 Boric acid BDBM39817 Boronic Acid
Boric acid BDBM39817 Boronic Acid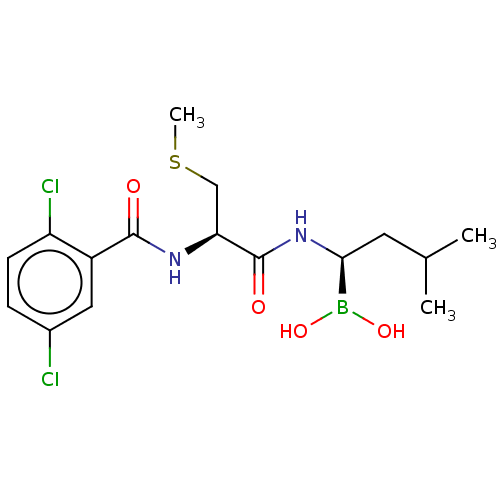 BDBM588478 (R)-N-(2,5-dichlorobenzoyl)-3- methylmercaptopropionamido-D-leucine boric acid US11542283, Compound IV-9
BDBM588478 (R)-N-(2,5-dichlorobenzoyl)-3- methylmercaptopropionamido-D-leucine boric acid US11542283, Compound IV-9
- ChEMBL_795274 (CHEMBL1937034) Displacement of [3H]-DHA from recombinant human beta2 adrenoceptor expressed in HEK293T cells in presence of 100 uM boric acid
- ChEMBL_795325 (CHEMBL1937181) Agonist activity at recombinant human beta2 adrenoceptor expressed in HEK293T cells assessed as increase in IBMX-induced cAMP accumulation using [3H]-cAMP after 30 mins by liquid scintillation counting in presence of 100 uM boric acid
- Inhibition Assay To prepare the substrate, 25-50 uCi of [3H]trioleoylglycerol (in toluene), 6.8 umol of unlabeled trioleoylglycerol and 0.6 mg of phospholipids (phosphatidylcholine/phosphatidylinositol 3:1 w/v) are mixed, dried with N2 and then taken up in 2 ml of 0.1 M KPi (pH 7.0) by ultrasound treatment (Branson 250, microtip, setting 1-2, 2x1 min with an interval of 1 min). After addition of 1 ml of KPi and renewed ultrasound treatment (4x30 sec on ice with intervals of 30 sec), 1 ml of 20% BSA (in KPi) is added (final concentration of trioleoylglycerol 1.7 mM). For the reaction, 100 ul of substrate solution are pipetted into 100 ul of HSL solution (HSL prepared as above, diluted in 20 mM KPi, pH 7.0, 1 mM EDTA, 1 mM DTT, 0.02% BSA, 20 ug/ml pepstatin, 10 ug/ml leupeptin) and incubated at 37 C. for 30 min. Addition of 3.25 ml of methanol/chloroform/heptane (10:9:7) and of 1.05 ml of 0.1 M K2CO3, 0.1 M boric acid (pH 10.5) is followed by thorough mixing.
- MIF Enzymatic Assay Assays were performed in small-volume clear-bottom black 96 or 384-well polystyrene plates (Greiner Bio-One). First, 2 μL of the enzyme solution containing 6 nM of MIF in DPBS, 0.025% w/v BSA, and 300 μM CHAPS was dispensed onto sample and negative control wells using Multidrop Combi with metallic tip cassettes (Thermo Fisher Scientific) previously treated with Sigmacote. Then, 2 μL of the same buffer without MIF was dispensed onto positive control wells. The reaction was started by addition of the following to all wells: 2 μL of substrate solution containing 3 mM ketonic pHPP in 200 mM boric acid, 25 mM sodium phosphate, 0.025% w/v BSA, and 300 μM CHAPS at pH 6.0. In order to remove bubbles, the plate was centrifuged in an Allegra 25R centrifuge (Beckman Coulter, Inc., Brea, CA) at 1000 rpm for 2 min at room temperature. Then, the plate was read in an EnVision. Final concentrations of enzyme and substrate were 3 nM and 1.5 mM, respectively. Initial rates were calculated for each well as the slope of the absorbance progress curve.
- Urease Inhibition Assay The inhibition studies of soybean urease were initiated with boric acid and boronic acids (butylboronic acid, 4-bromophenylboronic acid, and phenylboronic acid). Also, heavy metal ions (HgCl2, AgCl, and Cu(CH3COO)2) and sodium salts of mineral acids (NaF, NaCl, NaNO3, and Na2SO4) were investigated for their inhibitory effects. Stock solutions of inhibitors, except for 4-bromophenylboronicacid, were prepared in 0.05 M Tris-acetate buffer, pH 7.0, and were suitably diluted for experiments, whereas a stock solution of 4-bromophenylboronic acid was prepared in absolute ethanol and subsequently diluted in respective buffer. The activity assay was carried out at standard conditions as described earlier in the presence of varying concentrations of inhibitors. The yellow-orange colored solution was measured at 405 nm on a Unicam UV-2 spectrophotometer. The amount of NH3 liberated in the reaction mixture was estimated by calibrating Nessler's reagent with standard NH4Cl solution. One enzyme unit is defined as the amount of urease required to liberate 1 μmol of ammonia per minute under our test conditions (0.1 M urea, 0.05 M Trisacetatebuffer, pH 7.0, 37°C).
- HPP Tautomerase Assay Inhibition of the tautomerase activity of MIF was measured using the substrate 4-hydroxyphenyl pyruvic acid (HPP) in a procedure largely adapted from previous reports. A solution of HPP (10 mM) in acetate buffer (0.5 M ammonium acetate, pH adjusted to 6.0) was prepared and allowed to incubate overnight in the dark at room temperature to allow for equilibration of the keto and enol forms. Following the incubation period, the HPP solution was stored at 4 C. and used for no more than a week. For Ki determination, MIF protein (final concentration: ca. 50 nm) and inhibitor (multiple concentrations in DMSO, maintaining a final DMSO concentration of 1%) were incubated in borate buffer (0.5 M boric acid, pH 6.2) in a U-bottom 96-well plate (Falcon) for 20 minutes. A negative control (containing water and DMSO in lieu of protein and inhibitor, respectively) and a positive control (containing DMSO in lieu of inhibitor) were also prepared. The reaction began upon the addition of HPP solution (final concentration: 1.0 mM and 2.5 mM). The absorbance was monitored at 305 nm for the formation of the borate-enol complex using a Tecan INFINITE F500 plate reader over 175 seconds. Absorbance was measured three times for each [inhibitor]-[HPP] combination. Calculation of initial velocities and the nonlinear regression analyses for the enzyme kinetics were performed using the program Prism 6 (GraphPad), setting the Michaelis-Menten constant (Km) to 2.4. Reported Ki values represent the average value obtained from two assays performed on different days. (R)-ISO-1 (purchased from Santa Cruz Biotechnology) was used as a control.
- Inhibition of Arginase Inhibition of arginase I (ARG I) and arginase II (ARGG II) novel compounds is followed spectrophotometrically at 530 nm. The compound to be tested is dissolved in H2O and prepared 100 mM stock solution. 10 μl of the stock solution is diluted in 90 μl of the assay buffer that comprises 0.1M sodium phosphate buffer containing 130 mM NaCl, pH 7.4, to which is added ovalbumin (OVA) at a concentration of 1 mg/mi. Solutions of arginase I and II are prepared in 100 mM sodium phosphate buffer, pH 7.4 containing 1 mg/ml OVA to give an arginase stock solution at a final concentration of 100 ng/ml. To each well of a 96-well microtiter plate is add 40 μl of enzyme, 10 μl of an inventive compound and 10 μl of enzyme substrate (L-arginine+manganese sulfate). For wells that are used as positive controls, only the enzyme and its substrate are added, while wells used as negative controls contain only manganese sulfate. After incubating the microtiter plate at 37° C. for 60 minutes, 150 μl of a urea reagent obtained by combining equal proportions (1:1) of reagents A and B is added to each well of the microtiter plate to stop the reaction. The urea reagent is made just before use by combining Reagent A (10 mM o-phthaldialdehyde, and 0.4% polyoxyethylene (23) lauryl ether (w/v) in 1.8 M sulfuric acid) with Reagent B (1.3 mM primaquinone diphosphate, 0.4% polyoxyethylene (23) lauryl ether (w/v), 130 mM boric acid in 3.6 M sulfuric acid). After quenching the reaction mixture, the microtiter plate is allowed to stand for an additional 10 minutes at room temperature to allow color development. T
- Inhibition Assay of Arginase Inhibition of arginase I (ARG I) and arginase II (ARG II) by Formula I or Formula II compounds is followed spectrophotometrically at 530 nm. The compound to be tested was dissolved in DMSO at an initial concentration 50-fold greater than its final concentration in the cuvette. 10 μl of the stock solution was diluted in 90 μl of the assay buffer that comprises 0.1M sodium phosphate buffer containing 130 mM NaCl, pH 7.4, to which is added ovalbumin (OVA) at a concentration of 1 mg/ml. Solutions of arginase I and II were prepared in 100 mM sodium phosphate buffer, pH 7.4 containing 1 mg/ml of OVA to give an arginase stock solution at a final concentration of 100 ng/ml. To each well of a 96-well microtiter plate was added 40 μl of enzyme, 10 μl of an inventive compound and 10 μl of enzyme substrate (L-arginine+manganese sulfate). For wells that were used as positive controls, only the enzyme and its substrate were added, while wells used as negative controls contained only manganese sulfate. After incubating the microtiter plate at 37° C. for 60 minutes, 150 μl of a urea reagent obtained by combining equal proportions (1:1) of reagents A and B is added to each well of the microtiter plate to stop the reaction. The urea reagent is made just before use by combining Reagent A (10 mM o-phthaldialdehyde, and 0.4% polyoxyethylene (23) lauryl ether (w/v) in 1.8 M sulfuric acid) with Reagent B (1.3 mM primaquine diphosphate, 0.4% polyoxyethylene (23) lauryl ether (w/v), 130 mM boric acid in 3.6 mM sulfuric acid). After quenching the reaction mixture, the microtiter plate is allowed to stand for an additional 10 minutes at room temperature to allow the color to develop. The inhibition of arginase was computed by measuring the optical density (OD) of the reaction mixture at 530 nm and normalizing the OD value to percent inhibition observed in the control. The normalized OD is then used to generate a dose-response curve by plotting the normalized OD values against log [concentration] and using regression analysis to compute the IC50 values.
- Inhibition of Arginase Inhibition of arginase I (ARG I) and arginase II (ARG II) by Formula I or Formula II compounds is followed spectrophotometrically at 530 nm. The compound to be tested was dissolved in DMSO at an initial concentration 50-fold greater than its final concentration in the cuvette. 10 μl of the stock solution was diluted in 90 μl of the assay buffer that comprises 0.1M sodium phosphate buffer containing 130 mM NaCl, pH 7.4, to which is added ovalbumin (OVA) at a concentration of 1 mg/ml. Solutions of arginase I and II were prepared in 100 mM sodium phosphate buffer, pH 7.4 containing 1 mg/ml of OVA to give an arginase stock solution at a final concentration of 100 ng/ml.To each well of a 96-well microtiter plate was added 40 μl of enzyme, 10 μl of an inventive compound and 10 μl of enzyme substrate (L-arginine+manganese sulfate). For wells that were used as positive controls, only the enzyme and its substrate were added, while wells used as negative controls contained only manganese sulfate.After incubating the microtiter plate at 37° C. for 60 minutes, 150 μl of a urea reagent obtained by combining equal proportions (1:1) of reagents A and B is added to each well of the microtitcr plate to stop the reaction. The urea reagent is made just before use by combining Reagent A (10 mM o-phthaldialdehyde, and 0.4% polyoxyethylene (23) lauryl ether (w/v) in 1.8 M sulfuric acid) with Reagent B (1.3 mM primaquine diphosphate, 0.4% polyoxyethylene (23) lauryl ether (w/v), 130 mM boric acid in 3.6 mM sulfuric acid). After quenching the reaction mixture, the microtiter plate is allowed to stand for an additional 10 minutes at room temperature to allow the color to develop. The inhibition of arginase was computed by measuring the optical density (OD) of the reaction mixture at 530 nm and normalizing the OD value to percent inhibition observed in the control. The normalized OD is then used to generate a dose-response curve by plotting the the normalized OD values against log [concentration] and using regression analysis to compute the IC50 values.
- Inhibition of Tautomerase Activity of Human MIF Inhibition of the tautomerase activity of MIF was measured using 4-hydroxyphenyl pyruvic acid (HPP) as substrate, largely following previously reported protocols (Taylor et al., Biochemistry, 1999, 38, 7444-7452). HPP was dissolved in 0.5 M acetate buffer, pH 6.0 to a final concentration of 10 mM and incubated overnight at room temperature to allow equilibration of the keto and enol forms. MIF (6 μL) was premixed in 500 mM boric acid, pH 6.2 (142 μL) and transferred to a transparent U bottom 96-well plate to a final concentration of 50 nM MIF. At this concentration high signal-to-noise and linearity were observed after analysis of progress curves for enol production at different protein concentrations. Inhibitors were dissolved in DMSO to 10 mM and an initial screen was performed. For compounds that showed ca. 25% or greater inhibition at 10 μM, an inhibition constant, was measured.Compounds were placed into wells (2 μL) at 6 different concentrations and incubated for 30 minutes until the assay was started by addition of HPP (504) at two concentrations (1.0 and 2.5 mM). The negative control was MIF incubated with DMSO vehicle, which in all assays was 1% and did not influence tautomerase activity. MIF activity was monitored at 305 nm for formation of the borate-enol complex using an Infinite F500 plate reader (TECAN, Morrisville, N.C.) for 175 seconds. Calculation of initial velocities and the nonlinear regression analyses for the enzyme kinetics were repeated three times with the program Prism6 (GraphPad, La Jolla, Calif.).Data obtained for the Example compounds, obtained using the methods described in Example B, are provided in Table 3. Results are also included for a reference compound ISO-1 (Chang, K. F.; Al-Abed, Y. Bioorg. Med. Chem. Lett. 2006, 16, 3376-3379; Balachandran, S. et al. Bioorg. Med. Chem. Lett. 2009, 19, 4773-4776). X-ray crystal structures were obtained for complexes of 3a, 3b, and 3v (Examples 1, 2, and 21) with human MIF as described below in Example C, which confirm the binding of these inhibitors in the tautomerase active site.