_2274123 Inhibition of human HDAC using human HeLa cell nuclear extract measured after 60 mins by fluorescence assay
- ChEMBL_2373527 Inhibition of HDAC in human HeLa cells nuclear extract incubated for 30 mins by multiplate reader analysis
- ChEMBL_2373528 Inhibition of HDAC1 in human HeLa cells nuclear extract incubated for 30 mins by multiplate reader analysis
- ChEMBL_2460780 Inhibition of HDAC in human HeLa cells nuclear extract incubated for 30 mins by multiplate reader analysis
- ChEMBL_2525403 Inhibition of human HeLa cell nuclear extract purified DNA-PK using p53 peptide as substrate by ELISA
- ChEMBL_28280 (CHEMBL645821) concentration required to reduce AGT activity to 50% of control rate in HT-29 cell extract.
- ChEMBL_614728 (CHEMBL1114231) Inhibition of human HDAC in human HeLa cell nuclear extract after 15 mins by colorimetric assay
- ChEMBL_753553 (CHEMBL1799171) Inhibition of HDAC6 in human HeLa cell nuclear extract after 30 mins by fluorescence microplate reader
- ChEMBL_832314 (CHEMBL2066852) Inhibition of DNMT1 in human HeLa cell nuclear extract assessed as methylated substrate level by ELISA
- ChEMBL_966564 (CHEMBL2399492) Inhibition of HDAC isolated from human HeLa cell nuclear extract after 30 mins by fluorescence assay
- ChEMBL_977181 (CHEMBL2416774) Inhibition of HDAC in human HeLa cell extract using Fluor deLys as substrate by fluorimetric assay
- ChEMBL_1527400 (CHEMBL3636775) Inhibition of human HDAC in HeLa cell nuclear extract by fluorometric assay using Fluor de Lys substrate
- ChEMBL_1728624 (CHEMBL4143902) Inhibition of HDAC in human HeLa nuclear extract using Fluor de Lys as substrate by fluorimetric method
- ChEMBL_1839042 (CHEMBL4339257) Inhibition of HDAC (unknown origin) in human HeLa cell nuclear extract using Color de Lys as substrate
- ChEMBL_2150392 (CHEMBL5034854) Inhibition of human HDAC using human HeLa cell nuclear extract measured after 60 mins by fluorescence assay
- ChEMBL_2201002 (CHEMBL5113710) Inhibition of HDAC1 derived from human HeLa nuclear extract using COLOR DE LYS substrate by colorimetric assay
- ChEMBL_2488159 Inhibition of human HDAC extracted from human HeLa cell nuclear extract incubated for 30 mins by fluorometric analysis
- ChEMBL_1825529 (CHEMBL4325293) Inhibition of PI3Kdelta in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825530 (CHEMBL4325294) Inhibition of VPS34 in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1986966 (CHEMBL4620513) Inhibition of HDAC in human HeLa cell nuclear extract using fluor-de-lys as substrate by spectrofluorometric analysis
- ChEMBL_2047319 (CHEMBL4702018) Inhibition of HDAC in human HeLa nuclear extract using fluoroscence-labeled acetylated peptide as substrate by fluorometric assay
- ChEMBL_2206578 (CHEMBL5119286) Inhibition of HDAC in human HeLa nuclear extract incubated for 30 mins by fluorescence-based Glo-luminescence assay
- ChEMBL_2488178 Inhibition of HDAC3 in human HeLa cell nuclear extract incubated for 30 mins by fluorescence based microplate reader analysis
- ChEMBL_873195 (CHEMBL2183783) Inhibition of ATM isolated from human HeLa cell extract using glutathione S-transferase-p53N66 as substrate by ELISA
- ChEBML_1684531 Inhibition of AChE1 in Anopheles gambiae body extract using acetylthiocholine iodide as substrate measured over 60 secs by Ellman's method
- ChEMBL_143356 (CHEMBL751280) Inhibitory activity evaluated from soluble cell extract of Neuronal nitric oxide synthase and partially purified by DEAE-sepharose chromatography
- ChEMBL_1825526 (CHEMBL4325290) Binding affinity to PI3Kdelta in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825527 (CHEMBL4325291) Binding affinity to VPS34 in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825557 (CHEMBL4325321) Binding affinity to PI3Kalpha in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825558 (CHEMBL4325322) Binding affinity to PI3Kbeta in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825559 (CHEMBL4325323) Binding affinity to PI3Kgamma in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_2157455 (CHEMBL5042115) Inhibition of HDAC in human HeLa nuclear extract using fluorogenic substrate incubated for 30 mins by fluorescence based assay
- ChEMBL_2274135 Inhibition of human HDAC1 using human HeLa cell nuclear extract at 1 uM measured after 60 mins by fluorescence assay
- ChEMBL_2274136 Inhibition of human HDAC2 using human HeLa cell nuclear extract at 1 uM measured after 60 mins by fluorescence assay
- ChEMBL_2274137 Inhibition of human HDAC3 using human HeLa cell nuclear extract at 1 uM measured after 60 mins by fluorescence assay
- ChEMBL_1351241 (CHEMBL3271696) Inhibition of HDAC in human HeLa cell nuclear extract using acetylated lysine as substrate after 30 mins by spectrophotometric analysis
- ChEMBL_143357 (CHEMBL751281) Inhibitory activity evaluated from soluble cell extract of human Neuronal nitric oxide synthase and partially purified by DEAE-sepharose chromatography
- ChEMBL_143358 (CHEMBL751652) Inhibitory activity evaluated from soluble cell extract of human nNeuronal nitric oxide synthase and partially purified by DEAE-sepharose chromatography
- ChEMBL_1589349 (CHEMBL3830593) Inhibition of full length BRPF1 in human HUT78 cell nuclear/chromatin extract after 45 mins by chemoproteomic competition binding assay
- ChEMBL_164142 (CHEMBL771466) Compound concentration which displaces 50% of [125I]-labeled 2-5A probe bound to RNase L from mouse L cell extract
- ChEMBL_1684530 (CHEMBL4035009) Inhibition of AChE1 in Anopheles gambiae head extract using acetylthiocholine iodide as substrate measured over 60 secs by Ellman's method
- ChEMBL_1684531 (CHEMBL4035010) Inhibition of AChE1 in Anopheles gambiae body extract using acetylthiocholine iodide as substrate measured over 60 secs by Ellman's method
- ChEMBL_1700230 (CHEMBL4051212) Inhibition of HDAC 1 in human HeLa nuclear extract using HDAC substrate-3 measured after 60 mins by fluorometric analysis
- ChEMBL_1700231 (CHEMBL4051213) Inhibition of HDAC 2 in human HeLa nuclear extract using HDAC substrate-3 measured after 60 mins by fluorometric analysis
- ChEMBL_1700232 (CHEMBL4051214) Inhibition of HDAC 3 in human HeLa nuclear extract using HDAC substrate-3 measured after 60 mins by fluorometric analysis
- ChEMBL_2030958 (CHEMBL4685116) Inhibition of HDAC in human HeLa cell nuclear extract using fluorescence substrate incubated for 30 mins by fluorescence based assay
- ChEMBL_2032352 (CHEMBL4686510) Inhibition of tissue extract derived mEH (unknown origin) using [3H]trans-stilbene oxide as substrate by liquid scintillation counting method
- ChEMBL_2119132 (CHEMBL4828198) Inhibition of PI3Kdelta in human HL-60 cell extract measured after 2 hrs by kinobeads based chemoproteomic competition binding assay
- ChEMBL_2119133 (CHEMBL4828199) Inhibition of VPS34 in human HL-60 cell extract measured after 2 hrs by kinobeads based chemoproteomic competition binding assay
- ChEMBL_809835 (CHEMBL2014772) Inhibition of HDAC6 in human HeLa cells nuclear extract using Fluor-de-Lys as substrate after 30 mins by spectrophotometry
- ChEMBL_812542 (CHEMBL2014417) Inhibition of HDAC1 in human HeLa cells nuclear extract using Fluor-de-Lys as substrate after 30 mins by spectrophotometry
- ChEMBL_89193 (CHEMBL701145) Inhibitory activity evaluated for soluble cell extract of human Inducible nitric oxide synthase and partially purified by DEAE-sepharose chromatography
- ChEMBL_974420 (CHEMBL2412317) Inhibition of HDAC in human HeLa cell extract using fluor de Lys as substrate after 15 mins by fluorometric analysis
- ChEBML_1572320 Inhibition of HDAC in human HeLa nuclear extract using BOC-Ac-Lys-AMC as substrate incubated for 90 mins by fluorescence assay
- ChEMBL_1282340 (CHEMBL3100391) Inhibition of HDAC in human HeLa cell nuclear extract using fluor de Lys as substrate after 15 mins by fluorimetric analysis
- ChEMBL_1551034 (CHEMBL3761048) Inhibition of HDAC in human HeLa cell nuclear extract using BML-KI104 Fluor de Lys as substrate by fluorescence-based assay
- ChEMBL_1551259 (CHEMBL3761968) Inhibition of HDAC in human HeLa cells nuclear extract using Fluor de lys as substrate after 15 mins by fluorometric analysis
- ChEMBL_1570662 (CHEMBL3795030) Inhibition of HDAC in human HeLa nuclear extract using fluor de lys as substrate after 10 to 15 mins by spectrofluorometry
- ChEMBL_1772528 (CHEMBL4224640) Inhibition of chymotrypsin-like activity of 20S proteasome in human PC3 cell extract using Suc-LLVYaminoluciferin as substrate after 2 hrs
- ChEMBL_1772529 (CHEMBL4224641) Inhibition of chymotrypsin-like activity of 20S proteasome in human LNCAP cell extract using Suc-LLVYaminoluciferin as substrate after 2 hrs
- ChEMBL_1913617 (CHEMBL4416200) Binding affinity to CDPK4 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr by kinobeads based pull down assay
- ChEMBL_1913619 (CHEMBL4416202) Binding affinity to CDPK1 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr by kinobeads based pull down assay
- ChEMBL_1913623 (CHEMBL4416206) Binding affinity to CK1 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr by kinobeads based pull down assay
- ChEMBL_1919885 (CHEMBL4422730) Inhibition of HDAC in human HeLa nuclear extract using Fluor-de-lys as substrate measured after 60 mins by fluorescence assay
- ChEMBL_1919886 (CHEMBL4422731) Inhibition of HDAC in human HeLa cytosolic extract using Fluor-de-lys as substrate measured after 60 mins by fluorescence assay
- ChEMBL_2131671 (CHEMBL4841186) Inhibition of HDAC in human HeLa nuclear extract using Fluor de Lys as substrate incubated for 20 mins by fluorometric assay
- ChEMBL_2224455 (CHEMBL5137968) Inhibition of HDAC6 derived from human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2224456 (CHEMBL5137969) Inhibition of HDAC1 derived from human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2337887 Inhibition of HDAC in human nuclear extract using Ac-Arg-Gly-Lys(Ac)-AMC as substrate incubated overnight by fluorescence based assay
- ChEMBL_2492889 Inhibition of HDAC1 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2492890 Inhibition of HDAC2 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2492891 Inhibition of HDAC3 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2492892 Inhibition of HDAC6 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2492893 Inhibition of HDAC7 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_809834 (CHEMBL2014771) Inhibition of HDAC3-NCoR2 in human HeLa cells nuclear extract using Fluor-de-Lys as substrate after 30 mins by spectrophotometry
- ChEMBL_816565 (CHEMBL2025279) Inhibition of HDAC1 in human HeLa cell nuclear extract using Fluor de Lys as substrate after 15 mins by fluorometric analysis
- ChEMBL_816566 (CHEMBL2025280) Inhibition of HDAC6 in human HeLa cell nuclear extract using Fluor de Lys as substrate after 15 mins by fluorometric analysis
- ChEMBL_816567 (CHEMBL2025281) Inhibition of HDAC8 in human HeLa cell nuclear extract using Fluor de Lys as substrate after 15 mins by fluorometric analysis
- ChEMBL_873187 (CHEMBL2183418) Inhibition of DNA-PK isolated from human HeLa cell extract assessed as inhibition of p53 peptide fragment phosphorylation after 10 mins
- ChEMBL_1524328 (CHEMBL3632020) Inhibition of Stat3 dimer DNA binding activity in human U251MG cells nuclear extract after 1.5 hrs by EMSA using radiolabeled probe hSIE
- ChEMBL_1524329 (CHEMBL3632021) Inhibition of Stat3 dimer DNA binding activity in human U373MG cells nuclear extract after 1.5 hrs by EMSA using radiolabeled probe hSIE
- ChEMBL_1545338 (CHEMBL3751357) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (Ac)-AMC as substrate after 60 mins by fluorometric analysis
- ChEMBL_1545348 (CHEMBL3751367) Inhibition of HDAC1 in human HeLa cell nuclear extract using Boc-Lys (Ac)-AMC as substrate after 60 mins by fluorometric analysis
- ChEMBL_1545349 (CHEMBL3751368) Inhibition of HDAC6 in human HeLa cell nuclear extract using Boc-Lys (Ac)-AMC as substrate after 60 mins by fluorometric analysis
- ChEMBL_1545404 (CHEMBL3751629) Inhibition of HDAC8 in human HeLa cell nuclear extract using Boc-Lys (Ac)-AMC as substrate after 60 mins by fluorometric analysis
- ChEMBL_1572320 (CHEMBL3795851) Inhibition of HDAC in human HeLa nuclear extract using BOC-Ac-Lys-AMC as substrate incubated for 90 mins by fluorescence assay
- ChEMBL_1700233 (CHEMBL4051215) Inhibition of HDAC 8 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1700234 (CHEMBL4051216) Inhibition of HDAC 4 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1700235 (CHEMBL4051217) Inhibition of HDAC 5 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1700236 (CHEMBL4051218) Inhibition of HDAC 9 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1700238 (CHEMBL4051220) Inhibition of HDAC 11 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1774366 (CHEMBL4231358) Inhibition of HDAC in human HeLa nuclear extract using Ac-Lys(Ac)-pNA as substrate measured after 30 mins by fluorometric analysis
- ChEMBL_1875119 (CHEMBL4376408) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate measured after 30 mins by fluorescence assay
- ChEMBL_1919962 (CHEMBL4422807) Inhibition of HDAC2 in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate measured after 30 mins by fluorescence assay
- ChEMBL_1919963 (CHEMBL4422808) Inhibition of HDAC6 in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate measured after 30 mins by fluorescence assay
- ChEMBL_1919964 (CHEMBL4422809) Inhibition of HDAC8 in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate measured after 30 mins by fluorescence assay
- ChEMBL_2197604 (CHEMBL5110120) Inhibition of HDAC1/HDAC2 in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate and measured by fluorometric method
- ChEMBL_2224821 (CHEMBL5138334) Displacement of [3H]-acetylated histones HDAC6 derived from human K562 cell nuclear extract incubated for 10 mins by liquid scintillation counting method
- ChEMBL_2273494 Inhibition of human recombinant STAT3 assessed as reduction in DNA binding activity with HepG2 nuclear extract incubated for 1 hr by ELISA assay
- ChEMBL_1769411 (CHEMBL4221523) Inhibition of HDAC1/CoREST3 in HEK293 whole cell extract using fluorescent acetylated histone peptide as substrate after 60 mins by fluorescence based assay
- ChEMBL_2052178 (CHEMBL4707179) Inhibition of HDAC1 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate measured after 2 hrs by fluorescence based assay
- ChEMBL_2224822 (CHEMBL5138335) Displacement of [3H]-acetylated histones from HDAC1 derived from human K562 cell nuclear extract incubated for 10 mins by liquid scintillation counting method
- ChEMBL_2224823 (CHEMBL5138336) Displacement of [3H]-acetylated histones from HDAC3 derived from human K562 cell nuclear extract incubated for 10 mins by liquid scintillation counting method
- ChEMBL_2355383 Inhibition of HDAC in human HeLa cell nuclear extract using Kac fluorogenic peptide as substrate containing residues 379-382 of p53 by fluorescence assay
- ChEMBL_2452840 Inhibition of HDAC2 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_2452841 Inhibition of HDAC4 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_2452842 Inhibition of HDAC7 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_2452843 Inhibition of HDAC9 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_2452845 Inhibition of HDAC11 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_306888 (CHEMBL828694) In vitro inhibitory concentration against histone deacetylase of DU-145 prostate cell nuclear extract as deacetylation of biotinylated [3H]-acetyl histone H4 peptide
- ChEMBL_65296 (CHEMBL676785) Inhibitory activity evaluated from soluble cell extract of human endothelia constitutive enzyme (Endothelial nitric oxide synthase) and partially purified by DEAE-sepharose chromatography
- ChEMBL_1590578 (CHEMBL3829047) Inhibition of HDAC in human HeLa cell nuclear extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate after 30 mins by fluorescence assay
- ChEMBL_2310516 Inhibition of recombinant Top1 in Leishmania donovani Ag83 whole cell extract assessed as relaxation of supercoiled pBluescript SK(+) DNA measured by agarose gel electrophoresis analysis
- ChEMBL_629937 (CHEMBL1109181) Inhibition of NFkappa p50 isolated from nuclear extract of human HeLa cells assessed as blockade of binding to biotinylated consesus sequence by chemiluminescence assay
- ChEMBL_629938 (CHEMBL1109182) Inhibition of NFkappa p65 isolated from nuclear extract of human HeLa cells assessed as blockade of binding to biotinylated consesus sequence by chemiluminescence assay
- ChEMBL_749381 (CHEMBL1785171) Inhibition of NFkappa p65 in nuclear extract of human HeLa cells assessed as blockade of NFkappa p65 binding to biotinylated-consesus sequence by ELISA
- ChEMBL_809836 (CHEMBL2014773) Inhibition of HDAC8 in human HeLa cells nuclear extract using Arg-His-Lys(Ac)-Lys(Ac)-AMC as substrate after 30 mins by spectrophotometry
- ChEMBL_2050946 (CHEMBL4705645) Binding affinity to human full-length N-terminal His6-tagged Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract by isothermal titration calorimetry
- ChEMBL_2067854 (CHEMBL4723107) Inhibition of tyrosinase in human HBL cell extract using L-DOPA as substrate measured every 10 mins for 1 hr by MBTH based spectrophotometric method
- ChEMBL_2171587 (CHEMBL5056721) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as fluorogenic substrate measured after 1 hr by flourescence plate reader method
- ChEMBL_34793 (CHEMBL643770) In vitro inhibitory activity against Angiotensin I converting enzyme (ACE) isolated from rabbit lung extract using hippuryl-L-histidyl-L-leucine (HHL) as the substrate
- ChEMBL_653215 (CHEMBL1226418) Inhibition of eIF4A-mediated cap-dependent protein synthesis in FF-HCV-Ren mRNA transfected Swiss mouse Krebs2 cell extract by [35S]methionine metabolic labeling study
- ChEMBL_714888 (CHEMBL1663834) Inhibition of NF-kappaB p65 isolated from nuclear extract of human HeLa cells assessed as blockade of binding to biotinylated consesus sequence by chemiluminescence assay
- ChEMBL_769098 (CHEMBL1832655) Inhibition of STAT3 in mouse NIH3T3/vSrc nuclear extract assessed as disruption of the Stat3-DNA complex pre-incubated for 30 mins by EMSA analysis
- ChEMBL_1437098 (CHEMBL3381112) Inhibition of HDAC1/HDAC2 in human HeLa cell extract incubated for 5 mins prior to substrate addition measured after 30 mins by microtitre plate reader analysis
- ChEMBL_1634410 (CHEMBL3877202) Inhibition of immobilized N-LY294002 bead binding to BRD2 (unknown origin) expressed in HEK293T cell nuclear extract incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1634411 (CHEMBL3877203) Inhibition of immobilized N-LY294002 bead binding to BRD3 (unknown origin) expressed in HEK293T cell nuclear extract incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1763006 (CHEMBL4198253) Inhibition of HDAC1/2 in human K562 nuclear extract using (QSY-7)-RGGRGLGK(Ac)-GGARRHRK(TAMRA)NH2 as substrate incubated for 30 mins by fluorescence assay
- ChEMBL_2050947 (CHEMBL4705646) Binding affinity to human full-length N-terminal His6-tagged prephosphorylated Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract by isothermal titration calorimetry
- ChEMBL_2310567 Inhibition of recombinant Top1 in Antimony resistant Leishmania donovani BHU575 whole cell extract assessed as relaxation of supercoiled pBluescript SK(+) DNA measured by agarose gel electrophoresis analysis
- ChEMBL_984068 (CHEMBL2434426) Inhibition of GST-tagged full length XIAP (unknown origin) assessed as caspase 3/7 reactivation in S-100 cell extract after 4 hrs by fluorescence assay
- ChEMBL_1477045 (CHEMBL3428001) Inhibition of HDAC in human HeLa cell nuclear extract using Fluor de Lys as substrate incubated with compound for 30 mins by microtiter-plate reading flourimeter analysis
- ChEMBL_1763011 (CHEMBL4198258) Inhibition of HDAC in human HeLa nuclear extract preincubated for 10 mins followed by Boc-Lys(acetyl)-AMC substrate addition measured after 30 mins by fluorescence assay
- Enzyme Activity Assay α-Amylase activity was assayed with the chromogenic substrate RBB-starch. An enzyme aliquot was incubated (20 min, 26°C) with 0.3% RBB-starch in 0.1 M Britton-Robinson buffer at the pH optimum of the enzyme (6.5 for Aca s 4 and A. siro extract, 7.0 for D. farinae extract, 6.9 for PPA) or at pH 4.5-9.0 (pH profiling). The reaction was stopped with 0.2 M NaOH, the mixture was centrifuged (10000 g, 10 min), and the absorbance at 620 nm of the supernatant was measured against a control sample (incubated in the absence of enzyme/extract).
- ChEMBL_1555945 (CHEMBL3767300) Inhibition of HDAC1/HDAC2 in human HeLa cells nuclear extract preincubated for 5 mins before Boc-Lys (acetyl)-AMC substrate addition for 30 mins by microplate reader analysis
- ChEMBL_1835547 (CHEMBL4335680) Inhibition of TNF alpha stimulated NF-KappaB p65 in human HeLa nuclear extract assessed as decrease in NF-KappaB translocation to nucleus measured after 5 hrs by ELISA
- ChEMBL_1913618 (CHEMBL4416201) Binding affinity to CDPK4 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr in presence of ATP-competitive kinase inhibitor by kinobeads based pull down assay
- ChEMBL_1913620 (CHEMBL4416203) Binding affinity to CDPK1 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr in presence of ATP-competitive kinase inhibitor by kinobeads based pull down assay
- ChEMBL_2260085 (CHEMBL5215096) Inhibition of HDAC1 in human HeLa cell nuclear extract using Bos-Lys(acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition incubated for 30 mins
- ChEMBL_881914 (CHEMBL2212517) Inhibition of topoisomerase-1 in human U251 cells assessed as inhibition of hypoxia-induced HIF-1alpha accumulation in nuclear extract after 6 to 24 hrs by immunoblot analysis
- ChEBML_1769460 Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys-(Ac)-AMC as substrate preincubated for 15 mins followed by substrate addition measured after 60 mins by fluorescence assay
- ChEMBL_1432943 (CHEMBL3383918) Inhibition of 5-LO in human PMNL S100 extract using arachidonic acid as substrate incubated for 15 mins prior to substrate addition measured after 10 mins by HPLC analysis
- ChEMBL_1618726 (CHEMBL3860895) Inhibition of HDAC1/HDAC2 in human HeLa cell nuclear extract preincubated for 20 mins followed by addition of HDAC green as substrate measured after 60 mins by fluorescence analysis
- ChEMBL_2057508 (CHEMBL4712509) Inhibition of oligonucleotide [32P]-labelled 5'-AGCITCATTTCCCGTAAATCCCTA probe binding to STAT1 homodimer in mouse NIH3T3 nuclear extract preincubated for 30 mins followed by hSIE probe addition by EMSA analysis
- ChEMBL_2093845 (CHEMBL4775108) Inhibition of human HeLa nuclear extract derived ATM using glutathioneS-transferase-p53N66 as substrate preincubated for 10 mins followed by ATP addition and measured after 1 hr by ELISA
- ChEMBL_2114766 (CHEMBL4823707) Inhibition of DNA-PK isolated from human HeLa nuclear extract using full length His-tagged p53 as substrate measured after 75 mins in presence of ATP by HTRF assay
- ChEMBL_2277623 Inhibition of human HDAC4 in HeLa cells extract incubated for 5 mins followed by substrate addition and measured after 15 mins using Fluor-de-Lys as substrate by spectrofluorometric analysis
- ChEMBL_2277624 Inhibition of human HDAC2 in HeLa cells extract incubated for 5 mins followed by substrate addition and measured after 15 mins using Fluor-de-Lys as substrate by spectrofluorometric analysis
- ChEMBL_2277625 Inhibition of human HDAC7 in HeLa cells extract incubated for 5 mins followed by substrate addition and measured after 15 mins using Fluor-de-Lys as substrate by spectrofluorometric analysis
- ChEMBL_2277626 Inhibition of human HDAC8 in HeLa cells extract incubated for 5 mins followed by substrate addition and measured after 15 mins using Fluor-de-Lys as substrate by spectrofluorometric analysis
- ChEMBL_1444367 (CHEMBL3372358) Inhibition of HDAC in human HeLa cell extract using Boc-Lys (acetyl)-AMC as substrate incubated for 10 mins prior to substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_1634379 (CHEMBL3877171) Inhibition of immobilized N-LY294002 bead binding to C-terminal Flag-tagged BRD4 (unknown origin) expressed in HEK293T cell nuclear extract incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1769460 (CHEMBL4221572) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys-(Ac)-AMC as substrate preincubated for 15 mins followed by substrate addition measured after 60 mins by fluorescence assay
- ChEMBL_1784393 (CHEMBL4255910) Inhibition of STAT3 DNA binding activity in mouse NIH/3T3 nuclear extract preincubated for 30 mins followed by [32P]hSIE addition measured after 30 mins by electrophoretic mobility shift assay
- ChEMBL_1797560 (CHEMBL4269677) Inhibition of ATR in human HeLa cell nuclear extract using GST-fused p53N66 as substrate preincubated for 10 mins followed by ATP addition and measured after 1 hr by ELISA
- ChEMBL_1867771 (CHEMBL4368746) Inhibition of HDAC4 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_1867772 (CHEMBL4368747) Inhibition of HDAC5 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_1867773 (CHEMBL4368748) Inhibition of HDAC7 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_1867774 (CHEMBL4368749) Inhibition of HDAC9 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_1867775 (CHEMBL4368750) Inhibition of HDAC6 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_2057507 (CHEMBL4712508) Inhibition of oligonucleotide [32P]-labelled 5'-AGCITCATTTCCCGTAAATCCCTA probe binding to STAT1/STAT3 heterodimer in mouse NIH3T3 nuclear extract preincubated for 30 mins followed by hSIE probe addition by EMSA analysis
- ChEMBL_2197605 (CHEMBL5110121) Inhibition of HDAC1 in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(triflouroacetyl)-AMC as substrate incubated for 30 mins and measured by fluorescence assay
- ChEMBL_2197606 (CHEMBL5110122) Inhibition of HDAC6 in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(triflouroacetyl)-AMC as substrate incubated for 30 mins and measured by fluorescence assay
- ChEMBL_2197607 (CHEMBL5110123) Inhibition of HDAC8 in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(triflouroacetyl)-AMC as substrate incubated for 30 mins and measured by fluorescence assay
- Biological Assay For mTOR enzyme activity assays, mTOR protein was isolated from HeLa cell cytoplasmic extract by immunoprecipitation, and activity determined essentially as described previously using recombinant PHAS-1as a substrate (ref 21).
- ChEMBL_1763428 (CHEMBL4198675) Inhibition of HDAC1/2 in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_1769470 (CHEMBL4221582) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 10 mins followed by substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_1769471 (CHEMBL4221583) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_1781393 (CHEMBL4252910) Inhibition of HDAC in human HeLa cell extract using Fluor de Lys-Green as substrate preincubated for 5 mins followed by substrate addition and measured after 1 hr by fluorescence assay
- ChEMBL_1847057 (CHEMBL4347598) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_1936352 (CHEMBL4482111) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate preincubated for 10 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_1936353 (CHEMBL4482112) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2232525 (CHEMBL5146297) Inhibition of CDK2 (unknown origin) in baculovirus infected Sf9 insect cell extract using histone H1 as substrate incubated for 10 mins in presence of [gamma-32P]ATP by radiometric scintillation assay
- ChEMBL_830578 (CHEMBL2061433) Inhibition of human Tdp1 in Tdp1-deficient chicken DT40 whole cell extract using 5'-[32P]-labeled single-stranded DNA oligonucleotide containing 3'-phosphotyrosine as substrate after 15 mins by PAGE analysis
- ChEMBL_1551388 (CHEMBL3762509) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 15 mins followed by substrate addition measured after 60 mins by fluorescence-based assay
- ChEMBL_1683516 (CHEMBL4033995) Inhibition of HDAC1/2 in human HeLa cell nuclear extract using COLOR DE LYS as substrate pretreated for 5 mins followed by substrate addition measured after 30 mins by UV-absorption method
- ChEMBL_1893047 (CHEMBL4394968) Inhibition of HDAC in human HeLa cell nuclear extract using fluor-de-lys-green as substrate preincubated for 10 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2050942 (CHEMBL4705641) Binding affinity to human full-length N-terminal His6-tagged Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract after 30 mins using FAM-labelled compound by fluorescence polarization assay
- ChEMBL_2057493 (CHEMBL4712494) Inhibition of oligonucleotide [32P]-labelled 5'-AGCITCATTTCCCGTAAATCCCTA probe binding to STAT3 SH2 domain in mouse NIH3T3/v-Src nuclear extract preincubated for 30 mins followed by hSIE probe addition by EMSA analysis
- ChEMBL_2213936 (CHEMBL5127068) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by microplate reader analysis
- ChEMBL_2268733 Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence based analysis
- ChEMBL_2268738 Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 15 mins followed by substrate addition and measured after 60 mins by fluorescence based analysis
- ChEMBL_2273479 Inhibition of recombinant STAT3 (unknown origin) expressed in baculovirus in Sf9 insect cells assessed as reduction in DNA binding activity with NIH3T3 nuclear extract incubated for 30 mins by electrophoretic mobility shift assay
- ChEMBL_2488040 Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 15 mins followed by substrate addition and measured after 60 mins by fluorescence based analysis
- ChEMBL_1553268 (CHEMBL3767500) Inhibition of HDAC1/2 in human HeLa cell nuclear extract using color de Lys as substrate preincubated for 5 mins followed by substrate addition measured after 30 min by microtiter plate reader analysis
- ChEMBL_1750439 (CHEMBL4185199) Inhibition of HDAC1 in Plasmodium falciparum 3D7 nuclear extract using Ac-RGK(Ac)-AMC fluorogenic peptide as substrate preincubated for 1 hr followed by substrate addition measured after 10 min by fluorescence assay
- ChEMBL_1760362 (CHEMBL4195370) Inhibition of HDAC in human HeLa cell nuclear extract using fluorogenic Boc-Lys(acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorescence-based assay
- ChEMBL_2050943 (CHEMBL4705642) Binding affinity to human full-length N-terminal His6-tagged prephosphorylated Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract after 30 mins using FAM-labelled compound by fluorescence polarization assay
- ChEMBL_2050950 (CHEMBL4705649) Inhibition of FAM-Bim peptide binding to human full-length N-terminal His6-tagged Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract measured after 30 mins by fluorescence polarization assay
- ChEMBL_2031685 (CHEMBL4685843) Inhibition of class 1 HDAC in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2273504 Inhibition of recombinant STAT3 (unknown origin) expressed in Escherichia coli BL21 (DE3) cells assessed as reduction in DNA binding activity with SK-MEL-5 nuclear extract incubated for 30 mins by electrophoretic mobility shift assay
- ChEMBL_2298866 Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2496980 Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2496998 Inhibition of HDAC1 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2496999 Inhibition of HDAC2 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2497000 Inhibition of HDAC3 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_887090 (CHEMBL2214315) Inhibition of Apaf-1-caspase 9-cytochrome c-caspase 3 complex in human HEK293 cytosolic extract using afc-DEVD as substrate preincubated for 30 mins before substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_2018354 (CHEMBL4671932) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC or Boc-Lys (triflouroacetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2260034 (CHEMBL5215045) Inhibition of HDAC-1 (unknown origin) expressed in Escherichia coli BL21 (DE3) in HeLa nuclear extract using KI177 as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by Bradford reagent method
- ChEMBL_1738015 (CHEMBL4153765) Inhibition of ATM derived from human HeLa cell nuclear extract using glutathione S-transferase p53N66 as substrate preincubated for 10 mins followed by ATP addition and subsequent incubation for 1 hr measured after 1.5 hrs by ELISA
- ChEMBL_1895949 (CHEMBL4397984) Inhibition of HDAC in human HeLa cell nuclear extract assessed as decrease in deacetylation of FLUOR DE LYS Green substrate preincubated for 10 mins followed by substrate addition and measured after 30 mins by fluorescence based assay
- ChEMBL_2260035 (CHEMBL5215046) Inhibition of HDAC-6 (unknown origin) expressed in Escherichia coli BL21 (DE3) in HeLa nuclear extract using Boc-Lys(TFA)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by Bradford reagent method
- ChEMBL_2475467 Inhibition of C-terminal 6His-tagged recombinant human STAT3 (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 10 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475468 Inhibition of C-terminal 6His-tagged recombinant human STAT3 (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475469 Inhibition of C-terminal 6His-tagged recombinant human STAT3 (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 60 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- Enzyme Assay ATR for use in the in vitro enzyme assay was obtained from HeLa nuclear extract (CIL Biotech, Mons, Belgium) by immunoprecipitation with rabbit polycolonal antiserum raised to amino acids 400-480 of ATR (Tibbetts R S et, al, 1999, GenesDev.13:152-157).
- ChEMBL_2475470 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- RORgamma Gal4 Reporter Gene Assay (FF) Cells were incubated for additional 16 h before firefly (FF) luciferase activities were measured sequentially in the same cell extract using a Dual-Light-Luciferase-Assay system (Dyer et al., Anal. Biochem. 2000, 282:158). All experiments were done at least in triplicates.
- RORgamma Gal4 Reporter Gene Assay (REN) Cells were incubated for additional 16 h before renilla (REN) luciferase activities were measured sequentially in the same cell extract using a Dual-Light-Luciferase-Assay system (Dyer et al., Anal. Biochem. 2000, 282:158). All experiments were done at least in triplicates.
- ChEMBL_2475471 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 C328A mutant (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475472 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 C426A mutant (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475473 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 C468A mutant (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475474 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 C542S mutant (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- HDAC enzyme inhibitionAssay HDAC enzyme inhibition assays were performed using purified HDACs 1-10 essentially as described in Beckers et al., 2007, Int. J. Cancer., 121:1138-48 and Perez-Balado et al., 2007, J. Med. Chem., 50:2497-2505. Inhibition assays using nuclear extract were performed essentially as described in Herman et al., 2006, Nat. Chem. Biol., 2:551-558. Briefly, the purified HDACs or nuclear extract were incubated with an acetylated substrate in the absence of the compound to be assayed and with increasing concentrations of the compound. The rate of substrate deacetylation was measured under each condition, and half-maximal inhibitory concentration with regard to each HDAC was determined by standard means.
- Inhibition Assay HDAC inhibition assays can be performed, e.g., in a cell, in a cell extract, or in a cell-free mixture. Exemplary HDAC inhibition assays are described in P rez-Balado et al., 2007, J. Med. Chem., 50:2497-2505; Herman et al., 2006, Nat. Chem. Biol., 2:551-558; and Beckers et al., 2007, Int. J. Cancer, 121:1138-48.
- Enzyme inhibition assay The nucleus extract was extracted from HDAC8 enzyme was expressed in Escherichia coli. Boc-Lys (acetyl)-AMC was used as the substrate of HDAC. SAHA which is the HDAC inhibitor on market was used as a positive control. The compounds were diluted to six concentrations (25, 5, 1, 0.2, 0.04 and 0.008 uM/L) to investigate their ability of inhibiting HDAC activity.
- Fluor de Lys Assay Fluor de Lys is a fluorescence based HDAC activity assay comprising a combination of fluorogenic Histone deAcetylase Lysyl substrate and a developer. The kit is a highly sensitive and convenient alternative to radiolabeled, acetylated histones or peptide/HPLC methods for the assay of histone deacetylases. This assay is based on the ability of HeLa nuclear extract, which is enriched in HDAC activity, to mediate the deacetylation of the acetylated lysine side chain of the Fluor de Lys substrate. The assay procedure requires two steps. First, incubation of the HeLa nuclear extract with the Fluor de Lys substrate results in substrate deacetylation and thus sensitizes it to the second step. In the second step, treatment of the deacetylated substrate with the Fluor de Lys developer produces a fluorophore. The substrate-developer reaction, under normal circumstances goes to completion in less than 1 min at 25 C.
- Fluorescent Activity Assay In vitro HDAC assays were performed using a HDAC fluorescent activity assay kit (Biomol, UK) according to the manufacturer's instructions. Compounds were reduced prior to analysis; 1 mM compound was reduced with 30 mM DTT in DMSO overnight at room temperature, protected from light. Reactions were then set up in a 96-well plate. For each reaction 10 ul compound (5x required concentration in assay buffer) was mixed with 15 ml diluted HeIa Nuclear Extract (30-fold in assay buffer). Serial dilutions were set up for each compound. Reactions containing HeIa extract only and assay buffer only were also set up. 25 ul diluted Fluor de Lys substrate (100-fold in assay buffer) was added to each reaction, which were then incubated at 37C for 1 hour. The reaction was stopped by addition of 50 ul Fluor de Lys Developer (20-fold dilution in assay buffer, plus TSA diluted 100-fold). The reactions were then incubated at room temperature for 10 minutes before fluorescence was measured.
- Fluorometric Activity Assay A FLUOR DE LYS fluorometric activity assay kit (HDAC source: HeLa cell nuclear extract) and a FLUOR DE LYS HDAC1 fluorometric drug discovery assay kit (HDAC source: human recombinant HDAC1) were purchased from Enzo Life Sciences (Farmingdale, NY). HDAC inhibition assay was performed according to the instruction manuals. All samples were dissolved in dimethyl sulfoxide, except for 1 and 2, which were dissolved in the assay buffers provided. All assays were performed in triplicate, and errors were calculated as standard deviation.
- Biological Activity Assay The KDM1A demethylase enzyme activity can obtained from mammalian cells or tissues expressing KDM1A from an endogenous or recombinant gene and purified or assayed from a whole cell extract. These methods can be used to determine the concentration of the disclosed compounds can inhibit fifty percent of the enzyme activity (IC50). In one aspect, the disclosed compounds exhibit inhibition fifty percent of the KDM1A enzyme activity at a concentration of less than 500 nM, less than 100 nM, less than 50 nM or less than 10 nM.
- CB1 and CB2 Binding Inhibition by Compounds Purified from Milicia excelsa (African Teak) The organic extract (8 g) from the stem barks of Milicia excelsa, obtained using the methods described in Example 1, was divided and loaded separately onto two pre-packed flash columns (120 g silica, particle size 32-60 μm, 4 cm×19 cm), then the column was eluted with the gradient as described in Example 5. A Diels-Alder adduct of a chalcone and prenylphenyl moiety was isolated from one of the active fractions and identified as Sanggenon C/D/.
- Tyrosinase Inhibition Assay Briefly, mushroom tyrosinase (1250 units/mL) and 2 mM L-tyrosine (0.07 mL) were added to a solution of phosphate buffer (0.1 M, pH 6.8, 0.09 mL) containing the test sample. The test mixture (0.2 mL) was incubated for 10 min at 37°C and the absorption due to the formation of dopachrome was monitored at 475 nm. The same mixture except for the plant extract was used as a control. Arbutin (hydroquinone-O-β-glucopyranoside) was used as a positive control. Each treatment was replicated three times.
- PDE3A Enzyme Inhibition Assay For the determination of the in vitro effect of example compounds on the PDE3A reactions 2 μl of the respective example compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3A cell extract from Sf9 cells overexpressing human full length PDE3A (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3A cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:5000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H]adenosine 3′, 5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac).
- FASN Inhibition FASN activity of the SKBr3 cell extract was determined by measuring either NADPH oxidation or the amount of thiol-containing coenzyme A (CoA) released during the fatty acid synthase reaction. The dye CPM (7-diethylamino-3-(4′-maleimidyl-phenyl)-4-methylcoumarin) contains a thiol reactive group that increases its fluorescence emission on reaction with the sulfhydryl group of CoA. The biochemical activities shown in TABLE 31 were determined using the fluorescence measurement of CoA release via a procedure described in Chung C. C. et al. (Assay and Drug Development Technologies, 2008, 6(3), 361-374).
- Method of Biotinylated Wild-Type STING Protein Into pRSF1b (Novagen) having altered multiple cloning site was inserted Escherichia coli BirA, and transfected to ECOS JM109, whereby pRH8/FLAG-BirA was constructed. pET21HH/His-Avi-SUMO-FLAG-hTMEM173(139-379XH232R) (which was constructed by the method mentioned in the Example 36) and pRH8/FLAG-BirA for Avi tag biotinylation were simultaneously transformed to ECO (trade name) Competent E. coli BL21(DE3) to prepare His-Avi-SUMO-FLAG-hSTING (139-379, H232R)-expressing cell line. The expressing cell line was added to LB medium (10 g/L Tryptone, 5 g/L Yeast Extract, 5 g/L NaCl) containing ampicillin (100 μg/L) and kanamycin (50 μg/L), and the mixture was pre-cultured at 30° C., and expanded to TB medium (12 g/L Tryptone, 24 g/L Yeast Extract, 4 mL/L Glycerol, 2.3 g/L KH2PO4, 12.5 g/L K2HPO4) containing the same antibiotics, and the mixture was cultured at 37° C. When the turbidity of the culture solution reached 500 KU, the culture temperature was reduced to 16° C., 0.1 mM isopropylthiogalactoside and 50 μM (+)-biotin were added thereto, and the mixture was cultured for additional 16 hr.The culture solution was centrifuged, the obtained fungus bodies were suspended in Lysis Buffer (50 mM TrisHCl, 150 mM NaCl, 20 mM Imidazole, 1 mg/mL Lysozyme, 5 U/mL SEM Nuclease, recombinant, Complete EDTA-free, pH7.6), and the protein was extracted by ultrasonic fragmentation. The reagent was added thereto so that the salt concentration of the extract was adjusted to 300 mM NaCl, and the supernatant was collected by centrifugation. The obtained supernatant was passed through NiNTA superflow Cartridge equilibrated with Wash Buffer (50 mM TrisHCl, 300 mM NaCl, 20 mM Imidazole, pH7.6), and the Cartridge was washed with Wash Buffer, and eluted with Elution Buffer (50 mM TrisHCl, 300 mM NaCl, 250 mM Imidazole, pH7.6).
- Electrophoretic Mobility Shift Assay (EMSA) His10-Stat3 was expressed in Sf9 cells from a baculovirus encoding the recombinant protein. A nuclear extract of the Sf9 cells was incubated with 32P-labeled high affinity c-fos sis inducible element (hSIE) either alone or in the presence of inhibitor. After 20 min of incubation, samples were electrophoresed on polyacrylamide gels. The gels were dried, exposed to a phosphorimager screen and scanned. IC50 values were derived from plots of spot intensity versus phosphopeptide concentration. The affinity of peptides to the SH2 domain is measured by the intensity of the radioactivity of the Stat3-DNA complex band in the electrophoresis gel.
- Electrophoretic Mobility Shift Assay (EMSA) STAT proteins were expressed in Sf9 cells from a baculovirus encoding the recombinant protein. A nuclear extract of the Sf9 cells was incubated with 32P-labeled high affinity c-fos sis inducible element (hSIE) either alone or in the presence of inhibitor. After incubation, samples were electrophoresed on polyacrylamide gels. The gels were dried, exposed to a phosphorimager screen and scanned. IC50 values were derived from plots of spot intensity versus phosphopeptide concentration. The affinity of peptides to the SH2 domain is measured by the intensity of the radioactivity of the Stat-DNA complex band in the electrophoresis gel.
- PDE3B Enzyme Inhibition Assay The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of example compounds on the PDE3B reactions 2 μl of the respective example compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3B cell extract from Sf9 cells overexpressing human full length PDE3B (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3B cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:6000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H]adenosine 3′, 5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3B activity vs log compound concentration.
- In vitro HDACs Inhibition Fluorescence Assay In brief, 10 μL of HeLa nuclear extract was mixed with various concentrations of target compounds (50 μL), SAHA, using 100% and none HDACs groups as control group, and the mixture. After incubation at 37 °C for 10 min, fluorogenic substrate Boc-Lys (acetyl)-AMC (40 μL) was added and then the mixture was incubated at 37 °C for 30 min. The mixture was stopped by addition of 100 μL of developer containing trypsin and TSA afterward. Over the next incubation at 37 °C for 20 min, fluorescence intensity was measured using a microplate reader at excitation and emission wavelengths of 390 and 460 nm, respectively.
- PDE3A Enzyme Inhibition The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of example compounds on the PDE3A reactions 2 μl of the respective example compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3A cell extract from Sf9 cells overexpressing human full length PDE3A (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3A cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:5000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H]adenosine 3′, 5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3A activity vs log compound concentration.
- PDE3A Enzyme Inhibition The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of test substances on the PDE3A reactions 2 μl of the respective test compound solution in DMSO (serial dilutions) were is placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3A cell extract from Sf9 cells overexpressing human full length PDE3A (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3A cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:5000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H] adenosine 3′,5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3A activity vs log compound concentration.
- PDE3B Enzyme Inhibition The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of example compounds on the PDE3B reactions 2 μl of the respective example compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3B cell extract from Sf9 cells overexpressing human full length PDE3B (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3B cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:6000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H]adenosine 3′, 5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3B activity vs log compound concentration.
- PDE3B Enzyme Inhibition The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of test substances on the PDE3B reactions 2 μl of the respective test compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3B cell extract from Sf9 cells overexpressing human full length PDE3B (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3B cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:6000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H] adenosine 3′,5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3B activity vs log compound concentration.
- Biological Activity Assay The DLK dissociation constants (Kd) have been determined in the KINOMEscan KdELECT Service at DiscoveRx. A fusion protein of full length of human DLK (amino acids 1-859) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The DLK fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).
- In Vitro Enzyme Assay ATR for use in the in vitro enzyme assay was obtained from HeLa nuclear extract (CIL Biotech, Mons, Belgium) by immunoprecipitation with rabbit polyclonal antiserum raised to amino acids 400-480 of ATR (Tibbetts R S et al, 1999, Genes Dev. 13:152-157) contained in the following buffer (25 mM HEPES (pH7.4), 2 mM MgCl2, 250 mM NaCl, 0.5 mM EDTA, 0.1 mM Na3V04, 10% v/v glycerol, and 0.01% v/v Tween 20). ATR-antibody complexes were isolated from nuclear extract by incubating with protein A-Sepharose beads (Sigma, #P3476) for 1 hour and then through centrifugation to recover the beads. In the well of a 96-well plate, 10 ATR-containing Sepharose beads were incubated with 1 μg of substrate glutathione S-transferase-p53N66 (NH2-terminal 66 amino acids of p53 fused to glutathione {circumflex over ( )}-transferase was expressed in E. coli) in ATR assay buffer (50 mM HEPES (pH 7.4), 150 mM NaCl, 6 mM MgCl2, 4 mM MnCl2, 0.1 mM Na3V04, 0.1 mM DTT, and 10% (v/v) glycerol) at 37° C. in the presence or absence of inhibitor. After 10 minutes with gentle shaking, ATP was added to a final concentration of 3 μM and the reaction continued at 37° C. for an additional 1 hour. The reaction was stopped by addition of IOOμ PBS and the reaction was transferred to a white opaque glutathione coated 96-well plate (NUNC #436033) and incubated overnight at 4° C. This plate was then washed with PBS/0.05%>(v/v) Tween 20, blotted dry, and analyzed by a standard ELISA (Enzyme-Linked Immunosorbent Assay) technique with a phospho-serine 15 p53 (16G78) antibody (Cell Signaling Technology, #9286).
- Binding Assay Binding constants for compounds of the present invention against the JH2 domain were determined by the following protocol for a KINOMEscan assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH2domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP_003322.3) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2 (JH2domain-pseudokinase) fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).
- Biochemical Activity Assay Protein was expressed and purified from E. coli BL21 DE3 Rosetta 2 (EMD Millipore) cells using standard techniques. Cells were grown in 2x yeast extract tryptone medium and expression was initiated via the addition of isopropyl -D-1-thiogalactopyranoside. Expression proceeded overnight at 18 ° C. Cells were harvested by centrifugation and subsequently lysed via sonication. Insoluble fraction was removed by centrifugation. Maltose binding protein (MBP) fusion proteins were purified on a dextrin sepharose column (GE Healthcare) and the MBP tag was removed using tobacco etch virus protease overnight during dialysis. Protein was further purified on a heparin column (GE Healthcare) and eluted using a NaCl gradient. Column fraction were pooled and further purified on a Superdex 75 gel filtration column (GE Healthcare). Protein was quantified using 280 nm absorbance. Protein was then flash frozen in liquid nitrogen and stored at 80 ° C. until use.
- RSV Polymerase Assay (RSVpol) Standard RSV polymerase assays were conducted in the presence of 3 μL extract of RSV-infected cells in a reaction buffer containing 50 mM tris-acetate pH 8, 120 mM K-acetate, 4.5 mM MgCl2, 5% glycerol, 2 mM EDTA, 50 μg/ml BSA, and 3 mM DTT. Varying concentration of NTPs were used to initiate RNA synthesis for 120 minutes at 30 degrees, and radioactive 33P GTP (15 μCi) was used as tracer. The reaction was stopped by adding 50 mM EDTA, and RNA samples were purified through G-50 size exclusion spin columns and phenol-chloroform extraction. The radio-labeled RNA products were resolved by electrophoresis on a 6% polyacrylamide TBE gel, and visualized and quantitated after being exposed on a phosphorImager screen. Polymerase inhibition experiments (IC50s) were conducted the same way in the presence of increasing concentration of NTP analogs.
- TYK2 JH12 Domain Binding Assay Binding constants for compounds of the present invention against the JH2 domain were determined by the following protocol for a KINOMVEscan® assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH12domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP_003322.3) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2 (JH2domain-pseudokinase) fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).
- TYK2 JH2 Domain Binding Assay Binding constants for compounds disclosed herein against the JH2 domain are determined by the following protocol for a KINOMEscan® assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH2domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP 003322.3) and the DNA binding domain of NFkB is expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts are prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2 (JH2domain-pseudokinase) fusion protein is labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′) fused to an amplicon for qPCR readout, which is added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).
- TYK2 JH2 Domain Binding Assay Binding constants for the compounds described herein against the JH2 domain were determined by the following protocol for a KINOMEscan® assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH2domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP_003322.3) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2 (JH2domain-pseudokinase) fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).
- Nedd8 Conjugation Inhibitory Activity A purified NAE (heterodimer of APPBP1 and UBA3) solution was prepared in the following manner. The human APPBP1 gene (NCBI Reference Sequence number: NM_003905) region corresponding to amino acids 1 to 534 of human APPBP1 protein (NCBI Reference Sequence number: NP_003896, full length: 534 amino acids) was inserted in pBacPAK9 (produced by Clontech) to construct a plasmid pBacPAK9-APPBP1 for expressing APPBP1 full-length protein having a His tag and a TEV protease-recognition sequence at the N-terminus. Next, the human UBA3 gene (NCBI Reference Sequence number: NM_003968) region corresponding to amino acids 1 to 463 of human UBA3 protein (NCBI Reference Sequence number: NP_003959, full length: 463 amino acids) was inserted in pBacPAK9 to construct a plasmid pBacPAK9-UBA3 for expressing UBA3 full-length protein. The pBacPAK9-APPBP1 or pBacPAK9-UBA3, and BacPAK6 DNA were cotransfected into insect cells (Sf9, produced by Clontech) to produce a recombinant baculovirus containing APPBP1 or UBA3 gene. The APPBP1 gene recombinant baculovirus was mixed with the UBA3 gene recombinant baculovirus, and the resulting mixture was used to infect Sf9 cells. The baculovirus-infected Sf9 cells were incubated at 28° C. with shaking for 72 hours in Grace's Insect Medium (produced by Gibco), and the collected cells were suspended in a lysis buffer (50 mM Tris-HCl, 200 mM NaCl, and 10% glycerol (pH 7.4)), followed by sonication. The sonicated cell solution was centrifuged (40,000×g, for 30 minutes) to obtain the supernatant as a crude extract. The crude extract was fractionated on a HisTrap HP column (produced by GE Healthcare) and a TALON Superflow column (produced by Clontech), followed by the addition of a TEV protease. Then, a His-tag cleavage reaction was performed at 4° C. overnight. The resulting solution was subjected to TALON Superflow column chromatography, and the unadsorbed fraction was collected. This fraction was applied to a HiLoad 16/60 Superdex 75 prep grade column equilibrated with 50 mM Tris-HCl, 200 mM NaCl, and 10% glycerol (pH 7.4), and fractionated. A fraction containing an APPBP1/UBA3 complex was concentrated to obtain a purified NAE solution. The purification above was performed entirely at 4° C. The purified NAE solution was stored at −80° C. until use.A purified GST-UBC12 solution was prepared in the following manner. The human UBC12 gene (NCBI Reference Sequence number: NM_003969) region corresponding to amino acids 1 to 183 of human UBC12 protein (NCBI Reference Sequence number: NP_003960, full length: 183 amino acids) was inserted in pGEX-4T-2 (produced by GE Healthcare) to construct a plasmid pGEX-UBC12 for expressing UBC12 full-length protein having a GST tag at the N-terminus. The pGEX-UBC12 was introduced into Escherichia coli (BL21 (DE3), produced by Stratagene), followed by culture at 37° C. for 2 hours in the presence of 1 mM isopropyl-beta-D-thiogalactopyranoside (produced by Sigma-Aldrich). The collected Escherichia coli was suspended in PBS, followed by sonication. The sonicated cell solution was centrifuged (40,000×g, for 5 minutes) to obtain the supernatant as a crude extract. A Glutathione Sepharose 4B carrier (produced by GE Healthcare) was added to the crude extract, and eluted with 50 mM Tris-HCl (pH 7.9), 150 mM NaCl, and a 10 mM reduced glutathione solution, followed by dialysis with 50 mM HEPES (pH 7.5) and a 0.05% BSA solution to obtain a purified GST-UBC12 solution. The purified GST-UBC12 solution was divided and stored at −80° C. until use.The Nedd8 conjugation inhibitory activity was measured using an AlphaScreen assay system. Each of the purified NAE solution and GST-UBC12 solution was diluted with an assay buffer (50 mM HEPES (pH 7.5), 5 mM MgCl2, 1 mM DTT, 0.05% BSA) and added to a 384-well plate (#3673, produced by Corning) containing the test compound. After reaction at room temperature for 30 minutes, a solution obtained by diluting ATP and Biotin-Nedd8
- Biochemical Assay The FASN enzyme was isolated from SKBr3 cells. SKBr3 is a human breast cancer cell-line with high levels of FASN expression. It is estimated that FASN comprises about 25% of the cytosolic proteins in this cell line. SKBr3 cells were homogenized in a dounce homogenizer then centrifuged for 15 minutes at 4C. to remove particulate matter. The supernatant was then analyzed for protein content, diluted to the appropriate concentration, and used to measure FASN activity. The presence of FASN was confirmed by western blot analysis. A similar method for isolation of FASN from SKBr3 cells is described in Teresa, P. et al. (Clin. Cancer Res. 2009; 15(24), 7608-7615).FASN activity of the SKBr3 cell extract was determined by measuring either NADPH oxidation or the amount of thiol-containing coenzyme A (CoA) released during the fatty acid synthase reaction. The dye CPM (7-diethylamino-3-(4 -maleimidyl-phenyl)-4-methylcoumarin) contains a thiol reactive group that increases its fluorescence emission.
- HDAC Activity screening Histone Deacetylase (HDAC) Inhibition Assay using Boc-Lys(Ac)-AMC Substrate: Inhibition of HDAC has been implicated to modulate transcription and to induce apoptosis or differentiation in cancer cells. The fluorometric assay provides a fast and fluorescence based method that eliminates radioactivity, extractions or chromatography, as used in traditional assays. The assay is based on two steps. First, the HDAC fluorometric substrate, which comprises an acetylated lysine side chain, is incubated with a sample containing HDAC activity (Mouse Liver Extract). Deacetylation of the substrate sensitizes the substrate; in the second step, treatment with the Trypsin, stop solution produces a fluorophore that can be easily analyzed using fluorescence plate reader. Assay was done in 96-well black microplate and total volume of the assay was 100 mL. Mouse liver enzyme (10 mg/mL) was diluted 1:6 with HDAC buffer. Enzyme cocktail was made of 10 mL of diluted enzyme and 30 mL of HDAC buffer. Forty m
- ICMT Biochemical Assay An ICMT enzymatic assay was developed using Promega's Methyltransferase-GloTMreagents. In the assay, ICMT catalyzes the methylation of the N-Acetyl-S-farnesyl-L-cysteine by transferring a methyl group from SAM to the farnesylated cysteine and further converts the SAM to SAH. The methyltransferase activity is measured based on the amount of SAH produced from the reaction through the use of coupling enzymes that convert the SAH to ATP. The MTase-Glo detection solution then catalyzes the formation of light from ATP.For the IC50 determination, the compounds were incubated for 30 minutes with 0.04 ug/well ICMT membrane extract from Sf-9 cells. A final concentration of 2.0 μM and 20 μM of SAM and peptide were added and further incubated for 90 minutes at room temperature before adding the MTase Glo and detection reagent. Reaction signals were detected using microplate readers on luminescent mode (Satire Tecan). The IC50 was determined by nonlinear regression, using GraphPad Prism version, 5.03.
- In Vitro Inhibition Assay This work was performed at the MDS Pharma Services, Pharmacology Laboratories, Taiwan. The assay was an in vitro evaluation of the ability of an extract or a pure compound to inhibit the steroid 5alpha -reductase enzyme from metabolizing testosterone into dihydrotestosterone. This is an enzyme-immunoassay (EIA) for quantitative determination of testosterone in human serum or plasma. The significance of this type of inhibition is that it can lead to eradication of benign prostatic hyperplasia (BPH). Two distinct isozymes are found in mice, rats, monkeys and humans: type 1 and II. Each of these isozymes is differentially expressed in tissues and developmental stages. In human, type 1 steroid 5alpha -reductase is predominant in the sebaceous glands of most regions of skin, including scalp and liver and is responsible for approximately one third of circulating DHT. Inhibitors of steroid 5alpha -reductase may be of benefit in the treatment of androgenetic alopecia.
- Inhibition of Histone Deacetylase Enzymatic Assay For deacetylase assays, 20,000 cpm of the [3H]-metabolically labeled acetylated histone substrate (M. Yoshida et al., J. Biol. Chem. 265(28): 17174-17179 (1990)) is incubated with 30 ug of H446 nuclear extract or an equivalent amount of the cloned recombinant hHDAC-1 for 10 minutes at 37 C. The reaction is stopped by adding acetic acid (0.04 M, final concentration) and HCl (250 mM, final concentration). The mixture is extracted with ethyl acetate and the released [3H]-acetic acid was quantified by scintillation counting. For inhibition studies, the enzyme is preincubated with compounds at 4 C. for 30 minutes prior to initiation of the enzymatic assay. IC50 values for HDAC enzyme inhibitors are determined by performing dose response curves with individual compounds and determining the concentration of inhibitor producing fifty percent of the maximal inhibition. Alternatively, the following protocol is used to assay the compounds of the invention.
- Inhibitory Assay For the measurement of CH24H inhibitory activity, using the human CH24H lysate prepared in Experimental Example 2, the amount of 24-HC produced from cholesterol by catalytic activity of CH24H was measured in the presence of a test compound, and compared with that measured in the absence of the test compound. That is, a test compound solution at various concentrations was mixed with a reaction buffer (50 mM potassium phosphate containing 0.1% BSA and Complete, EDTA-free, pH 7.4) and human CH24H lysate. Then, [14C] cholesterol (53 mCi/mmol specific activity, 15 μM) was added, and CH24H reaction was performed at 37° C. for 5 hr. After completion of the reaction, a quenching solution consisting of chloroform/methanol/distilled water (2:2:1 v/v) was added, and the resulting 24-HC was extracted by shaking. The extract was applied to silica gel thin layer chromatography (ethyl acetate:toluene=4:6), and the obtained 14C-24HC fraction was measured with BAS2500 (Fujifilm Corporation).
- Inhibitory Assay The compounds of the present invention obtained in Production Examples were examined for the inhibitory effect on human and rat VAP-1 enzyme (SSAO) by the following method.The VAP-1 enzyme (SSAO) activity in both human and rat was measured by a radiochemical-enzyme assay using 14C-benzylamine as an artificial substrate. Human or rat VAP-1 was cloned from the cDNA library and expressed in a cell. The cell extract was preincubated with a test compound solution (final concentration 1×10−7-1×10−10 mol/L) at room temperature for 20 minutes. Then, 14C-benzylamine (final concentration 1×10−5 mol/L) was added, and the mixture was incubated at a final volume of 200 μL at 37° C. for 2 hours. The enzyme reaction was stopped by addition of 2 mol/L (200 μL) citric acid. The oxidation product was extracted with 1 mL toluene/ethyl acetate (1:1), and the radioactivity thereof was measured by a liquid scintillation counter.
- Binding Assay The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA)]. Extracts were used directly in binding assays without any enzyme purification steps at a ≥10,000-fold overall stock dilution (final DNA-tagged enzyme concentration<0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points.
- Biochemical Assay The FASN enzyme was isolated from SKBr3 cells. SKBr3 is a human breast cancer cell-line with high levels of FASN expression. It is estimated that FASN comprises about 25% of the cytosolic proteins in this cell line. SKBr3 cells were homogenized in a dounce homogenizer then centrifuged for 15 minutes at 4° C. to remove particulate matter. The supernatant was then analyzed for protein content, diluted to the appropriate concentration, and used to measure FASN activity. The presence of FASN was confirmed by western blot analysis. A similar method for isolation of FASN from SKBr3 cells is described in Teresa, P. et al. (Clin. Cancer Res. 2009; 15(24), 7608-7615). FASN activity of the SKBr3 cell extract was determined by measuring either NADPH oxidation or the amount of thiol-containing coenzyme A (CoA) released during the fatty acid synthase reaction. The dye CPM (7-diethylamino-3-(4'-maleimidyl-phenyl)-4-methylcoumarin) contains a thiol reactive group that increases its fluorescence emission on reaction with the sulfhydryl group of CoA.
- DLK Kd Determinations A fusion protein of full length of human DLK (amino acids 1-859) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The DLK fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′, SEQ ID NO:1) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).Streptavidin-coated magnetic beads (Dynal M280) were treated with a biotinylated small molecule ligand for 30 minutes at room temperature to generate affinity resins the binding assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific binding.
- In Vitro Assay In vitro HDAC assays were performed using a HDAC fluorescent activity assay kit (Biomol, UK) according to the manufacturer's instructions. Compounds were reduced prior to analysis; 1 mM compound was reduced with 30 mM DTT in DMSO overnight at room temperature, protected from light. Reactions were then set up in a 96-well plate. For each reaction 10 μl compound (5x required concentration in assay buffer) was mixed with 15 ml diluted HeIa Nuclear Extract (30-fold in assay buffer). 25 μl diluted Fluor de Lys substrate (100-fold in assay buffer) was added to each reaction, which were then incubated at 37 C for 1 hour. The reaction was stopped by addition of 50 μl Fluor de Lys Developer (20-fold dilution in assay buffer, plus TSA diluted 100-fold). The reactions were then incubated at room temperature for 10 minutes before fluorescence was measured using a CytoFluor Il Fluorescence Multiwell Plate Reader and CytoFluor Il software with filters set at 360 nM for excitation and 460 nM for emission.
- In Vitro Assay Nuclear extract preparations and DNA-binding activity/electrophoretic mobility shift assay (EMSA) were carried out as previously described (Zhang X, et al., (2010) Biochem Pharmacol 79:1398-409; Zhang X, et al., (2012) Proc Natl Acad Sci USA 109:9623-8). The 32P-labeled oligonucleotide probes used were hSIE (high affinity sis-inducible element from the c-fos gene, m67 variant, 5′-AGCTTCATTTCCCGTAAATCCCTA) (SEQ ID NO: 1) that binds Stat1 and Stat3 and MGFe (mammary gland factor element from the bovine β-casein gene promoter, 5′-AGATTTCTAGGAATTCAA) (SEQ ID NO: 2) for Stat1 and Stat5 binding. Except where indicated, nuclear extracts were pre-incubated with compound for 30 min at room temperature prior to incubation with the radiolabeled probe for 30 min at 30° C. before subjecting to EMSA analysis. Where appropriate, bands corresponding to Stat3:DNA complexes were scanned and quantified for each concentration of compound using ImageJ and plotted as percent of control (DMSO) against concentration of compound, from which the IC50 values were derived.
- TDP2 Assays with Whole Cell Extracts Ten-million cells (1 x 107), either human, chicken DT40 wild type, or knockout for TDP2 and complemented with human TDP2, were collected, washed, and centrifuged. Cell pellets were then resuspended in 100 uL of CelLytic M cell lysis reagent (SIGMA-Aldrich C2978). After 15 min on ice, lysates were centrifuged at 12 000g for 10 min, and supernatants were transferred to a new tube. Protein concentrations were determined using a Nanodrop spectrophotometer (Invitrogen), and whole cell extracts were stored at -80 °C. The TY19 single-stranded DNA oligonucleotide containing a 5'-phosphotyrosine (see above) was incubated at 1 nM with 10-20 ug/mL of whole cell extract to obtain 30-40% cleavage in the absence or presence of inhibitor for 15 min at RT in the reaction buffer containing 50 mM Tris-HCl, pH 7.5, 80 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 40 ug/mL BSA, and 0.01% Tween 20. Reactions were terminated and samples analyzed.
- Biochemical Activity Assay The FASN enzyme was isolated from SKBr3 cells. SKBr3 is a human breast cancer cell-line with high levels of FASN expression. It is estimated that FASN comprises about 25% of the cytosolic proteins in this cell line. SKBr3 cells were homogenized in a dounce homogenizer then centrifuged for 15 minutes at 4° C. to remove particulate matter. The supernatant was then analyzed for protein content, diluted to the appropriate concentration, and used to measure FASN activity. The presence of FASN was confirmed by western blot analysis. ASN activity of the SKBr3 cell extract was determined by measuring either NADPH oxidation or the amount of thiol-containing coenzyme A (CoA) released during the fatty acid synthase reaction. The dye CPM (7-diethylamino-3-(4′-maleimidyl-phenyl)-4-methylcoumarin) contains a thiol reactive group that increases its fluorescence emission on reaction with the sulfhydryl group of CoA. CoA is a byproduct of the FASN reaction, 8 molecules of CoA are released for every molecule of palmitate produced. Reaction of the CoA thiol group with CPM results in fluorescence emission at 405/530 nM.
- Biological Activity Assay Assaying the inhibition of KDM1A can be determined in vitro, in cultured cells, and in animals. There are a variety of spectrophotometric methods to detect the results of demethylation of methylated lysines, viz., detecting the products of KDM1A demethylase oxidative activity on a peptide fragment of at least 18 amino acid representing the N-terminus of the histone H3 substrate that contains a monomethyl at the fourth lysine residue. Hydrogen peroxide, one product of the KDM1A demethylase reaction, reacts with horseradish peroxidase and dihydroxyphenoxazine (ADHP) to produce the fluorescent compound resorufin (excitation=530-560 nm:emission=590 nm). The KDM1A demethylase enzyme activity can obtained from mammalian cells or tissues expressing KDM1A from an endogenous or recombinant gene and purified or assayed from a whole cell extract. These methods can be used to determine the concentration of the disclosed compounds can inhibit fifty percent of the enzyme activity (IC50). In one aspect, the disclosed compounds exhibit inhibition fifty percent of the KDM1A enzyme activity at a concentration of less than 500 nM, less than 100 nM, less than 50 nM or less than 10 nM.
- STING-Binding Assay The above-mentioned biotinylated STING protein (wild-type (WT)) was prepared by the following method. The culture solution was centrifuged, the obtained fungus bodies were suspended in Lysis Buffer (50 mM TrisHCl, 150 mM NaCl, 20 mM Imidazole, 1 mg/mL Lysozyme, 5 U/mL SEM Nuclease, recombinant, Complete EDTA-free, pH7.6), and the protein was extracted by ultrasonic fragmentation. The reagent was added thereto so that the salt concentration of the extract was adjusted to 300 mM NaCl, and the supernatant was collected by centrifugation. The obtained supernatant was passed through NiNTA superflow Cartridge equilibrated with Wash Buffer (50 mM TrisHCl, 300 mM NaCl, 20 mM Imidazole, pH7.6), and the Cartridge was washed with Wash Buffer, and eluted with Elution Buffer (50 mM TrisHCl, 300 mM NaCl, 250 mM Imidazole, pH7.6). The eluate was passed through HiLoad 26/60 Superdex 200 pg column equilibrated with Storage Buffer (50 mM TrisHCl, 150 mM NaCl, pH7.6), and the eluted fraction was collected as biotinylated His-Avi-SUMO-FLAG-hSTING (139-379, H232R). The protein concentration was measured using BCA protein assay kit, and the fraction was cryopreserved at −80° C. until used.
- TYK2 JH2 Domain Binding Assay The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA)]. Extracts were used directly in binding assays without any enzyme purification steps at a ≥10,000-fold overall stock dilution (final DNA-tagged enzyme concentration <0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO
- Inhibition Assays Class I PI3-Ks can be either purchased (p110α/p85α, p110β/p85α, p110δ/p85α from Upstate, and p110γ from Sigma) or expressed as previously described (Knight et al., 2004). IC50 values are measured using either a standard TLC assay for lipid kinase activity (described below) or a high-throughput membrane capture assay. Kinase reactions are performed by preparing a reaction mixture containing kinase, inhibitor (2% DMSO final concentration), buffer (25 mM HEPES, pH 7.4, 10 mM MgCl2), and freshly sonicated phosphatidylinositol (100 μg/mL). Reactions are initiated by the addition of ATP containing 10 μCi of γ-32P-ATP to a final concentration of 10 or 100 μM and allowed to proceed for 5 minutes at room temperature. For TLC analysis, reactions are then terminated by the addition of 105 μL 1N HCl followed by 160 μL CHCl3:MeOH (1:1). The biphasic mixture is vortexed, briefly centrifuged, and the organic phase is transferred to a new tube using a gel loading pipette tip precoated with CHCl3. This extract is spotted on TLC plates and developed for 3-4 hours in a 65:35 solution of n-propanol:1M acetic acid.
- VEGFR tyrosine kinase inhibition In a representative synthetic procedure (compound 3), a mixture of 1 (330 mg, 2 mM) and 3-F3C-C6H4-NHNH2 (370 mg, 2.1 mM) in anisole (20 mL) was stirred at reflux for 10 h. The resulting dark yellow mixture was cooled down, diluted with 50 mL of EtOAc and washed with 2 · 25 mL of saturated aqueous NaHCO3. Organic phase was dried over anhydrous Na2SO4 and concentrated. Solid residue was triturated with 3 · 10 mL of a 3:1 mixture (v/v) of Et2O and EtOAc to give triazole 2 (352 mg, 58% yield). Intermediate 2 (304 mg, 1 mM) was treated with 4-Py-COCl (170 mg, 1.2 mM) in 10 mL of pyridine at room temperature. The resulting dark yellow solution was stirred at room temperature for 12 h, poured into 25 mL of saturated NaHCO3, extracted with 3 · 20 mL of EtOAc. The combined organic extract was washed with 3 · 10 mL of water, dried over anhydrous Na2SO4 and concentrated. Solid residue was triturated with 3 · 10 mL of Et2O. Toluene (50 mL) followed by P4S10 (2.22 g, 5 mM) was added to the r
- Chromatin Immunoprecipitation (CHIP) ChIP assay was performed using EZ ChIP Kit (Millipore, Billerica, Mass., USA) as per manufacturer's instruction. Briefly, after crosslinking with formaldehyde (1%), cell were lysed using SDS Lysis Buffer containing Protease Inhibitor Cocktail and nuclear extract were obtained. Then, crosslinked DNA was sheared using sonication; sheared DNA size was checked on agarose gel and ranged, in all cases, between 300-500 bp. An amount of 1/20 of the shared chromatin was kept as an input. Sheared chromatin was incubated with rabbit polyclonal Phospho-Stat5 (Tyr694) Antibody (Cell Signaling, Danvers, Mass., USA) at dilution 1:50 or rabbit polyclonal IgG at appropriate concentration. Incubation was performed at 4° C. overnight. The antibody/chromatin complex was precipitated using Protein G Agarose beads. After elution, the crosslink of protein/DNA was reversed, and DNA was purified using Spin Columns. PCR was performed using platinum Taq (Invitrogen Canada, Burlington, ON, Canada). Two sets of primers were designed to amplify two DNA segments that contain STAT5-binding sites located 1672 bp and 428 bp upstream the start sites of C-Myc and Cyclin-D1, respectively. The STAT5-binding site in the C-MYC promoter is characterized by a 4N spacer and has the sequence (ttcccccgaa), whereas the one in the Cyclin D1 promoter is characterized by a 3N spacer and has the sequence (ttcttggaa).
- Ecto-5'-Nucleotidase Inhibition Assay The ecto-5'-nucleotidase inhibition assay was performed according to our previously reported protocol [Raza et al., Med. Chem., 8:1133-1139]. The compounds were analyzed at final concentration of 0.1 mM prepared in assay buffer ((2 mM MgCl2, 10 mM Tris HCl and 1 mM CaCl2, pH 7.4). (2 mM MgCl2, 10 mM Tris HCl and 1 mM CaCl2, pH 7.4). The total 100 μL containing 10 μL of sample, 10 μL of h-e5'NT (6.94 μg/mL) protein extract or r-e5'NT (7.17 μg/mL) and 70 μL of assay buffer. Then the reaction mixture was allowed to incubate for 10 min at 37 °C. After incubation, 10 μL of substrate AMP (adenosine monophosphate) with final concentration 500 μM was added to start the enzymatic reaction. The mixture was again allowed to incubate for 30 min at 37 °C. Then the mixture was placed in water bath at 99 °C for 20 min to stop the enzymatic reaction by thermal denaturation of protein. The aliquots of 50 μL of this reaction mixture were transferred into CE mini vials and injected hydrodynamically into the capillary under pressure of 0.5 psi for 5 s, followed by the application of 15 kV voltage for separation of substrate and product peaks.
- Ecto-5'-Nucleotidase Inhibition Assay The enzymatic assay of both human and rat ecto-5'-nucleotidase were performed with slight modifications in the previously described method. The stock solution of compounds were prepared with concentration of 10mM in 10% DMSO and their activity were determined by diluting them further in assay buffer (1 mM CaCl2, 2 mM MgCl2 and 10 mM Tris HCl (pH 7.4) to 1mM concentration. The final assay volume 100 μL contained 70 μL of assay buffer (2 mM MgCl2, 1 mM CaCl2 and 10 mM Tris HCl, pH 7.4), 10 μL of compound and 10 μL of human e5'NT (6.94 μg/mL) protein extract or rat e5'NT (7.17 μg/mL). Then the reaction mixture was incubated for 10 min at 37 °C. Afterwards, 10 μL of AMP substrate was added to initiate the reaction. The reaction was stopped by providing temperature of 99 °C for 20 min. Aliquots of 50 μL of each reaction mixture was transferred into CE mini vial and hydrodynamically injected into the capillary by using pressure of 0.5 psi for 5s. The electrosmatic separation of substrate and product peaks occurred by applying voltage of 15 kV. The concentration of product i.e. adenosine was estimated by calculating the area under its absorbance peak at 260 nm.
- FASN Inhibition Assay Determination of FASN biochemical activity: The FASN enzyme was isolated from SKBr3 cells. SKBr3 is a human breast cancer cell-line with high levels of FASN expression. It is estimated that FASN comprises about 25% of the cytosolic proteins in this cell line. SKBr3 cells were homogenized in a dounce homogenizer then centrifuged for 15 minutes at 4° C. to remove particulate matter. The supernatant was then analyzed for protein content, diluted to the appropriate concentration, and used to measure FASN activity. The presence of FASN was confirmed by western blot analysis. A similar method for isolation of FASN from SKBr3 cells is described in Teresa, P. et al. (Clin. Cancer Res. 2009; 15 (24), 7608-7615). ASN activity of the SKBr3 cell extract was determined by measuring either NADPH oxidation or the amount of thiol-containing coenzyme A (CoA) released during the fatty acid synthase reaction. The dye CPM (7-diethylamino-3-(4′-maleimidyl-phenyl)-4-methylcoumarin) contains a thiol reactive group that increases its fluorescence emission on reaction with the sulfhydryl group of CoA. The biochemical activities shown in Tables C-1-C-3 were determined using the fluorescence measurement of CoA release via a procedure described in Chung C.C. et al. (Assay and Drug Development Technologies, 2008, 6 (3), 361-374).
- Inhibition Assay Class I PI3-Ks can be either purchased (p110α/p85α, p110β/p85α, p110 /p85α from Upstate, and p110γ from Sigma) or expressed as previously described (Knight et al., 2004). IC50 values are measured using either a standard TLC assay for lipid kinase activity (described below) or a high-throughput membrane capture assay. Kinase reactions are performed by preparing a reaction mixture containing kinase, inhibitor (2% DMSO final concentration), buffer (25 mM HEPES, pH 7.4, 10 mM MgCl2), and freshly sonicated phosphatidylinositol (100 ug/ml). Reactions are initiated by the addition of ATP containing 10 uCi of γ-32P-ATP to a final concentration 10 or 100 uM and allowed to proceed for 5 minutes at room temperature. For TLC analysis, reactions are then terminated by the addition of 105 ul 1N HCl followed by 160 ul CHCl3:MeOH (1:1). The biphasic mixture is vortexed, briefly centrifuged, and the organic phase is transferred to a new tube using a gel loading pipette tip precoated with CHCl3. This extract is spotted on TLC plates and developed for 3-4 hours in a 65:35 solution of n-propanol:1M acetic acid. The TLC plates are then dried, exposed to a phosphorimager screen (Storm, Amersham), and quantitated. For each compound, kinase activity is measured at 10-12 inhibitor concentrations representing two-fold dilutions from the highest concentration tested (typically, 200 uM).
- Spectrophotometrical In Vitro Assay The activity of compounds 1-19 and 21-23 as inhibitors of ACMSD1 was determined by measuring the conversion of 3OH-Anthranilic Acid into product (i.e., ACMS) in a spectrophotometrical in vitro assay.The pre-assay mixture consisting of 3-hydroxyanthranilic acid (3OH-HA), 3-hydroxyanthranilic acid, 3,4-diOxygenase (HAO), and a dialyzed crude extract of E. coli BL21 (DE3) cells expressing the recombinant enzyme, was incubated at 25° C. with monitoring of the increase in absorbance at 360 nm due to the formation of ACMS from 3OH-HA. After the reaction was completed within 2 mins, an aliquot of ACMSD1 solution (prepared and purified from Pichia Pastoris overexpressing the recombinant enzyme) was added, and the decrease in absorbance at 360 nm was followed at 15 second intervals. The effect of ACMS concentration on the enzyme activity was investigated by varying 3OH-HA concentration from 2 to 20 μM. Kinetic parameters were calculated from the initial velocity data by using the Lineweaver-Burk plot.The rate of the decrease in absorbance caused by ACMSD1 was calculated by subtracting that of the control reaction mixture without ACMSD from that described above. One unit of ACMSD activity was indicated as the amount of enzyme that converts 1 mmol of ACMS per minute at 25° C. The absence or a reduction of ACMSD1 activity (e.g., by using ACMSD inhibitors) results in a slow ACMS-spontaneous degradation (i.e., cyclization to form quinolic acid).
- TYK2 JH2 Domain Binding Assay Binding constants for compounds of the present invention against the JH2 domain were determined by the following protocol for a KINOMEscan assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH2domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP_003322.3) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2(JH2domain-pseudokinase) fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).Streptavidin-coated magnetic beads (Dynal M280) were treated with a biotinylated small molecule ligand for 30 minutes at room temperature to generate affinity resins for the binding assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific binding.The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA). Extracts were used directly in binding assays without any enzyme purification steps at a ≥10,000-fold overall stock dilution (final DNA-tagged enzyme concentration <0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. All reactions were performed in polypropylene 384-well plates. Each was a final volume of 0.02 mL. Assays were incubated with shaking for 1 hour at room temperature. Then the beads were pelleted and washed with wash buffer (1×PBS, 0.05% Tween 20) to remove displaced kinase and test compound. The washed based were re-suspended in elution buffer (1×PBS, 0.05% Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. qPCR reactions were assembled by adding 2.5 μL of kinase eluate to 7.5 μL of qPCR master mix containing 0.15 μM amplicon primers and 0.15 μM amplicon probe. The qPCR protocol consisted of a 10 minute hot start at 95° C., followed by 35 cycles of 95° C. for 15 seconds, 60° C. for 1 minute.
- ACMSD1 Inhibition Assay The pre-assay mixture consisting of 3-hydroxyanthranilic acid (3OH-HA), 3-hydroxyanthranilic acid, 3,4-diOxygenase (HAO), and a dialyzed crude extract of E. coli BL21 (DE3) cells expressing the recombinant enzyme, was incubated at 25° C. with monitoring of the increase in absorbance at 360 nm due to the formation of ACMS from 3OH-HA. After the reaction was completed within 2 mins, an aliquot of ACMSD1 solution (prepared and purified from Pichia Pastoris overexpressing the recombinant enzyme) was added, and the decrease in absorbance at 360 nm was followed at 15 second intervals. The effect of ACMS concentration on the enzyme activity was investigated by varying 3OH-HA concentration from 2 to 20 μM. Kinetic parameters were calculated from the initial velocity data by using the Lineweaver-Burk plot. The rate of the decrease in absorbance caused by ACMSD1 was calculated by subtracting that of the control reaction mixture without ACMSD from that described above. One unit of ACMSD activity was indicated as the amount of enzyme that converts 1 mmol of ACMS per minute at 25° C. The absence or a reduction of ACMSD1 activity (e.g., by using ACMSD inhibitors) results in a slow ACMS-spontaneous degradation (i.e., cyclization to form quinolic acid). The enzymatic activity was determined at a HAA concentration of 10 μM in the presence of the compounds in Table 1 below. The compounds were tested at the concentration of about 5 μM and 10 μM and the IC50 was calculated for compounds showing inhibitory activity higher than 50%.
- Luciferase Assay Estrogen receptor-negative CV-1 kidney cells are maintained in Dulbecco's modified Eagle's medium with 4.5 g/L glucose supplemented with 10% fetal bovine serum and 100 units/ml penicillin-streptomycin at 37° C. in a humidified 5% CO2 atmosphere. The cells are then plated in 6-well dishes at a density of 2×10^5 cells per well in phenol-red free Dulbecco's modified Eagle's medium containing 10% charcoal-dextran-stripped fetal bovine serum. CV-1 cells are transfected using LipofectAMINE reagent according to the manufacturer's protocol. Transfections containing 1.5 μg of reporter plasmid (containing ERE-tk-luciferase containing a single ERE cloned upstream of the thymidine kinase promoter and luciferase gene) and 0.5 μg of either ERα or ERβ expression vector (containing CMV-ERα or CMV-ERβ full length coding sequence respectively). The next day, cells receive no treatment (controls) or are treated with estradiol alone (1 nM) or estradiol plus a compound of the invention (at varying concentrations). After 16-24 hours, cells are harvested and assayed for luciferase activity. At the outset, cell monolayers are washed twice with ice-cold phosphate-buffered saline and incubated for 15 minutes in 250 μl of 1× cell culture lysis reagent (Promega, Madison, Wis.). Cell extracts are transferred to a fresh tube and assayed using the luciferase assay system (Promega). For each assay, 10 μl of extract is diluted with 90 μl of 1× cell culture lysis reagent. Luminescence is read using an AutoLumat LB953 luminometer.
- spectrophotometrical in vitro assay The pre-assay mixture consisting of 3-hydroxyanthranilic acid (30H-HA), 3-hydroxyanthranilic acid, 3,4-diOxygenase (HAO), and a dialyzed crude extract of E. coli BL21 (DE3) cells expressing the recombinant enzyme, was incubated at 25° C. with monitoring of the increase in absorbance at 360 nm due to the formation of ACMS from 3OH-HA. After the reaction was completed within 2 mins, an aliquot of ACMSD1 solution (prepared and purified from Pichia Pastoris overexpressing the recombinant enzyme) was added, and the decrease in absorbance at 360 nm was followed at 15 second intervals. The effect of ACMS concentration on the enzyme activity was investigated by varying 30H-HA concentration from 2 to 20 μM. Kinetic parameters were calculated from the initial velocity data by using the Lineweaver-Burk plot.The rate of the decrease in absorbance caused by ACMSD1 was calculated by subtracting that of the control reaction mixture without ACMSD from that described above. One unit of ACMSD activity was indicated as the amount of enzyme that converts 1 mmol of ACMS per minute at 25° C. The absence or a reduction of ACMSD1 activity (e.g., by using ACMSD inhibitors) results in a slow ACMS-spontaneous degradation (i.e., cyclization to form quinolic acid).The enzymatic activity was determined at a HAA concentration of 10 μM in the presence of the compounds in Table 1 below. The compounds were tested at the concentration of about 5 μM and 10 μM and the IC50 was calculated for compounds showing inhibitory activity higher than 50%.
- Inhibition Assay Class I PI3-Ks can be either purchased (p110α/p85α, p110β/p85α, p110δ/p85α from Upstate, and p110γ from Sigma) or expressed as previously described (Knight et al., 2004). IC50 values are measured using either a standard TLC assay for lipid kinase activity (described below) or a high-throughput membrane capture assay. Kinase reactions are performed by preparing a reaction mixture containing kinase, inhibitor (2% DMSO final concentration), buffer (25 mM HEPES, pH 7.4, 10 mM MgCl2), and freshly sonicated phosphatidylinositol (100 μg/ml). Reactions are initiated by the addition of ATP containing 10 μCi of γ-32P-ATP to a final concentration 10 or 100 μM and allowed to proceed for 5 minutes at room temperature. For TLC analysis, reactions are then terminated by the addition of 105 l 1N HCl followed by 160 μl CHCl3:MeOH (1:1). The biphasic mixture is vortexed, briefly centrifuged, and the organic phase is transferred to a new tube using a gel loading pipette tip precoated with CHCl3. This extract is spotted on TLC plates and developed for 3-4 hours in a 65:35 solution of n-propanol:1M acetic acid. The TLC plates are then dried, exposed to a phosphorimager screen (Storm, Amersham), and quantitated. For each compound, kinase activity is measured at 10-12 inhibitor concentrations representing two-fold dilutions from the highest concentration tested (typically, 200 μM). For compounds showing significant activity, IC50 determinations are repeated two to four times, and the reported value is the average of these independent measurements.
- Inhibition Assay Class I PI3-Ks can be either purchased (p110α/p85α, p110β/p85α, p110δ/p85α from Upstate, and p110γ from Sigma) or expressed as previously described (Knight et al., 2004). IC50 values are measured using either a standard TLC assay for lipid kinase activity (described below) or a high-throughput membrane capture assay. Kinase reactions are performed by preparing a reaction mixture containing kinase, inhibitor (2% DMSO final concentration), buffer (25 mM HEPES, pH 7.4, 10 mM MgCl2), and freshly sonicated phosphatidylinositol (100 μg/ml). Reactions are initiated by the addition of ATP containing 10 μCi of γ-32P-ATP to a final concentration of 10 or 100 μM and allowed to proceed for 5 minutes at room temperature. For TLC analysis, reactions are then terminated by the addition of 105 μL 1N HCl followed by 160 μL CHCl3:MeOH (1:1). The biphasic mixture is vortexed, briefly centrifuged, and the organic phase is transferred to a new tube using a gel loading pipette tip precoated with CHCl3. This extract is spotted on TLC plates and developed for 3-4 hours in a 65:35 solution of n-propanol:1M acetic acid. The TLC plates are then dried, exposed to a phosphorimager screen (Storm, Amersham), and quantitated. For each compound, kinase activity is measured at 10-12 inhibitor concentrations representing two-fold dilutions from the highest concentration tested (typically, 200 μM). For compounds showing significant activity, IC50 determinations are repeated two to four times, and the reported value is the average of these independent measurements.
- SPR Binding Measurements All SPR measurements with compounds were performed using a Biacore 3000 (GE Healthcare) at 25° C. Biotinylated ASGPR was immobilized typically at 2000-3000 resonance units (Ru) using either SA sensor chips (GE Healthcare) or custom sensor chips with Neutravidin (Pierce Biochemical) immobilized by standard amine coupling to CM5 sensor chips (GE Healthcare). The running buffer was HBS (10 mM HEPES, 150 mM NaCl), 20 mM CaCl2, 0.01% p20, 3% DMSO or 50 mM tris, 150 mM NaCl, 50 mM CaCl2), 0.01% p20, 3% DMSO pH 7.5. Compounds were diluted into running buffer at a concentration of 900 uM and serially diluted 3 fold to 3.7 uM. Compound solutions were injected at 50 ul/min for 1 min followed by a 1 min dissociation in duplicate for each concentration. For the multimeric conjugates (dimers, trimers), the conjugates were diluted in running buffer to concentrations of 100 nM or 10 nM and serially diluted. Conjugates were injected for 2 min and off rates were detected for 300 or 600 sec. After completion of off phase data the compounds were displaced using an injection of 900 uM GalNAc returning the receptor surface to the free state. All data was processed using Scrubber2 (Biologic Software, Inc.) to zero, align, reference and correct for excluded volume effects. KDs were determined by fitting the steady state binding responses for the compounds and single conjugated molecules in Scrubber2. KD for multimeric conjugates showing kinetic responses were processed in Scrubber2 and fit in BiaEval (GE Healthcare) to extract the on and off rate parameters in order to calculate KD. Values reflect standard deviations from multiple experiments.
- SPR binding assay All SPR measurements with compounds were performed using a Biacore 3000 (GE Healthcare) at 25° C. Biotinylated ASGPR was immobilized typically at 2000-3000 resonance units (Ru) using either SA sensor chips (GE Healthcare) or custom sensor chips with Neutravidin (Pierce Biochemical) immobilized by standard amine coupling to CM5 sensor chips (GE Healthcare). The running buffer was HBS (10 mM HEPES, 150 mM NaCl), 20 mM CaCl2, 0.01% p20, 3% DMSO or 50 mM tris, 150 mM NaCl, 50 mM CaCl2), 0.01% p20, 3% DMSO pH 7.5. Compounds were diluted into running buffer at a concentration of 900 uM and serially diluted 3 fold to 3.7 uM. Compound solutions were injected at 50 ul/min for 1 min followed by a 1 min dissociation in duplicate for each concentration. For the multimeric conjugates (dimers, trimers), the conjugates were diluted in running buffer to concentrations of 100 nM or 10 nM and serially diluted. Conjugates were injected for 2 min and off rates were detected for 300 or 600 sec. After completion of off phase data the compounds were displaced using an injection of 900 uM GalNAc returning the receptor surface to the free state. All data was processed using Scrubber2 (Biologic Software, Inc.) to zero, align, reference and correct for excluded volume effects. KDs were determined by fitting the steady state binding responses for the compounds and single conjugated molecules in Scrubber2. KD for multimeric conjugates showing kinetic responses were processed in Scrubber2 and fit in BiaEval (GE Healthcare) to extract the on and off rate parameters in order to calculate KD. Values reflect standard deviations from multiple experiments.
- TYK2 JH2 Domain Binding Assay Binding constants for compounds of the present invention against the JH2 domain were determined by the following protocol for a KINOMEscan assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH2domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP_003322.3) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2(JH2domain-pseudokinase) fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).Streptavidin-coated magnetic beads (Dynal M280) were treated with a biotinylated small molecule ligand for 30 minutes at room temperature to generate affinity resins the binding assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific binding.The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA)]. Extracts were used directly in binding assays without any enzyme purification steps at a ≥10,000-fold overall stock dilution (final DNA-tagged enzyme concentration <0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions performed in polypropylene 384-well plates. Each was a final volume of 0.02 mL. Assays were incubated with shaking for 1 hour at room temperature. Then the beads were pelleted and washed with wash buffer (1×PBS, 0.05% Tween 20) to remove displaced kinase and test compound. The washed based were re-suspended in elution buffer (1×PBS, 0.05% Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. qPCR reactions were assembled by adding 2.5 μL of kinase eluate to 7.5 μL of qPCR master mix containing 0.15 μM amplicon primers and 0.15 μM amplicon probe. The qPCR protocol consisted of a 10 minute hot start at 95° C., followed by 35 cycles of 95° C. for 15 seconds, 60° C. for 1 minute.
- TYK2 JH2 Domain Binding Assay Binding constants for compounds of the present invention against the JH2 domain were determined by the following protocol for a KINOMEscan assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH2domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP_003322.3) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2(JH2domain-pseudokinase) fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).Streptavidin-coated magnetic beads (Dynal M280) were treated with a biotinylated small molecule ligand for 30 minutes at room temperature to generate affinity resins the binding assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific binding.The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA)]. Extracts were used directly in binding assays without any enzyme purification steps at a ≥10,000-fold overall stock dilution (final DNA-tagged enzyme concentration <0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions performed in polypropylene 384-well plates. Each was a final volume of 0.02 mL. Assays were incubated with shaking for 1 hour at room temperature. Then the beads were pelleted and washed with wash buffer (lx PBS, 0.05% Tween 20) to remove displaced kinase and test compound. The washed based were re-suspended in elution buffer (lx PBS, 0.05% Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. qPCR reactions were assembled by adding 2.5 μL of kinase eluate to 7.5 μL of qPCR master mix containing 0.15 μM amplicon primers and 0.15 μM amplicon probe. The qPCR protocol consisted of a 10 minute hot start at 95° C., followed by 35 cycles of 95° C. for 15 seconds, 60° C. for 1 minute.
- DNAPK Enzyme Potency Assay (DNA-PK Enz) The inhibitory activity of compounds against DNAPK was determined by TR-FRET measuring a fluorescent labelled peptide substrate converting to a phosphorylated product. Fluorescently tagged peptide substrate were purchased from Thermo Fisher Scientific. 12 point half-log compound concentration-response curves, with a top concentration of 100 μM were generated from 10 mM stocks of compound solubilised in DMSO using an Echo 555 (Labcyte Inc., Sunnyvale, CA). All assays were preformed in white Greiner 1536 well low volume plates (Greiner Bio-One, UK), in a total reaction volume of 3 μL and 1% (v/v) final DMSO concentration. Enzymes and substrates were added separately to the compound plates and incubated at room temperature. The kinase reaction was then quenched by the addition of 3 μL of stop buffer. Stopped assay plates were read using a BMG Pherastar. IC50 values were calculated using a Genedata Screener software (Genedata, Inc., Basel, Switzerland).Full length human DNAPK protein was purified from HeLa cell extract by ion exchange. Initially DNAPK protein was incubated with compound for 30 minutes at room temperature in reaction buffer (50 mM Hepes pH 7.5, 0.01% Brij-35, 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, 2 g/mL Calf Thymus DNA). The reaction was then initiated by the addition of ATP and fluorescently tagged peptide substrate (Fluorescein-EPPLSQEAFADLWKK, Thermo Fisher Scientific). The kinase reaction (18 μM ATP, 35 pM DNAPK, 1.6 μM peptide substrate) was quenched after 40 minutes by the addition of 3 μL of stop buffer (20 mM Tris pH7.5, 0.02% sodium azide, 0.01% Nonidet-P40, 20 m EDTA, 4 nM Tb anti-phospho-p53 [Ser15] Antibody. The reaction was incubated for a further hour and the plates were read on a BMG Pherastar.
- FAAH Assay All reagents were purchased from Sigma-Aldrich unless specified. Human and Rat Fatty Acid Amide Hydrolase (FAAH) genes used in assay have been described by Patricelli et al. (Biochemistry. 1998, 37(43), 15177-87). The transmembrane domain-deleted Fatty Acid Amide Hydrolase (FAAH) genes were cloned into pET15b (Novagen, #69661) (human FAAH)/pET 28a (Novagen, #69864-3) (rat FAAH gene) plasmids and expressed in E coli BL21 DE3. Chaperone protein groEL-groES in pGRO7 plasmid (Takara Bio Inc, Japan) was co-expressed with Fatty Acid Amide Hydrolase (FAAH) to improve solubility of the protein expressed in E coli. The protein was expressed and enriched as described in Mileni et al. (Proc Natl Acad Sci USA. 2008, 105(35),12820-4). Briefly, the bacterial cultures in Luria Broth (2 L) were induced with arabinose (2 mM) and Isopropyl (β-D-1-thiogalactopyranoside, IPTG (1 mM), for 20 h at room temperature. The cultures were centrifuged at 1200×g for 10 min and the cell pellet was resuspended in 100 mL buffer containing of 20 mM NaPi (pH 7.4), 100 mM NaCl, Benzonase (500u), Aprotinin (1 μg/mL) and Leupeptin (1 μg/mL). Cells were lyzed by sonication (Amp 20%, Pulse 15 s×15, on ice) and the cell debris removed by centrifugation at 5000×g for 20 min. The supernatant was enriched via ultracentrifugation at 100,000×g for 1 h and the cell pellet was resuspended in 16 mL buffer containing 20 mM NaPi (pH 7.8), 500 mM NaCl, 1% Triton X-100. The resuspended cell extract was subjected to ultra-centrifugation at 100,000×g for 1 h and the enriched supernatant was used in the in vitro assay. All protein extraction steps were carried out on ice or at 4° C.
- TYK2 JH2 Domain Binding Assay Binding constants for compounds of the present invention against the JH2 domain were determined by the following protocol for a KINOMEscan assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH2 domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP_003322.3) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2 (JH2 domain-pseudokinase) fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).Streptavidin-coated magnetic beads (Dynal M280) were treated with a biotinylated small molecule ligand for 30 minutes at room temperature to generate affinity resins the binding assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific binding.The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA)]. Extracts were used directly in binding assays without any enzyme purification steps at a ≥10,000-fold overall stock dilution (final DNA-tagged enzyme concentration<0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions performed in polypropylene 384-well plates. Each was a final volume of 0.02 mL. Assays were incubated with shaking for 1 hour at room temperature. Then the beads were pelleted and washed with wash buffer (lx PBS, 0.05% Tween 20) to remove displaced kinase and test compound. The washed based were re-suspended in elution buffer (lx PBS, 0.05% Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. qPCR reactions were assembled by adding 2.5 μL of kinase eluate to 7.5 μL of qPCR master mix containing 0.15 μM amplicon primers and 0.15 μM amplicon probe. The qPCR protocol consisted of a 10 minute hot start at 95° C., followed by 35 cycles of 95° C. for 15 seconds, 60° C. for 1 minute.Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. The Kds were determined using a compound top concentration of 30,000 nM. Kd measurements were performed in duplicate.Binding constants (Kds) were calculated with a standard dose-response curve using the Hill equation.
- TYK2 JH2 Domain Binding Assay Binding constants for compounds of the present invention against the JH2 domain were determined by the following protocol for a KINOMEscan assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH2domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP_003322.3) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2 (JH2domain-pseudokinase) fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′ (SEQ ID NO: 1)) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).Streptavidin-coated magnetic beads (Dynal M280) were treated with a biotinylated small molecule ligand for 30 minutes at room temperature to generate affinity resins the binding assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific binding.The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA)]. Extracts were used directly in binding assays without any enzyme purification steps at a ≥10,000-fold overall stock dilution (final DNA-tagged enzyme concentration <0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions performed in polypropylene 384-well plates. Each was a final volume of 0.02 mL. Assays were incubated with shaking for 1 hour at room temperature. Then the beads were pelleted and washed with wash buffer (1×PBS, 0.05% Tween 20) to remove displaced kinase and test compound. The washed based were re-suspended in elution buffer (1×PBS, 0.05% Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. qPCR reactions were assembled by adding 2.5 μL of kinase eluate to 7.5 μL of qPCR master mix containing 0.15 μM amplicon primers and 0.15 μM amplicon probe. The qPCR protocol consisted of a 10 minute hot start at 95° C., followed by 35 cycles of 95° C. for 15 seconds, 60° C. for 1 minute.Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. The Kds were determined using a compound top concentration of 30,000 nM. Kd measurements were performed in duplicate.
- TYK2 JH2 Domain Binding Assay Binding constants for compounds of the present invention against the JH2 domain were determined by the following protocol for a KINOMEscan assay (DiscoveRx). A fusion protein of a partial length construct of human TYK2 (JH2domain-pseudokinase) (amino acids G556 to D888 based on reference sequence NP_003322.3) and the DNA binding domain of NFkB was expressed in transiently transfected HEK293 cells. From these HEK 293 cells, extracts were prepared in M-PER extraction buffer (Pierce) in the presence of Protease Inhibitor Cocktail Complete (Roche) and Phosphatase Inhibitor Cocktail Set II (Merck) per manufacturers' instructions. The TYK2 (JH2domain-pseudokinase) fusion protein was labeled with a chimeric double-stranded DNA tag containing the NFkB binding site (5′-GGGAATTCCC-3′) fused to an amplicon for qPCR readout, which was added directly to the expression extract (the final concentration of DNA-tag in the binding reaction is 0.1 nM).Streptavidin-coated magnetic beads (Dynal M280) were treated with a biotinylated small molecule ligand for 30 minutes at room temperature to generate affinity resins the binding assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific binding.The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA)]. Extracts were used directly in binding assays without any enzyme purification steps at a ≥10,000-fold overall stock dilution (final DNA-tagged enzyme concentration <0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions performed in polypropylene 384-well plates. Each was a final volume of 0.02 mL. Assays were incubated with shaking for 1 hour at room temperature. Then the beads were pelleted and washed with wash buffer (1×PBS, 0.05% Tween 20) to remove displaced kinase and test compound. The washed based were re-suspended in elution buffer (1×PBS, 0.05% Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. qPCR reactions were assembled by adding 2.5 μL of kinase eluate to 7.5 μL of qPCR master mix containing 0.15 μM amplicon primers and 0.15 μM amplicon probe. The qPCR protocol consisted of a 10 minute hot start at 95° C., followed by 35 cycles of 95° C. for 15 seconds, 60° C. for 1 minute.Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. The Kds were determined using a compound top concentration of 30,000 nM. Kd measurements were performed in duplicate.
- 5α-Reductase Activity Assay The reaction mixture contained a final volume of 1 mL: 1 mM dithiothreitol, sodium phosphate buffer 40 mM, at pH 6.5 for human prostate, 2 mM NADPH, 2 nM [1,2,6,7-3H]T [Cabeza et al., Steroids 60:630-5]. The reaction in duplicate was started when it was added to the enzymatic fraction (500 μg protein in a volume of 80 μL) incubated at 37°C for 60 min [Hirosumi et al., J. Steroid Biochem. Mol. Biol., 52:357-63] and stopped by mixing with 1 mL of dichloromethane; this was considered as the end point. Incubation without tissue was used as a control. The mixture (incubation medium/dichloromethane) was agitated on a vortex for 1 min and the dichloromethane phase was separated and placed in another tube. This procedure was repeated four more times. The dichloromethane extract was evaporated to dryness under a nitrogen stream and suspended in 50 mL of methanol that was spotted on high-performance thin layer chromatography (HPTLC) Keiselgel 60 F254 plates. T and DHT were used as carriers, and were applied in different lanes on both lateral sides of the plates (T, T + DHT, and DHT). The plates were developed in chloroform-acetone, 9:1, and were air-dried; the chromatography was repeated twice more. The steroid carriers were detected using phosphomolybdic acid reagent (DHT) and with an ultraviolet (UV) lamp (254 nm) (T). After the plates were segmented into pieces of 1 cm each, these were cut off and the strips soaked in 5 mL of Ultima Gold (Packard). The radioactivity was determined in a scintillation counter (Packard Tri-Carb 2100 TR). The radioactivity content in the segment corresponding to T and DHT carriers was identified. Radioactivity with identical chromatographic behavior to the DHT standard was considered as the DHT transformation. Control incubations, chromatography separations, and identifications were carried out in the same manner as described above, except that the tubes did not contain tissue.
- DNAPK Enzyme Potency Assay (DNA-PK Enz) The inhibitory activity of compounds against DNAPK was determined by TR-FRET measuring a fluorescent labelled peptide substrate converting to a phosphorylated product. fluorescently tagged peptide substrate were purchased from Thermo Fisher Scientific. 12 point half-log compound concentration-response curves, with a top concentration of 100 μM were generated from 10 mM stocks of compound solubilised in DMSO using an Echo 555 (Labcyte Inc., Sunnyvale, Calif.). All assays were preformed in white Greiner 1536 well low volume plates (Greiner Bio-One, UK), in a total reaction volume of 3 μL and 1% (v/v) final DMSO concentration. Enzymes and substrates were added separately to the compound plates and incubated at room temperature. The kinase reaction was then quenched by the addition of 3 μL of stop buffer. Stopped assay plates were read using a BMG Pherastar. IC50 values were calculated using a Genedata Screener software (Genedata, Inc., Basel, Switzerland).Full length human DNAPK protein was purified from HeLa cell extract by ion exchange. Initially DNAPK protein was incubated with compound for 30 minutes at room temperature in reaction buffer (50 mM Hepes pH 7.5, 0.01% Brij-35, 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, 2 μg/ml Calf Thymus DNA). The reaction was then initiated by the addition of ATP and fluorescently tagged peptide substrate (Fluorescein-EPPLSQEAFADLWKK, Thermo Fisher Scientific). The kinase reaction (18 μM ATP, 35 μM DNAPK, 1.6 μM peptide substrate) was quenched after 40 minutes by the addition of 3 μL of stop buffer (20 mM Tris pH7.5, 0.02% sodium azide, 0.01% Nonidet-P40, 20 m EDTA, 4 nM Tb anti-phospho-p53 [Ser15] Antibody. The reaction was incubated for a further hour and the plates were read on a BMG Pherastar.Data was analysed and IC50 values were calculated using Genedata Screener software (Genedata, Inc., Basel, Switzerland). The pIC50 values were calculated as the negative logarithm of the molar concentration of compound required for 50% reduction in measured response.
- Enzyme Kinetic Assay To determine the inhibition selectivity for inhibitor candidates, human TNAP, PLAP or IAP were added to microtiter plates followed by addition of the substrate pNPP (0.5 mM) and activity was measured in 1 M DEA-HCl buffer, pH 9.8 or in 1 M Tris-HCl buffer, pH 7.5, containing 1 mM MgCl2 and 20 μM ZnCl2, in the presence of potential inhibitors (0-30 μM). TNAP, PLAP and IAP activities were adjusted to an approximate λA405 nm, equivalent to 1, measured after 30 min. Residual AP activity in the presence of inhibitors was expressed as percentage of the control activity. To investigate the mechanism of inhibition, double reciprocal plots of enzyme activity (expressed as mA405 nm min−1) vs. substrate concentration were constructed, in the presence of various concentrations of added inhibitors (0-30 μM). The y-axis intercepts of the 1/v vs. 1/[S] plots, were then plotted vs. [I] to graphically extract Ki values as the x-intercept in this plot. The numerical values from y- and x-intercepts were derived via linear regression analysis, using software Prism 3.02 (GraphPad Software, CA). These analyses were performed, using pNPP as a substrate in 1 M DEA-HCl buffer, pH 9.8, as well as in 1 M Tris-HCl buffer, pH 7.5, to determine Ki at optimal and physiological pH respectively. Inhibitors were further tested and sorted based on their kinetic properties at pH 7.4 using PPi, the relevant natural substrate of TNAP. In this part of the study, pyrophosphate sodium salt (99% ACS reagent, Sigma-Aldrich, St Louis, Mo.) was used as a substrate. Amounts of released phosphate were measured using the Biomol Green Reagent (Biomol Research Laboratories, Inc., Plymouth Meeting, Pa.). Finally, to document the potency of selected inhibitors in physiological media, TNAP inhibition by compounds of Formula I-IV (0-30 μM) was studied at pH 7.4, during catalysis of 0.1 mM pNPP, in the presence of increasing concentrations of Na2HPO4 (0-10 mM) and pyrophosphate (0-40 mM).
- SPR Assay Surface Plasmon Resonance (SPR) assays have been developed to test the affinity (KD) of VHL or CRBN-based compounds to respective recombinant ligase complex/domain.The assays are based on surface plasmon resonance (SPR), which enables to measure the changes of the local refractive index due to changes of molecular mass on a gold chip surface in the case of a binding event and in a flowing system. To detect binding between both partners, the respective E3 ligase is immobilized to the chip surface, while the test compounds are flown over the chip surface at a steady velocity. The detected changes in the RU response are indicative of the binding event and are concentration dependent.For the SPR experiments, either a commercially available VHL complex (Merck, 23-044; composed of 5 units: VHL, Elongin B, Elongin C, Cul2, and Rbx1) exhibiting a his-tag at the Cul2 subunit or an internally produced biotin-tagged mouse CRBN thalidomide binding domain (mCRBN-TBD) was used. These tags provide the anchor for the capturing process to either an NTA or Streptavidin coated chip surface (immobilization level of 3,000-5,000 RU). Because of the rather complex structure of the VHL complex, it was additionally coupled to the chip surface by amino coupling to prevent any protein loss by disruption of the complex in the flowing system.To detect binding of compounds and extract dissociation constants KD for the tested compounds to the immobilized E3 ligase, concentration response curves of the compounds were recorded. Compounds were usually tested in 10-pt dilutions up to 20 μM final concentration in assay buffer and were flown over the chip at 30 μL/min. The contact time for each cycle includes 90 s for association and 200 s for dissociation of compounds. Every test cycle was read out as a sensorgram that was referenced to the sensor surface that does not present the target protein.
- Primer Extension Assay (((Guanine-9-yl) propan-2-oxy) methyl) phosphonic acid diphosphate (PMPGpp) acts as a chain terminator and competes with dGTP.Purified human telomerase was incubated with d(TTAGGG)3 (SEQ ID NO: 1) and deoxy-nucleoside triphosphates for 90 minutes at 37° C. All reactions were done in the presence of 200 dTTP and 10 μM dATP (50,000 cpm/pmol a-33P-dATP). Activity was determined by incubating affinity purified telomerase extract with 1 μM primer [d(TTAGGG)3] (SEQ ID NO: 1), 200 μM dTTP, 50 μM dGTP, 10 μM dATP, 10 μCi [α-33P]dATP (2000-4000 Ci/mmol) in a buffer containing 50 mM (N-2-hydroxyethyl piperazine-N′-3 propanesulfonic acid)-NaOH pH 8.5, 1 mM MgCl2, 1 mM DTT, 5% glycerol, 0.5 mM EGTA and 100 mM KOAc in a final reaction volume of 40 μl. Primer extension products were analyzed on a 15% polyacrylamide gel containing 7 M urea. A characteristic telomerase ladder has multiple bands. If a compound is not a substrate then (TTAGGG)3TTA (21 nt) (SEQ ID NO: 2). If a compound is a chain terminator then (TTAGGG)3TTAG* (22 nt) (SEQ ID NO: 3).FIG. 1 is a photograph of a 15% polyacrylamide gel containing 7 M urea showing the primer extension products in the presence of compounds. Reactions containing 50 or 100 μM dGTP, (lanes 1 and 2) show a characteristic 6 nucleotide ladder. Lanes 8-11 contain increasing concentrations of PMPGpp (ID#142692) in addition to 50 μM dGTP. Lane 12 contains only 50 μM PMPGpp (ID#142692). Lanes 3-7 show a comparison using the known chain terminator, 3′-azido-dGTP.IC 50s for (R)-PMPGpp and (S)-PMPGpp are shown in Table 3. (R)-(((Guanine-9-yl) propan-2-oxy) methyl) phosphonic acid diphosphate is efficiently recognized by human telomerase and added to the 3′ end of a telomeric primer resulting in chain termination. The molecule competes with dGTP and when measured in a cell free assay, using purified telomerase and dGTP concentrations of 50 μM, shows an IC50 of about 1.0 μM.
- TYK2 JH2 Domain Binding Assay The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA)]. Extracts were used directly in binding assays without any enzyme purification steps at a ≥10,000-fold overall stock dilution (final DNA-tagged enzyme concentration <0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions performed in polypropylene 384-well plates. Each was a final volume of 0.02 mL. Assays were incubated with shaking for 1 hour at room temperature. Then the beads were pelleted and washed with wash buffer (1×PBS, 0.05% Tween 20) to remove displaced kinase and test compound. The washed based were re-suspended in elution buffer (1×PBS, 0.05% Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. qPCR reactions were assembled by adding 2.5 μL of kinase eluate to 7.5 μL of qPCR master mix containing 0.15 μM amplicon primers and 0.15 μM amplicon probe. The qPCR protocol consisted of a 10 minute hot start at 95° C., followed by 35 cycles of 95° C. for 15 seconds, 60° C. for 1 minute.
- TYK2 JH2 Domain Binding Assay The binding reaction was assembled by combining 16 μl of DNA-tagged kinase extract, 3.8 μl liganded affinity beads, and 0.18 μl test compound (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA)]. Extracts were used directly in binding assays without any enzyme purification steps at a >10,000-fold overall stock dilution (final DNA-tagged enzyme concentration <0.1 nM). Extracts were loaded with DNA-tag and diluted into the binding reaction in a two step process. First extracts were diluted 1:100 in 1× binding buffer (PBS/0.05% Tween 20/10 mM DTT/0.1% BSA/2 μg/ml sonicated salmon sperm DNA) containing 10 nM DNA-tag. This dilution was allowed to equilibrate at room temperature for 15 minutes and then subsequently diluted 1:100 in 1× binding buffer. Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions performed in polypropylene 384-well plates. Each was a final volume of 0.02 mL. Assays were incubated with shaking for 1 hour at room temperature. Then the beads were pelleted and washed with wash buffer (lx PBS, 0.05% Tween 20) to remove displaced kinase and test compound. The washed based were re-suspended in elution buffer (lx PBS, 0.05% Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. qPCR reactions were assembled by adding 2.5 μL of kinase eluate to 7.5 μL of qPCR master mix containing 0.15 μM amplicon primers and 0.15 μM amplicon probe. The qPCR protocol consisted of a 10 minute hot start at 95° C., followed by 35 cycles of 95° C. for 15 seconds, 60° C. for 1 minute.
- Fluorescence Assay α-Synuclein recombinant protein was produced in E. coli. BL21(DE3)RIL E. coli were transformed with a pRK172 bacterial expression plasmid containing the human α-synuclein coding sequence. Freshly transformed BL21 colonies were inoculated into 2 L baffled flasks containing 250 mL sterilized TB (1.2% bactotryptone, 2.4% yeast extract, 0.4% glycerol, 0.17 M KH2PO4, 0.72 M K2HPO4) with 50 μg/ml ampicillin, and incubated overnight at 37° C. with shaking. Overnight cultures were pelleted by centrifugation at 3,900×g for 10 min at 25° C. Bacterial pellets were resuspended in 20 mL osmotic shock buffer (30 mM Tris-HCl, 2 mM EDTA, 40% sucrose, pH 7.2) by gentle vortexing and incubated at room temperature for 10 minutes. The cell suspension was then centrifuged at 8,000×g for 10 min at 25° C. and the pellet was resuspended in 22.5 mL cold H2O before adding 9.4 μL 2 M MgCl2 to each tube. The suspension was incubated on ice for 3 min prior to centrifugation at 20,000×g for 15 min at 4° C. The supernatant was transferred to a fresh tube, streptomyocin was added to a final concentration of 10 mg/mL, and then centrifuged at 20,000×g for 15 min at 4° C. The supernatant from this step was collected and dithiothreitol (DTT) and Tris-HCl were added to final concentrations of 1 mM and 20 mM respectively, before boiling for 10 min to precipitate heat-sensitive proteins, which were pelleted at 20,000×g for 15 minutes at 4° C. The supernatant was collected and filtered through a 0.45 μm surfactant free cellulose acetate filter (Corning, Corning, NY) before loading onto a 1 mL DEAE Sepharose column equilibrated in 20 mM Tris- HCl pH 8, 1 mM EDTA, and 1 mM DTT. The DEAE column was washed with 20 mM Tris- HCl pH 8, 1 mM EDTA, 1 mM DTT before eluting α-synuclein protein in 20 mM Tris-HCl, pH 8, buffer with 1 mM EDTA, 1 mM DTT and 0.3 M NaCl. The purified α-synuclein protein was dialyzed overnight in 10 mM Tris-HCl, pH 7.6, 50 mM NaCl, and 1 mM DTT. Preparations contained greater than 95% α-synuclein protein as determined by SDS-PAGE and BCA assay with a typical yield of 30 mg protein per 250 ml culture.
- Mammalian One Hybrid (M111) Assay Determination of a ligand mediated Gal4 promoter driven transactivation to quantify ligand binding mediated activation of FXR was performed as follows.The cDNA part encoding the FXR ligand binding domain was cloned into vector pCMV-BD (Stratagene) as a fusion to the yeast GAL4 DNA binding domain under the control of the CMV promoter. The amino acid boundaries of the ligand binding domain were amino acids 187-472 of Database entry NM 005123 (RefSeq). The plasmid pFR-Luc (Stratagene) was used as the reporter plasmid, containing a synthetic promoter with five tandem repeats of the yeast GAL4 binding sites, driving the expression of the Photinus pyralis (American firefly) luciferase gene as the reporter gene. In order to improve experimental accuracy the plasmid pRL-CMV (Promega) was cotransfected. pRL-CMV contains the constitutive CMV promoter, controlling the expression of the Renilla reniformis luciferase. All Gal4 reporter gene assays were done in HEK293 cells (obtained from DSMZ, Braunschweig, Germany) grown in MEM with L-Glutamine and Earle's BSS supplemented with 10% fetal bovine serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 100 units Penicilin/Streptavidin per mL at 37° C. in 5% CO2. Medium and supplements were obtained from Invitrogen. For the assay, 5×105 cells were plated per well in 96 well plates in 100 μL per well MEM without Phenol Red and L-Glutamine and with Earle's BSS supplemented with 10% charcoal/dextran treated FBS (HyClone, South Logan, Utah), 0.1 mM nonessential amino acids, 2 mM glutamine, 1 mM sodium pyruvate, and 100 units Penicilin/Streptavidin per mL, incubated at 37° C. in 5% CO2. The following day the cells were >90% confluence. Medium was removed and cells were transiently transfected using 20 μL per well of an OptiMEM-polyethylene-imine-based transfection-reagent (OptiMEM, Invitrogen; Polyethyleneimine, Aldrich Cat No. 40,827-7) including the three plasmids described above. MEM with the same composition as used for plating cells was added 2-4 h after addition of transfection mixture. Then compound stocks, prediluted in MEM were added (final vehicle concentration not exceeding 0.1%). Cells were incubated for additional 16 h before firefly and renilla luciferase activities were measured sequentially in the same cell extract using a Dual-Light-Luciferase-Assay system (Dyer et al., Anal. Biochem. 2000, 282, 158-161). All experiments were done in triplicates.
- Mammalian One Hybrid (M1H) Assay Determination of a ligand mediated Gal4 promoter driven transactivation to quantify ligand binding mediated activation of FXR was performed as follows.The cDNA part encoding the FXR ligand binding domain was cloned into vector pCMV-BD (Stratagene) as a fusion to the yeast GAL4 DNA binding domain under the control of the CMV promoter. The amino acid boundaries of the ligand binding domain were amino acids 187-472 of Database entry NM_005123 (RefSeq). The plasmid pFR-Luc (Stratagene) was used as the reporter plasmid, containing a synthetic promoter with five tandem repeats of the yeast GAL4 binding sites, driving the expression of the Photinus pyralis (American firefly) luciferase gene as the reporter gene. In order to improve experimental accuracy the plasmid pRL-CMV (Promega) was cotransfected. pRL-CMV contains the constitutive CMV promoter, controlling the expression of the Renilla reniformis luciferase. All Gal4 reporter gene assays were done in HEK293 cells (obtained from DSMZ, Braunschweig, Germany) grown in MEM with L-Glutamine and Earle's BSS supplemented with 10% fetal bovine serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 100 units Penicilin/Streptavidin per mL at 37° C. in 5% CO2. Medium and supplements were obtained from Invitrogen. For the assay, 5×105 cells were plated per well in 96 well plates in 100 μL per well MEM without Phenol Red and L-Glutamine and with Earle's BSS supplemented with 10% charcoal/dextran treated FBS (HyClone, South Logan, Utah), 0.1 mM nonessential amino acids, 2 mM glutamine, 1 mM sodium pyruvate, and 100 units Penicilin/Streptavidin per mL, incubated at 37° C. in 5% CO2. The following day the cells were >90% confluence. Medium was removed and cells were transiently transfected using 20 μL per well of a OptiMEM-polyethylene-imine-based transfection-reagent (OptiMEM, Invitrogen; Polyethyleneimine, Aldrich Cat No. 40,827-7) including the three plasmids described above. MEM with the same composition as used for plating cells was added 2-4 h after addition of transfection mixture. Then compound stocks, prediluted in MEM were added (final vehicle concentration not exceeding 0.1%). Cells were incubated for additional 16 h before firefly and renilla luciferase activities were measured sequentially in the same cell extract using a Dual-Light-Luciferase-Assay system (Dyer et al., Anal. Biochem. 2000, 282, 158-161). All experiments were done in triplicates.
- Mammalian One Hybrid (M1H) Assay The cDNA part encoding the FXR ligand binding domain was cloned into vector pCMV-BD (Stratagene) as a fusion to the yeast GAL4 DNA binding domain under the control of the CMV promoter. The amino acid boundaries of the ligand binding domain were amino acids 187-472 of Database entry NM_005123 (RefSeq). The plasmid pFR-Luc (Stratagene) was used as the reporter plasmid, containing a synthetic promoter with five tandem repeats of the yeast GAL4 binding sites, driving the expression of the Photinus pyralis (American firefly) luciferase gene as the reporter gene. In order to improve experimental accuracy the plasmid pRL-CMV (Promega) was cotransfected. pRL-CMV contains the constitutive CMV promoter, controlling the expression of the Renilla reniformis luciferase. All Gal4 reporter gene assays were done in HEK293 cells (obtained from DSMZ, Braunschweig, Germany) grown in MEM with L-Glutamine and Earle's BSS supplemented with 10% fetal bovine serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 100 units Penicilin/Streptavidin per mL at 37° C. in 5% CO2. Medium and supplements were obtained from Invitrogen. For the assay, 5×105 cells were plated per well in 96 well plates in 100 μL per well MEM without Phenol Red and L-Glutamine and with Earle's BSS supplemented with 10% charcoal/dextran treated FBS (HyClone, South Logan, Utah), 0.1 mM nonessential amino acids, 2 mM glutamine, 1 mM sodium pyruvate, and 100 units Penicilin/Streptavidin per mL, incubated at 37° C. in 5% CO2. The following day the cells were >90% confluence. Medium was removed and cells were transiently transfected using 20 μL per well of an OptiMEM-polyethylene-imine-based transfection-reagent (OptiMEM, Invitrogen; Polyethyleneimine, Aldrich Cat No. 40, 827-7) including the three plasmids described above. MEM with the same composition as used for plating cells was added 2-4 h after addition of transfection mixture. Then compound stocks, prediluted in MEM were added (final vehicle concentration not exceeding 0.1%). Cells were incubated for additional 16 h before firefly and renilla luciferase activities were measured sequentially in the same cell extract using a Dual-Light-Luciferase-Assay system (Dyer et al., Anal. Biochem. 2000, 282, 158-161). All experiments were done in triplicates.
- RapidFire MPro inhibition assay The assay was performed according to the published procedure. Briefly, compounds were seeded into assay-ready plates (Greiner 384PP, cat# 781280) using an ECHO 650T dispenser and DMSO was back-filled for a uniform concentration in assay plates (DMSO concentration < 1%, final volume = 500 nL.). A 15 uM enzyme stock solution is prepared in 20 mM HEPES, pH 7.5 and 300 mM NaCl, and subsequently diluted to a working solution of 300 nM Mpro in assay buffer (20 mM HEPES, pH 7.5 and 50 mM NaCl) before the addition of 25 uL to each well using a Multidrop Combi (Thermo Scientific). After a quick centrifugation step (1000 rpm, 15 s) the plate is incubated for 15 min at room temperature. The reaction is initiated with the addition of 25 uL of 4 uM 11-mer (TSAVLQSGFRK-NH2, initially custom synthesized by the Schofield group, GLBiochem, used until March 2021), or 10 uM 37-mer (ALNDFSNSGSDVLYQPPQTSITSAVLQSGFRKMAFPS-NH2, GLBiochem, used after March 2021), dissolved in assay buffer. After centrifugation (1000 rpm, 14 s) the reaction is incubated for 10 min (11-mer) or 5 min (37-mer) at room temperature before quenching with 10 % formic acid. The reactions are analysed with MS using RapidFire (RF) 365 high-throughput sampling robot (Agilent) connected to an iFunnel Agilent 6550 accurate mass quadrupole time-of-flight (Q-TOF) mass spectrometer using electrospray. All compounds are triaged by testing the % inhibition at 5 and 50 uM final concentration. Dose response curves uses an 11-point range of 100--0.0017 uM inhibitor concentrations. RapidFire integrator software (Agilent) was used to extract the charged states from the total ion chromatogram data followed by peak integration. For the 11-mer peptide the m/z (+1) charge states of both the substrate (1191.67 Da) and cleaved N-terminal product TSAVLQ (617.34 Da) were used and the 37-mer peptide the m/z (+2) charge states of the substrate (3960.94 Da) and m/z (+1) of the cleaved C-terminal product SGFRKMAFPS (1125.57 Da). Percentage conversion (product peak integral / (product peak integral + substrate peak integral))*100) and percentage inhibitions were calculated and normalised against DMSO control with deduction of any background signal in Microsoft Excel. IC50s were calculated using Levenberg Marquardt algorithm used to fit a restrained Hill equation to the dose-response data with both GraphPad PRISM and CDD.
- RORgamma Gal4 Reporter Gene Assay (FRET) Determination of a ligand mediated Gal4 promoter driven transactivation to quantify ligand binding to RORγ was performed as follows: DNA encoding three different RORγ protein fragments was cloned into vector pCMV-BD (Stratagene). Expression was under control of a CMV promoter and as fusion to the DNA-binding domain of the yeast protein GAL4. The amino acid boundaries of the three proteins and the respective database entries are listed in Table 2. Other vectors used were pFR-Luc (Stratagene) as regulated reporter plasmid. pFR-Luc contains a synthetic promoter with five tandem repeats of the yeast GAL4 binding sites that control expression of the Photinus pyralis (American firefly) luciferase gene. In order to improve experimental accuracy the plasmid pRL-CMV was cotransfected. pRL-CMV contains the constitutive CMV promoter, controlling the expression of the Renilla reniformis luciferase. All Gal4 reporter gene assays were done in 293T cells (DSMZ (German Collection of Microorganisms and Cell Cultures), Braunschweig, Germany, ACC635) grown in Minimum Essential Medium (MEM) with Phenol Red. The medium is supplemented with 10% fetal bovine serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 1% Glutamax and 100 units Penicilin/Streptavidin per mL at 37° C. in 5% CO2.For the assay, 5×105 cells were plated per well in 96 well plates in 100 μL per well, incubated over night at 37° C. in 5% CO2. The following day, medium was discarded and the cells were transiently transfected using 20 μL per well of a OptiMEM PEI-based transfection-reagent (Sigma-Aldrich, 408727) including the three plasmids described above. About 4 h after addition of the transfection solution, fresh Minimal Essential Medium (MEM, same composition as used for plating cells, but without serum) was added. Then compound stocks, prediluted in MEM (same composition as used for plating cells) were added (final vehicle concentration not exceeding 0.1%).Cells were incubated for additional 16 h before firefly (FF) and renilla (REN) luciferase activities were measured sequentially in the same cell extract using a Dual-Light-Luciferase-Assay system (Dyer et al., Anal. Biochem. 2000, 282:158). All experiments were done at least in triplicates.Applying the Gal4 reporter gene assay as described above, the Examples of the present invention usually have an inhibition activity (IC50 FF resp. IC50 RENnorm) ranging from below 10 nM to about 20 μM, and typically, from about 10 nM to about 1 μM. The RORγ modulating compounds of the invention desirably have an inhibition in the Gal4 reporter gene assay ranging from below 10 nM to about 1 μM. Table 3 list the pIC50-value of typical examples of compounds of the invention that have an RORγ activity in the Gal4 reporter gene assay for firefly (FF) and renilla normalised (RENnorm) luciferase measurements (nt=not tested). It is understood that the data illustrated below may have reasonable variation depending on the specific conditions and procedures used by the person conducting the test. The efficacy was determined in comparison to the RORγt inhibitor T0901317 (equals 100%) and the pIC50-value is underlined, when the efficacy of the compound is below 50% of the reference.
- BCA Protein Assay Escherichia coli BL21*DE3 pET30a-Ec yeaWX #1 (Ec YeaWX) strain was generated as described below. The contiguous Escherichia coli coding sequence yeaW (equivalent to uniprot ID P0ABR7.1 (YeaW) (SEQ ID NO: 2)) and yeaX (equivalent to uniprot ID P76254.1 (YeaX) (SEQ ID NO: 3)) were PCR amplified from Escherichia coli strain K-12 substr. BW25113 genomic DNA. PCR primers (YeaW_Nde I_fwd2-SEQ ID NO: 4; YeaX_rev2-SEQ ID NO: 5) were designed to create a 5′ NdeI restriction site including the ATG start codon of yeaW and create a PstI restriction site just 3′ of the yeaX TAG stop codon.The bacteria were grown aerobically in 50 mL LB broth (Difco #244620; 10 g/L Tryptone, 5 g/L yeast extract, 10 g/L NaCl, 50 μg/mL kanamycin), in a 500 mL Erlenmeyer flask. The cultures were inoculated from glycerol stock of BL21*DE3 pET30a-Ec yeaWX #1 strain. Strains were cultured all day at 37° C. with 250 rpm shaking. Two 300 mL Minimal M9 Medium (6 g/L Na2HPO4, 3 g/L KH2PO4, 0.5 g/L NaCl, 1 g/L NH4Cl, 0.1 mM CaCl2, 1 mM MgSO4, 0.2% Dextrose, 1 mg/L Thiamine, 50 μg/mL kanamycin), in 1 L Erlenmeyer flasks, were inoculated with 5 mL of the LB broth day culture and cultured overnight at 37° C. with 250 rpm shaking. The overnight cultures were used to inoculate twelve 1 L cultures of Minimal M9 media in 2.8 L fluted Erlenmeyer flasks to an OD 600 nm of 0.05 (typically approximately 28 mLs), which were grown at 37° C. with 250 rpm shaking until an OD600 of approximately 0.4 was reached. Expression of YeaWX was induced with 1 mM IPTG and the induced cultures were further grown overnight at 37° C. with 250 rpm shaking. The biomass was pelleted by centrifugation at 6000×g for 12 minutes at 4° C. The cell pellet was suspended in 240 mL of ice-cold 1× Phosphate Buffered Saline (Ca2+ and Mg2+ free). Ninety micrograms of Lysozyme (Sigma #L6876 Lot #SLBG8654V; Sigma-Aldrich Corp., St. Louis, Mo.) was added and incubated with 320 rpm shaking for 30 minutes at 4° C. Lysis was achieved via French press with a 4° C. prechilled 1″ diameter chamber at 1000 psi (high ratio; internal PSI equivalent 16000). The lysate was centrifuged at 6,000×g for 12 minutes at 4° C. to pellet extra debris. Glycerol was added to the centrifuged lysate supernatant at a final concentration of 15% A protein concentration of the centrifuged lysate supernatant was determined by a BCA Protein Assay Kit (Pierce #23225), typically in the 2.5 to 4.5 mg/ml range. The centrifuged Ec YeaWX lysate supernatant was aliquoted into 20 mL volumes and stored frozen at −80° C.Ec YeaWX lysate was diluted to 2.0 mg/mL protein with 1× Dulbecco's phosphate buffered saline (DPBS) plus 15% glycerol. Nicotinamide adenine dinucleotide phosphate (NADPH) was added to 250 μM. One hundred and fifty microliters of Ec YeaWX lysate was dispensed into a deep-well plate (polypropylene, 2 mL volume, Corning Axygen catalogue #P-DW-20-C). Candidate IC50 compounds from TABLE 1 and vehicle control (respective vehicle control of DMSO or water), or control compounds (IC50 control, 8-Quinolinol hemisulfate salt (Sigma Catalog #55100)) were added at a 1:100 dilution (e.g., 1.5 μL per well). The plates were agitated on a plate shaker for 1 minute. d9-carnitine chloride (1.5 μL of 5 mM) was added to all wells to reach a final d9-carnitine chloride concentration of 50 μM.