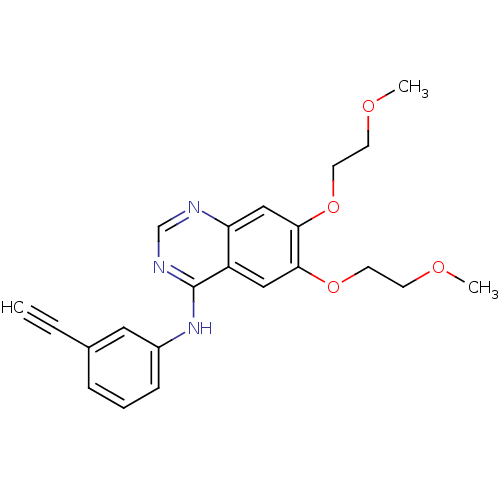
 ERLOTINIB HYDROCHLORIDE US9730934, Erlotinib US9409845, Table 1, Compound 22: erlotinib OSI-774 US10189853, erlotinib WO2022090481, Example erlotinib BDBM5446 Erotinib CHEMBL553 US11524945, Compound Erlotinib N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine US10507209, Compound Erlotinib Erlotinib Tarceva N-(3-Ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine Monohydrochloride cid_176870
ERLOTINIB HYDROCHLORIDE US9730934, Erlotinib US9409845, Table 1, Compound 22: erlotinib OSI-774 US10189853, erlotinib WO2022090481, Example erlotinib BDBM5446 Erotinib CHEMBL553 US11524945, Compound Erlotinib N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine US10507209, Compound Erlotinib Erlotinib Tarceva N-(3-Ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine Monohydrochloride cid_176870 ERLOTINIB HYDROCHLORIDE Erlotinib OSI-774 CP-358774-01 BDBM50311470 CHEMBL1079742
ERLOTINIB HYDROCHLORIDE Erlotinib OSI-774 CP-358774-01 BDBM50311470 CHEMBL1079742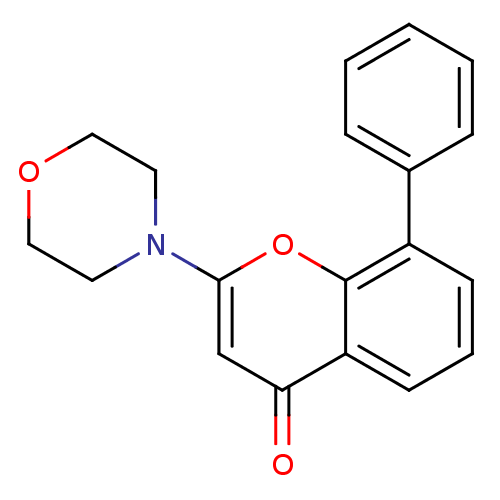 BDBM12915 CHEMBL98350 US9505780, LY294002 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one 2-(morpholin-4-yl)-8-phenyl-4H-chromen-4-one LY294002 US10308662, Compound LY294002
BDBM12915 CHEMBL98350 US9505780, LY294002 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one 2-(morpholin-4-yl)-8-phenyl-4H-chromen-4-one LY294002 US10308662, Compound LY294002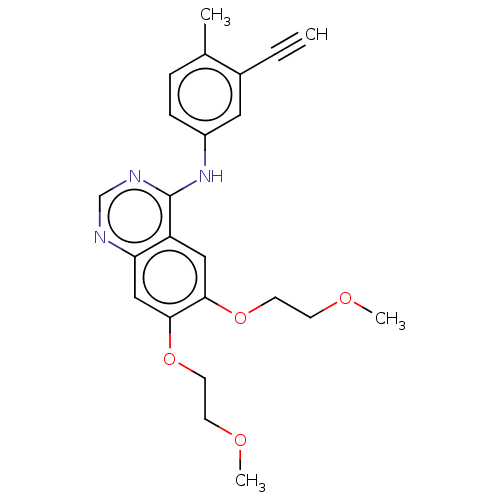 US11524945, Compound Erlotinib-4-methylphenyl analog (E4ME) BDBM582529
US11524945, Compound Erlotinib-4-methylphenyl analog (E4ME) BDBM582529
- Li, Z; Xu, M; Xing, S; Ho, WT; Ishii, T; Li, Q; Fu, X; Zhao, ZJ Erlotinib effectively inhibits JAK2V617F activity and polycythemia vera cell growth. J Biol Chem 282: 3428-32 (2007)
- Abbott, BM; Thompson, PE PDE2 inhibition by the PI3 kinase inhibitor LY294002 and analogues. Bioorg Med Chem Lett 14: 2847-51 (2004)
- Chen, KF; Pao, KC; Su, JC; Chou, YC; Liu, CY; Chen, HJ; Huang, JW; Kim, I; Shiau, CW Development of erlotinib derivatives as CIP2A-ablating agents independent of EGFR activity. Bioorg Med Chem 20: 6144-53 (2012)
- Chiosis, G; Rosen, N; Sepp-Lorenzino, L LY294002-geldanamycin heterodimers as selective inhibitors of the PI3K and PI3K-related family. Bioorg Med Chem Lett 11: 909-13 (2001)
- Zhang, Y; Tortorella, MD; Liao, J; Qin, X; Chen, T; Luo, J; Guan, J; Talley, JJ; Tu, Z Synthesis and Evaluation of Novel Erlotinib-NSAID Conjugates as More Comprehensive Anticancer Agents. ACS Med Chem Lett 6: 1086-90 (2015)
- Walker, EH; Pacold, ME; Perisic, O; Stephens, L; Hawkins, PT; Wymann, MP; Williams, RL Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 6: 909-19 (2000)
- Yang, X; Hou, Z; Wang, D; Mou, Y; Guo, C Design, synthesis and biological evaluation of novel heptamethine cyanine dye-erlotinib conjugates as antitumor agents. Bioorg Med Chem Lett 30: (2020)
- Jacobs, MD; Black, J; Futer, O; Swenson, L; Hare, B; Fleming, M; Saxena, K Pim-1 ligand-bound structures reveal the mechanism of serine/threonine kinase inhibition by LY294002. J Biol Chem 280: 13728-34 (2005)
- Chen, X; Du, Y; Sun, H; Wang, F; Kong, L; Sun, M Synthesis and biological evaluation of novel tricyclic oxazine and oxazepine fused quinazolines. Part 1: erlotinib analogs. Bioorg Med Chem Lett 24: 884-7 (2014)
- Biochemical Assay Thus, while these compounds were extensively used in studying ILK-mediated cellular and disease processes, their reported inhibitory effects are probably due to unknown artifacts or indirect binding events. Next, we turned our attention to previously reported studies on kinase profiling and quantitative chemical proteomics. These studies suggested that a widely known lung cancer drug erlotinib, which targets EGFR, might also bind to ILK as an off target. By performing a robust fluorescence-based binding assay, we found that the FDA approved drug Erlotinib (TARCEVA) indeed binds potently to purified recombinant ILK at KD 0.43M, which is very close to the affinity of Erlotinib to EGFR measured at the same experimental conditions (KD 0.31 μM). Another erlotinib-like EGFR inhibitor Gefitinib exhibited 10-fold weaker binding affinity to ILK (KD 4.51 μM) yet 3-fold stronger affinity to EGFR (KD 0.11 μM) than erlotinib.
- ChEMBL_1634412 (CHEMBL3877204) Inhibition of immobilized N-LY294002 bead binding to PI3Kalpha (unknown origin) expressed in HEK293T cells incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1634413 (CHEMBL3877205) Inhibition of immobilized N-LY294002 bead binding to PI3Kbeta (unknown origin) expressed in HEK293T cells incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1634414 (CHEMBL3877206) Inhibition of immobilized N-LY294002 bead binding to PI3Kdelta (unknown origin) expressed in HEK293T cells incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1634410 (CHEMBL3877202) Inhibition of immobilized N-LY294002 bead binding to BRD2 (unknown origin) expressed in HEK293T cell nuclear extract incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1634411 (CHEMBL3877203) Inhibition of immobilized N-LY294002 bead binding to BRD3 (unknown origin) expressed in HEK293T cell nuclear extract incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1634379 (CHEMBL3877171) Inhibition of immobilized N-LY294002 bead binding to C-terminal Flag-tagged BRD4 (unknown origin) expressed in HEK293T cell nuclear extract incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1538415 (CHEMBL3737962) Inhibition of EGFR T790M/del (746 to 750) deletion mutant phosphorylation in erlotinib-resistant human PC9 cells preincubated for 1 hr followed by stimulation with EGF for 8 mins by electrochemiluminescent immunoassay
- DNA-PK Enzyme Inhibitory Assay The assays of DNA-PK were performed by Reaction Biology Corporation, One Great Valley Parkway, Suite 2 Malvern, PA 19355 USA. All Compounds were dissolved in DMSO (negative control solvent) and tested for their ability to inhibit human DNA-PK enzyme activity. All synthesized compounds were tested in a 10-dose IC50 profile with 4-fold serial dilutions starting at concentration 100 μM. The control compound, LY294002 was tested in a 10-dose IC50 profile with 3-fold serial dilutions starting at 10 μM concentration. Reactions were carried out using 20 μM peptide substrate [EPPLSQEAFADLWKK], 10 μg/ml DNA and 10 μM ATP using the HTRF assay format.
- Kinase Inhibition Assay Reagents and Materials:WT EGFR (Carna, Cat. No. 08-115), EGFR [L858R] (Carna, Cat. No. 08-502), EGFR [L858R/T790M] (Carna, Cat. No. 08-510), ATP (Sigma, Cat. No. A7699-1G), DMSO (Sigma, Cat. No. D2650), 96-well plate (Corning, Cat. No. 3365), 384-well plate (Greiner, Cat. No. 784076), HTRF Kinase TK Kit (Cisbio, Cat. No. 62TK0PEJ), Erlotinib (Selleckchem, Cat. No. S7787), EGFR [d746-750] (Life Technologies, Cat. No. PV6178), 5× Kinase Buffer A (Life Technologies, Cat. No. PV3186), Kinase Tracer 199 (Life Technologies, Cat. No. PV5830), LanthaScreen Eu-anti-GST antibody (Life Technologies, Cat. No. PV5594).Specific Experimental Protocol:Compound preparation: the test compound was dissolved in DMSO to make a 20 mM stock solution. Then, it was diluted in DMSO with a 3-fold series gradient dilution for 10 times. The dilutions were diluted 10 fold with buffer when dosing.WT EGFR and EGFR [L858R/T790M] kinase assay: WT EGFR or EGFR [L858R/T790M] kinase was mixed with different concentrations of pre-diluted compounds for 10 minutes in 5× Kinase Buffer A in duplicate. The corresponding substrate and ATP were added and reacted at room temperature for 20 minutes (in which a negative and a positive control were set: the negative control is blank and the positive control is erlotinib). After the reaction, the detection reagent (the reagent in the HTRF Kinase TK kit) was added, and after incubation at room temperature for 30 minutes, the enzyme activity in the presence of the compounds of the present disclosure at each concentration was measured by an Evnvision microplate reader, and the inhibition of the enzyme by the compound at each concentrations were calculated. The inhibitions of the enzyme activity by the compounds at different concentrations were then fitted using Graphpad 5.0 software according to the four-parameter equation, and the IC50 values were calculated.
- Kinase Assay JAK2 and JAK2 [V617F] Kinase Assay: In 5× Kinase Buffer A, JAK2 or JAK2 [V617F] kinase was mixed with pre-diluted compounds at different concentrations in duplicate for 10 minutes. The corresponding substrate and ATP were added and reacted at room temperature for 20 minutes (in which negative and positive controls were set: the negative control was a blank control and the positive control was erlotinib). After the reaction was completed, a detection reagent (the reagent in the HTRF Kinase TK kit) was added. After incubation at room temperature for 30 minutes, the enzyme activities in the presence of the compounds disclosed herein at each concentration were measured by an Evnvision microplate reader, and inhibitory activities of compounds at different concentrations on the enzyme activity were calculated. The inhibitory activities of compounds at different concentrations on enzyme activity were then fitted according to the four-parameter equation using Graphpad 5.0 software, and the IC50 values were calculated.
- Kinase Inhibition Assay WT EGFR and EGFR[L858R/T790M] kinase detection: In 5× kinase buffer A, WT EGFR or EGFR[L858R/T790M] kinase was mixed with different concentrations of compounds prepared by pre-dilution for 10 minutes. Each concentration was tested in duplicate. The corresponding substrate and ATP were added, and reaction was performed at room temperature for 20 minutes (negative and positive controls were provided: the negative control was a blank control, and the positive control was erlotinib). After completion of the reaction, detection reagents (reagents in the HTRF Kinase TK kit) were added. After incubating for 30 minutes at room temperature, enzyme activity in the presence of various concentrations of the compounds of the present disclosure was determined by Evnvision microplate reader, and inhibitory activity of different concentrations of the compounds on enzyme activity was calculated. The inhibitory activity of different concentrations of the compounds on enzyme activity was then fitted by Graphpad 5.0 software according to the four-parameter equation, and the IC50 value was calculated.
- Inhibition of PI3K alpha Quantification of ATP to ADP conversion as a measure of PI3Kα activity. Active PI3Kα (Life Technologies), in the presence or absence of PI3Kα inhibitor, was reacted with PIP2:PS (Life Technologies), a substrate specifically optimized for use with Class I PI3 kinases, and ultrapure ATP (Promega). The conversion of ATP to ADP by PI3Kα was measured as luminescence signal via Promega ADP-Glo kinase activity assay. Assay was validated using published PI3Kα inhibitors LY294002, PI-103, BYL719, GDC0198 and also DMSO vehicle control.Compounds were prepared at 100× final concentration as a 12-point, 1:3 serial-dilution in DMSO series, with DMSO control as 12th point. Compound was then diluted in (25 mM HEPES pH 7.5, 1 mM EGTA, 0.3% CHAPS) prior to addition to PI3Kα. Active PI3Kα diluted to 0.24 ng/μL (1.1 nM) in (50 mM HEPES pH 7.5, 6 mM MgCl2, 1 mM EGTA, 200 mM NaCl, 0.03% CHAPS, 8 mM DTT) was incubated with compound for 0 hr and 3 hr prior to the start of the reaction. 25 μM PIP2:PS and 60 μM ATP were diluted from stock in (25 mM HEPES pH 7.5, 1 mM EGTA, 0.3% CHAPS) and added to initiate the PI3Kα reaction. Reaction time was 30 minutes. ATP to ADP conversion was measured in Luminescence Counts on DTX880 Plate Reader (Beckman Coulter). Compound IC50s were reported using GraphPad Prism software. Analytical method was non-linear regression, 4-parameter curve fit with bottom fit to validated PI3Kα inhibitor reference controls and no top fit (floating top).
- pAKT Protocol Inhibition of the PI3K-AKT-mTOR pathway was measured by quantifying the loss of (Ser-473) pAKT using AlphaScreen (Perkin Elmer). B103 (Rat Neuroblastoma) cells were seeded in serum containing medium (High Glucose DMEM (-Phenol Red)+10% FBS+2× Glutamax+1 mM Sodium Pyruvate+10 mM HEPES+1× Non-Essential Amino Acids+1× Pen/Strep) on a 96-well tissue culture treated plate and grown for 20 hours. Cells were then serum starved in serum free medium (High Glucose DMEM (-Phenol Red)+1× Glutamax+1 mM Sodium Pyruvate+1× Pen/Strep) for 6 hours prior to a 2-hour pretreatment with inhibitors of the pathway, including reference inhibitor LY294002. These inhibitors were prepared at a 200× final concentration as a 6-point, 1:3 serial dilution in DMSO series, with DMSO as the 7th point. The inhibitors were then diluted in experimental medium (High Glucose DMEM (-Phenol Red)+1× Glutamax+1 mM Sodium Pyruvate+1× Pen/Strep+25 mM HEPES+0.1% BSA) and combined with the cells at 1× final concentration in 0.5% DMSO. The cells were then stimulated for 20 minutes with (2.5 μg/mL) insulin, an activator of the PI3K-AKT-mTOR pathway and a demonstrated (Ser-473) pAKT agonist. Cells were promptly lysed using Perkin Elmer proprietary lysis buffer and the (Ser-473) pAKT and total AKT contained in the lysate was measured by AlphaScreen. In AlphaScreen, donor beads were coated with streptavidin to capture one of the antibodies, which is biotinylated. Acceptor beads were coated with Protein A to immobilize the other antibody. In the presence of target protein, the two antibodies bring the donor and acceptor beads close together, generating signal. The amount of light emission is directly proportional to the amount of target protein present in the sample. For each inhibitor tested: the ratio of measured (Ser-473) pAKT/totalAKT was plotted in GraphPad Prism as a 7-point, non-linear regression, 4-parameter curve with bottom constrained to reference control bottom and unconstrained top anchored to DMSO.
- In Vitro Enzyme Inhibitory Activity Samples:Controls: Gefitinib, erlotinib hydrochloride, purchased from Anqing worldchem Co., LTD.; lapatinib ditosylate, purchased from Taizhou Xingcheng Chempharm Co., Ltd.; CI-1033 hydrochloride, purchased from Shanghai hanxiangchem, Co., Ltd.; andThe present compounds: lab-made, their chemical names and structural formulae are shown in the preparation examples.Assay Procedures:The abbreviations used in the following assay have the following meanings:HEPES: hydroxyethyl piperazine ethanesulfonic acid;Brij-35: polyoxyethylene lauryl ether;DTT: dithiothreitol;Coating Reagent #3: #3 coating agent;EDTA: ethylene diamine tetraacetic acid, purchased from Sigma Co. Ltd.;FAM labeled peptide: fluorescein labeled peptide 22 (GL Biochem);ATP: adenosine triphosphate (Sigma);DMSO: dimethyl sulfoxide;EGFR: human epidermal growth factor receptor (Carna);HER2: human epidermal growth factor receptor 2 (Carna);HER4: human epidermal growth factor receptor 4 (Carna).1. Formulating the agents to be used in the assay(1) 1.25-fold MnCl2-free kinase buffer (62.5 mM HEPES, PH 7.5, 0.001875% Brij-35, 12.5 mM MgCl2, 2.5 mM DTT);(2) 1.25-fold MnCl2-containing kinase buffer (62.5 mM HEPES, pH 7.5, 0.001875% Brij-35, 12.5 mM MgCl2, 12.5 mM MnCl2, 2.5 mM DTT);(3) Stop buffer (100 mM HEPES, pH 7.5, 0.015% Brij-35, 0.2% Coating Reagent #3, 50 mM EDTA);(4) 2.5-fold kinase solutions (to the 1.25-fold kinase buffers were added the corresponding kinases to formulate 2.5-fold EGFR, HER2, HER4 kinase solutions);(5) 2.5-fold peptide solutions (to the 1.25-fold kinase buffers were added FAM labeled peptide and ATP to formulate the peptide solutions);(6) 5-fold compound solutions (using 100% DMSO to formulate 50-fold compound solutions having different concentration gradients, and diluting with water by 10 times to obtain 5-fold compound solutions having different concentration gradients);2. Adding 5 μL of a 5-fold compound solution to a 384-well plate;3. Adding 10 μL of a 2.5-fold kinase solution to incubate for 10 min;4. Then adding 10 μL of a 2.5-fold peptide solution, and reacting at 28° C. for 1 h; and5. Finally, adding 25 μL of stop buffer to terminate the reaction, and reading the data with Caliper.6. Curve fitting to obtain an IC50 value.The calculated inhibition ratio (%)=(the maximum conversion rate−the conversion rate)/(the maximum conversion rate−the minimum conversion rate)×100The curve fitting was conducted with the Xlfit software to obtain IC50 values.