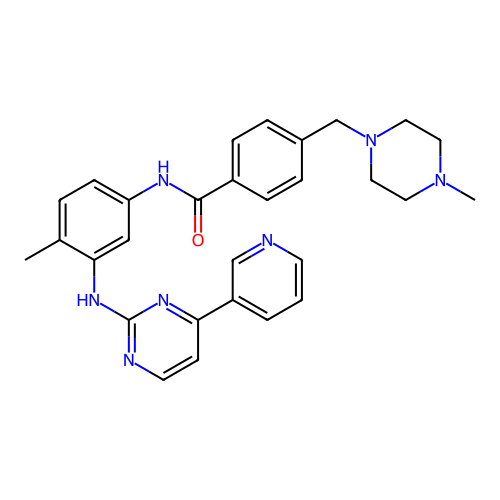
 IMATINIB BDBM31341
IMATINIB BDBM31341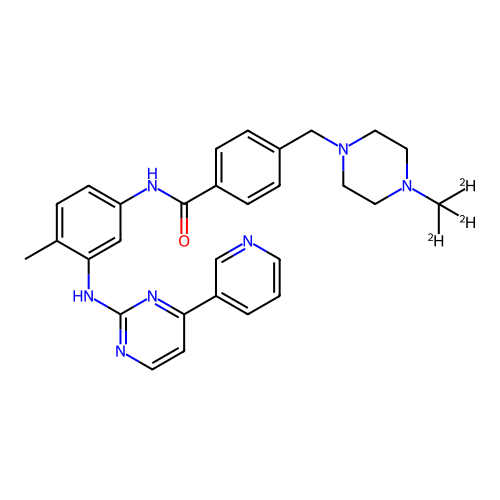 imatinib-CD3 BDBM50434581 CHEMBL2386595
imatinib-CD3 BDBM50434581 CHEMBL2386595 Imatinib STI571 Imatinib, 21 US20250129067, Compound Imatinib US11649218, Example Imatinib CHEMBL941 Gleevec cid_5291 4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide US11725005, Compound imatinib N-(4-methyl-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}phenyl)-4-[(4-methylpiperazin-1-yl)methyl]benzamide US10906896, Cpd imatinib med.21724, Compound 6 US9255107, Imatinib BDBM13530 STI-571
Imatinib STI571 Imatinib, 21 US20250129067, Compound Imatinib US11649218, Example Imatinib CHEMBL941 Gleevec cid_5291 4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide US11725005, Compound imatinib N-(4-methyl-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}phenyl)-4-[(4-methylpiperazin-1-yl)methyl]benzamide US10906896, Cpd imatinib med.21724, Compound 6 US9255107, Imatinib BDBM13530 STI-571
- Manley, PW; Stiefl, N; Cowan-Jacob, SW; Kaufman, S; Mestan, J; Wartmann, M; Wiesmann, M; Woodman, R; Gallagher, N Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib. Bioorg Med Chem 18: 6977-86 (2010)
- Schoepf, AM; Salcher, S; Obexer, P; Gust, R Overcoming imatinib resistance in chronic myelogenous leukemia cells using non-cytotoxic cell death modulators. Eur J Med Chem 185: (2020)
- Pan, X; Dong, J; Shao, R; Su, P; Shi, Y; Wang, J; He, L Expanding the structural diversity of Bcr-Abl inhibitors: Hybrid molecules based on GNF-2 and Imatinib. Bioorg Med Chem Lett 25: 4164-8 (2015)
- Parkkila, S; Innocenti, A; Kallio, H; Hilvo, M; Scozzafava, A; Supuran, CT The protein tyrosine kinase inhibitors imatinib and nilotinib strongly inhibit several mammalian alpha-carbonic anhydrase isoforms. Bioorg Med Chem Lett 19: 4102-6 (2009)
- Sorrenti, V; Pittalà, V; Romeo, G; Amata, E; Dichiara, M; Marrazzo, A; Turnaturi, R; Prezzavento, O; Barbagallo, I; Vanella, L; Rescifina, A; Floresta, G; Tibullo, D; Di Raimondo, F; Intagliata, S; Salerno, L Targeting heme Oxygenase-1 with hybrid compounds to overcome Imatinib resistance in chronic myeloid leukemia cell lines. Eur J Med Chem 158: 937-950 (2018)
- Crespan, E; Radi, M; Zanoli, S; Schenone, S; Botta, M; Maga, G Dual Src and Abl inhibitors target wild type Abl and the AblT315I Imatinib-resistant mutant with different mechanisms. Bioorg Med Chem 18: 3999-4008 (2010)
- Manley, PW; Blasco, F; Mestan, J; Aichholz, R The kinetic deuterium isotope effect as applied to metabolic deactivation of imatinib to the des-methyl metabolite, CGP74588. Bioorg Med Chem 21: 3231-9 (2013)
- Peng, Z; Maxwell, DS; Sun, D; Bhanu Prasad, BA; Pal, A; Wang, S; Balatoni, J; Ghosh, P; Lim, ST; Volgin, A; Shavrin, A; Alauddin, MM; Gelovani, JG; Bornmann, WG Imatinib analogs as potential agents for PET imaging of Bcr-Abl and c-KIT expression at a kinase level. Bioorg Med Chem 22: 623-32 (2013)
- In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants.
- Houghton, PJ; Germain, GS; Harwood, FC; Schuetz, JD; Stewart, CF; Buchdunger, E; Traxler, P Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res 64: 2333-7 (2004)
- Yang, M; Xi, Q; Jia, W; Wang, X Structure-based analysis and biological characterization of imatinib derivatives reveal insights towards the inhibition of wild-type BCR-ABL and its mutants. Bioorg Med Chem Lett 29: (2019)
- Salah, E; Ugochukwu, E; Barr, AJ; von Delft, F; Knapp, S; Elkins, JM Crystal structures of ABL-related gene (ABL2) in complex with imatinib, tozasertib (VX-680), and a type I inhibitor of the triazole carbothioamide class. J Med Chem 54: 2359-67 (2011)
- Ghosh, AK; Mishevich, JL; Kovela, S; Shaktah, R; Ghosh, AK; Johnson, M; Wang, YF; Wong-Sam, A; Agniswamy, J; Amano, M; Takamatsu, Y; Hattori, SI; Weber, IT; Mitsuya, H Exploration of imatinib and nilotinib-derived templates as the P2-Ligand for HIV-1 protease inhibitors: Design, synthesis, protein X-ray structural studies, and biological evaluation. Eur J Med Chem 255: (2023)
- Schoepf, AM; Salcher, S; Obexer, P; Gust, R Identification and development of non-cytotoxic cell death modulators: Impact of sartans and derivatives on PPARγ activation and on growth of imatinib-resistant chronic myelogenous leukemia cells. Eur J Med Chem 195: (2020)
- Mahboobi, S; Dove, S; Sellmer, A; Winkler, M; Eichhorn, E; Pongratz, H; Ciossek, T; Baer, T; Maier, T; Beckers, T Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: a series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rbeta, and histone deacetylases. J Med Chem 52: 2265-79 (2009)
- Lu, Y; Mao, F; Li, X; Zheng, X; Wang, M; Xu, Q; Zhu, J; Li, J Discovery of Potent, Selective Stem Cell Factor Receptor/Platelet Derived Growth Factor Receptor Alpha (c-KIT/PDGFRa) Dual Inhibitor for the Treatment of Imatinib-Resistant Gastrointestinal Stromal Tumors (GISTs). J Med Chem 60: 5099-5119 (2017)
- Ren, X; Pan, X; Zhang, Z; Wang, D; Lu, X; Li, Y; Wen, D; Long, H; Luo, J; Feng, Y; Zhuang, X; Zhang, F; Liu, J; Leng, F; Lang, X; Bai, Y; She, M; Tu, Z; Pan, J; Ding, K Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated breakpoint cluster region-Abelson (Bcr-Abl) kinase and overcomes clinically acquired mutation-induced resistance against imatinib. J Med Chem 56: 879-94 (2013)
- ChEMBL_836767 (CHEMBL2075189) TP_TRANSPORTER: drug resistance(Imatinib mesylate) in BCRP-expressing SaoS2 cells
- ChEMBL_1743176 (CHEMBL4158926) Inhibition of chymotrypsin-like activity of 20S proteasome in imatinib-resistant human SUP-B15 cells using Suc-LLVY aminoluciferin as substrate after 2 hrs by proteasome-Glo assay
- ChEMBL_1922293 (CHEMBL4425249) Inhibition of imatinib-resistant BCR-ABL T315I mutant (unknown origin) expressed in mouse BA/F3 cells assessed as decrease in cell viability after 48 hrs by trypan blue exclusion assay
- ChEMBL_1922299 (CHEMBL4425255) Inhibition of imatinib-resistant PDGFR D842V mutant (unknown origin) expressed in mouse BA/F3 cells assessed as decrease in cell viability after 48 hrs by CellTiter 96 aqueous one solution cell proliferation assay
- ChEMBL_2370075 Inhibition of KIT (delta 560 to 576 residues) deletion/V654A double mutant autophosphorylation at Y703 in human GIST-430/654 harboring KIT (delta 560 to 576 residues) deletion mutation at exon 11 and imatinib resistance mutation V654A in exon 13
- PDGFRβ HTRF Assay Assay in white ProxiPlate 384-wellStep 1. Dispensing inhibitors/DMSO and low control: Using the ECHO 555 acoustic dispenser, spot desired compound serial dilutions in DMSO, NEAT DMSO to represent the uninhibited enzyme control, and 10 μM final [imatinib] to the represent the 100% inhibited enzyme controlStep 2. PDGFRβ E+I pre-incubation: Add 2 μL 2× protein solution to columns 1-24 using the Multidrop Combi. Centrifuge at 1000 rpm for 1 min. Incubate 30 min at RTStep 2. Enzymatic reaction: Add 2 μL substrate solution to columns 1-24 to initiate the reaction using the Multidrop Combi; cover/seal the assay plate to reduce evaporation. Centrifuge at 1000 rpm for 1 min. Incubate at room temperature for 3 hours.Final concentrations of components in PDGFRp cascade assay:50 mM Hepes, pH 7.510 mM MgCl20.01% Brij-351 mM EGTA2 mM DTT0.01% Ovalbumin50 μM inactive PDGFRβ0.5 μM TK-substrate biotin peptide62.5 nM SA-XL-665TK antibody-Eu3+-cryptate (diluted by 1/3 final from stock)800 μM ATP≤1% DMSOStep 3. Quench/Detection: Add 2 μl 3× quench/detection solution to columns 1-24 using the Multidrop Combi; cover/seal the plate. Centrifuge 1 min 1000 rpm. Incubate at RT for 60 min. Read the plate in PHERAstar (or similar instrument) on HTRF setting at excitation 337 nmdual emission665/620 nm ratio.
- Biochemical Assay for mTOR TR-FRET assays for protein kinases uses a long-lifetime lanthanide Terbium or Europium chelates as the donor species which overcome interference by compound autofluorescence or light scatter from precipitated compounds, by introducing a delay after excitation by a flashlamp excitation source. Results are often expressed as a ratio of the intensities of the acceptor and donor fluorophores. The ratiometric nature of such a value corrects for differences in assay volumes between wells, as well as corrects for quenching effects due to colored compounds.Binding Assays are based on the binding and displacement of an Alexa Fluor 647-labeled, ATP-competitive kinase inhibitors to the kinase of interest. Invitrogen's “Kinase Tracers” have been developed to address a wide range of kinase targets and are based on ATP-competitive kinase inhibitors, making them suitable for detection of any compounds that bind to the ATP site or to an allosteric site altering the conformation of the ATP site. Inhibitors that bind the ATP site include both Type I kinase inhibitors, which bind solely to the ATP site, and Type II inhibitors (e.g., Gleevec/Imatinib, Sorafenib, BIRB-796), which bind to both the ATP site and a hydrophobic site exposed in the DFG-out (non-active) conformation. Type III inhibitors are compounds that do not compete with ATP are loosely referred to as allosteric inhibitors. A study of 15 diverse Type III inhibitors demonstrated that all but one compound was detected in the binding assay with equivalent potency to activity assays. The sole exception was a substrate-competitive compound, and thus not a true allosteric inhibitor.In contrast to most fluorescence-based kinase activity assays, LanthaScreen Eu3+ Kinase Binding Assays can be read continuously, which facilitates evaluation of compounds with slow binding kinetics. Also, unlike most activity assays, binding assays can be performed using either active or non-activated kinase preparations, which enables characterization of compounds that bind preferentially to non-activated kinases, such as Gleevec /imatinib and some allosteric inhibitors.In the Lanthascreen™ kinase binding assay, the donor (Eu3+-anti-GST antibody) is excited at 340 nm and will transfer its energy to the acceptor (Alexa Fluor 647-labeled ATP-competitive kinase inhibitor=Tracer-314). The emission from the Tracer-314 (Alexa Fluor 647 inhibitor) can be monitored with a filter centered at 665 nm because it is located between the emission peaks of the donor, which is measured at 615/620 nm. The binding of both, the Tracer-314 and Eu3+-anti-GST antibody, to the kinase results in a high degree of FRET from the Eu3+-donor fluorophore to the Alexa-Fluor 647-acceptor fluorophore on the Tracer-314. Binding of an inhibitor to the kinase competes for binding with the tracer, resulting in a loss of FRET.50 nL of compound dilutions were dispensed onto white 384-well small volume polystyrene plate as described in section 2.2. Then 5 μL of GST-mTOR and Europium-anti-GST antibody followed by 5 μL of tracer-314 (final assay volume 10 μL) are incubated at RT. The standard reaction buffer for the Lanthascreen™ kinase binding assay contained 50 mM HEPES pH 7.5, 5 mM MgCl2, 1 mM EGTA, 0.01% Pluronic F-127. Plates are read 60 mins later in a Synergy2 reader using an integration time of 0.2 microseconds and a delay of 0.1 microseconds.To calculate the emission ratio, the signal emitted at 665 nm from the acceptor (Alexa Fluor 647-labeled Tracer-314) is divided by the signal emitted at 620 nm from the donor (Eu3+ anti-GST antibody)Control for the 0% inhibition was given by the solvent vehicle of the compounds (90% DMSO in H2O). Control for the relative 100% inhibition was performed by adding 10 μM in the mix containing GST-mTOR and Europium anti-GST antibody. An additional control for the absolute 0% inhibition is given by Eu3+ anti-GST antibody without GST-mTOR.
- LanthaScreen Eu Kinase Binding Assay Binding of an Alexa Fluor™ conjugate or “tracer” to a kinase is detected by addition of an Eu-labeled anti-tag antibody. Binding of the tracer and antibody to a kinase result in a high degree of FRET, whereas displacement of the tracer with a kinase inhibitor results in a loss of FRET. This assay is carried out by mixing the compound tested with the reagents and reading, no development step is required. Life Technologies' Kinase Tracers are based on ATP-competitive kinase inhibitors, making them suitable for detection of any compounds that bind to the ATP site. Inhibitors that bind the ATP site include both Type I kinase inhibitors, which bind solely to the ATP site, and Type II inhibitors (e.g., Gleevec /Imatinib, Sorafenib, BIRB-796), which bind to both the ATP site and a second site often referred to as the allosteric site. The following protocol is used to carry out this assay: The Test Compounds are screened in 1% DMSO (final) in the well. For 10-point titrations, 3-fold serial dilutions are conducted from the starting concentration. All Kinase/Antibody Mixtures are diluted to a 2× working concentration in the specified kinase buffer. The 4× AlexaFluor labeled Tracer is prepared in Kinase Buffer. Assay Protocol Bar-coded, low volume, white 384-well plate (Greiner Cat. #784207) 1. 160 nL100× Test Compound in 100% DMSO 2. 3.84 μL—Kinase Buffer 3. 8.0 μL2× Kinase/Antibody Mixture 4. 4.0 μL—4× Tracer 5. 30-second plate shake 6. 60-minute incubation at room temperature7. Read on fluorescence plate reader and analyze the data. The following controls are made for each individual kinase and are located on the same plate as the kinase:0% Displacement Control: the maximum Emission Ratio is established by the 0% Displacement Control wells, which do not contain known inhibitor in the reaction and therefore exhibits no displacement of the tracer. 100% Displacement Control: the minimum Emission Ratio is established by the 100% Displacement Control wells, which contain the highest concentration of the known inhibitor used in that assay. Known Inhibitor Control Protocol: a known inhibitor control standard curve, 10-point titration, is run for each individual kinase on the same plate as the kinase to ensure the inhibitor is displaced within an expected IC50 range previously determined.