 US20240246972, Compound Staurosporine US10189849, staurosporine US11091485, Compound Staurosporine US11542261, Compound staurosporine US11814388, Compound Staurosporine US20240025908, Example Staurosporine BDBM139540 US10307427, Staurosporine US8889696, Staurosporine US9586965, Control Staurosporine US10329300, Staurosporine US20230312583, Compound staurosporine US9051313, Staurosporine
US20240246972, Compound Staurosporine US10189849, staurosporine US11091485, Compound Staurosporine US11542261, Compound staurosporine US11814388, Compound Staurosporine US20240025908, Example Staurosporine BDBM139540 US10307427, Staurosporine US8889696, Staurosporine US9586965, Control Staurosporine US10329300, Staurosporine US20230312583, Compound staurosporine US9051313, Staurosporine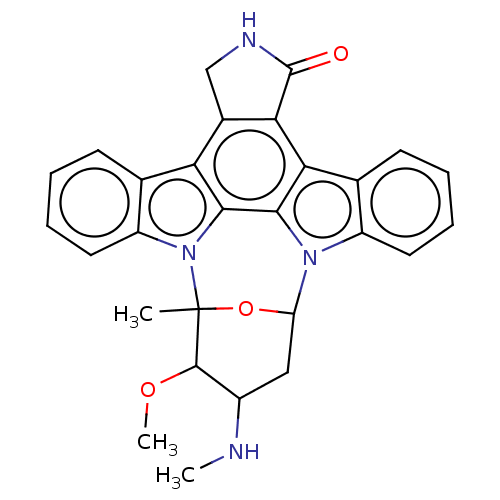 US10927120, Compound staurosporine US9920060, Staurosporine BDBM130909 US10683289, Example Staurosporine US8822500, Stauro- sporine
US10927120, Compound staurosporine US9920060, Staurosporine BDBM130909 US10683289, Example Staurosporine US8822500, Stauro- sporine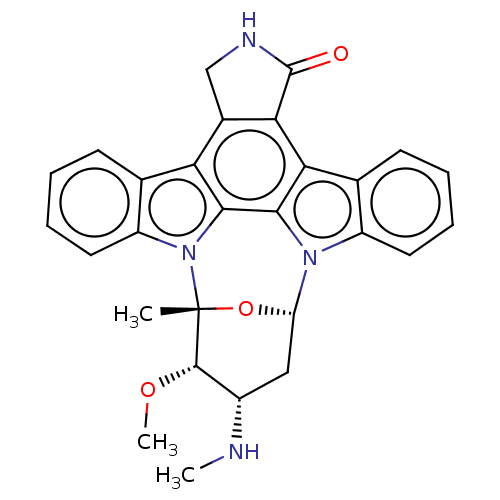 Staurosporine cid_451705 US20240300958, Compound Staurosporine BDBM31096 CHEMBL290084
Staurosporine cid_451705 US20240300958, Compound Staurosporine BDBM31096 CHEMBL290084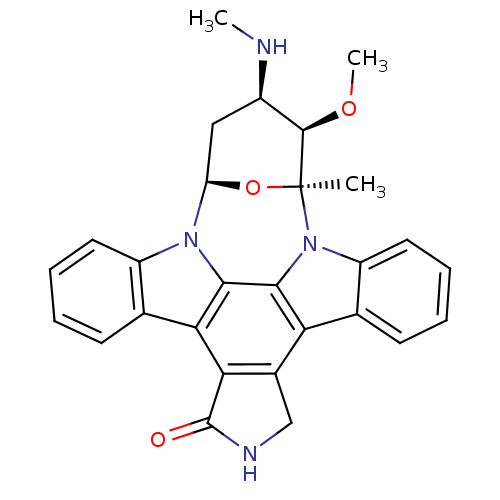 (2S,3R,4R,6R)-3-methoxy-2-methyl-4-(methylamino)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.1^{2,6}.0^{7,28}.0^{8,13}.0^{15,19}.0^{20,27}.0^{21,26}]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one Staurosporine US9226923, Staurosporine US9206188, Staurosporine Staurosporin, 4 CHEMBL388978 US12398142, Compound Staurosporine US20240002365, Compound staurosporine US11958831, Example Staurosporine US12410173, Compound Staurosporine BDBM2579 US12350272, Compound Staurosporine US20240309004, Compound STAUROSPORINE Staurosporine, 8 US20240254124, Compound Staurosporine
(2S,3R,4R,6R)-3-methoxy-2-methyl-4-(methylamino)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.1^{2,6}.0^{7,28}.0^{8,13}.0^{15,19}.0^{20,27}.0^{21,26}]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one Staurosporine US9226923, Staurosporine US9206188, Staurosporine Staurosporin, 4 CHEMBL388978 US12398142, Compound Staurosporine US20240002365, Compound staurosporine US11958831, Example Staurosporine US12410173, Compound Staurosporine BDBM2579 US12350272, Compound Staurosporine US20240309004, Compound STAUROSPORINE Staurosporine, 8 US20240254124, Compound Staurosporine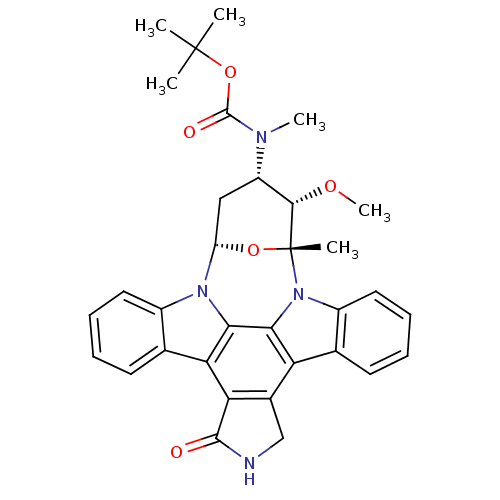 CHEMBL338449 Staurosporine derivative BDBM50283884
CHEMBL338449 Staurosporine derivative BDBM50283884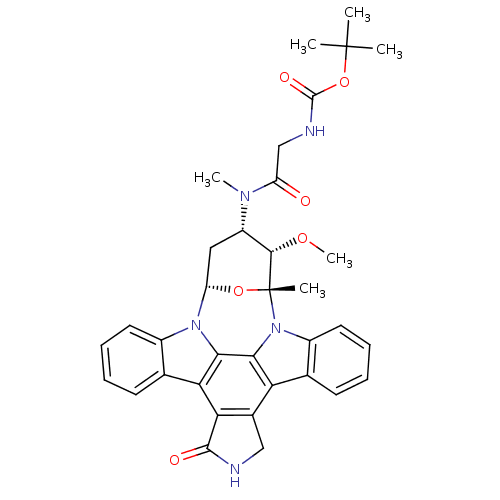 Staurosporine derivative BDBM50283901 CHEMBL335931
Staurosporine derivative BDBM50283901 CHEMBL335931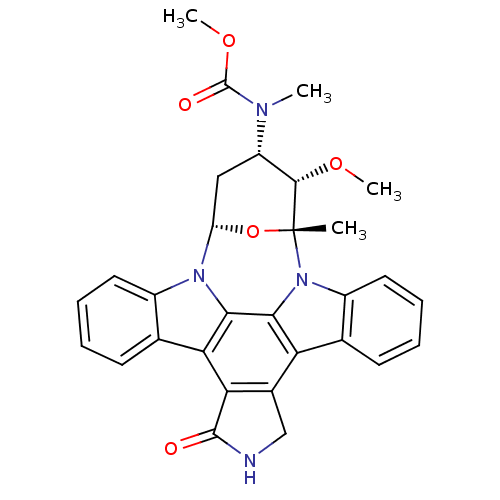 Staurosporine derivative CHEMBL130049 BDBM50283889
Staurosporine derivative CHEMBL130049 BDBM50283889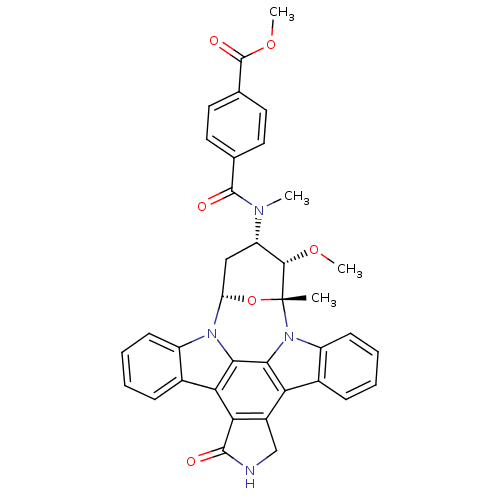 Staurosporine derivative CHEMBL336192 BDBM50283878
Staurosporine derivative CHEMBL336192 BDBM50283878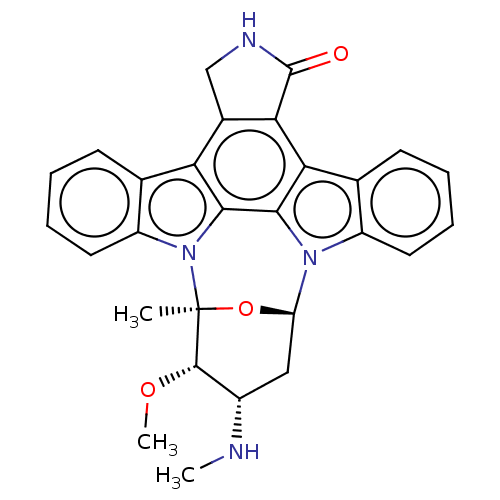 BDBM32337 MLS000028832 cid_5311103 SMR000058536 STAUROSPORINE
BDBM32337 MLS000028832 cid_5311103 SMR000058536 STAUROSPORINE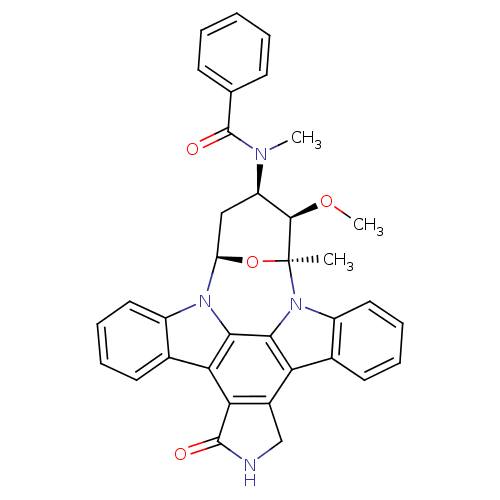 CHEMBL608533 PKC-412 BDBM50326053 US20240132489, Compound Staurosporine
CHEMBL608533 PKC-412 BDBM50326053 US20240132489, Compound Staurosporine CHEMBL162 Isoindolinone Urea derivative 3-methoxy-2-methyl-4-methylamino-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14,19,21(26),22,24,27-nonaen-16-one BDBM50059889 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21,23,25-nonaen-16-one 12-[3-methoxy-6-methyl-4-methylamino-(2S,3R,4R,6S)-tetrahydro-2H-2-pyranyl]-6,7,12,13-tetrahydro-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazol-5-one staurosporin (staurosporine)3-methoxy-2-methyl-4-methylamino-(2S,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2S,3S,4S,6R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21,23,25-nonaen-16-one AM-2282 15,16-dimethoxy-15-methyl-17-methylamino-(15S)-4,14,20-triazaheptacyclo[12.12.2.02,6.07,28.08,13.020,27.021,26]octacosa-1(27),2(6),7(28),8(13),9,11,21(26),22,24-nonaen-3-one 3-methoxy-2-methyl-4-methylamino-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14,19,21(26),22,24,27-nonaen-16-one 8,12-Epoxy-1H,8H-2,7b,12a-triazadibenzo(a,g)cyclonona(cde)trinden-1-one,2,3,9,10,11,12-hexahydro-9-methoxy-8-methyl-10-(methylamino)-, (8alpha,9beta,10beta,12alpha)-(+)- 3-methoxy-2-methyl-4-methylamino-(2S,3R,4R,6R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2S,3R,4R,6R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-4-methylamino-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 2-Amino-4-(2-amino-2-phenyl-ethyl)-6-(2-hydroxy-phenyl)-nicotinonitrile 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 12-[3-methoxy-2,6-dimethyl-4-methylamino-(2S,3R,4R,6S)-tetrahydro-2H-2-pyranyl]-6,7,12,13-tetrahydro-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazol-5-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one(Staurosporine) Indolocarbazole nitrogen derivative 3-methoxy-2-methyl-4-methylamino-(2S,3R,4R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14,19,21(26),22,24,27-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14,19,21(26),22,24,27-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2S,3R,4R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one
CHEMBL162 Isoindolinone Urea derivative 3-methoxy-2-methyl-4-methylamino-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14,19,21(26),22,24,27-nonaen-16-one BDBM50059889 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21,23,25-nonaen-16-one 12-[3-methoxy-6-methyl-4-methylamino-(2S,3R,4R,6S)-tetrahydro-2H-2-pyranyl]-6,7,12,13-tetrahydro-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazol-5-one staurosporin (staurosporine)3-methoxy-2-methyl-4-methylamino-(2S,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2S,3S,4S,6R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21,23,25-nonaen-16-one AM-2282 15,16-dimethoxy-15-methyl-17-methylamino-(15S)-4,14,20-triazaheptacyclo[12.12.2.02,6.07,28.08,13.020,27.021,26]octacosa-1(27),2(6),7(28),8(13),9,11,21(26),22,24-nonaen-3-one 3-methoxy-2-methyl-4-methylamino-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14,19,21(26),22,24,27-nonaen-16-one 8,12-Epoxy-1H,8H-2,7b,12a-triazadibenzo(a,g)cyclonona(cde)trinden-1-one,2,3,9,10,11,12-hexahydro-9-methoxy-8-methyl-10-(methylamino)-, (8alpha,9beta,10beta,12alpha)-(+)- 3-methoxy-2-methyl-4-methylamino-(2S,3R,4R,6R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2S,3R,4R,6R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 3-methoxy-4-methylamino-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 2-Amino-4-(2-amino-2-phenyl-ethyl)-6-(2-hydroxy-phenyl)-nicotinonitrile 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-16-one 12-[3-methoxy-2,6-dimethyl-4-methylamino-(2S,3R,4R,6S)-tetrahydro-2H-2-pyranyl]-6,7,12,13-tetrahydro-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazol-5-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one(Staurosporine) Indolocarbazole nitrogen derivative 3-methoxy-2-methyl-4-methylamino-(2S,3R,4R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14,19,21(26),22,24,27-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2R,3S,4S,6S)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14,19,21(26),22,24,27-nonaen-16-one 3-methoxy-2-methyl-4-methylamino-(2S,3R,4R)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8(13),9,11,14(28),15(19),20(27),21(26),22,24-nonaen-16-one
- Yang, SM; Malaviya, R; Wilson, LJ; Argentieri, R; Chen, X; Yang, C; Wang, B; Cavender, D; Murray, WV Simplified staurosporine analogs as potent JAK3 inhibitors. Bioorg Med Chem Lett 17: 326-31 (2007)
- Caravatti, G; Meyer, T; Fredenhagen, A; Trinks, U; Mett, H; Fabbro, D Inhibitory activity and selectivity of staurosporine derivatives towards protein kinase C Bioorg Med Chem Lett 4: 399-404 (1994)
- Esvan, YJ; Giraud, F; Pereira, E; Suchaud, V; Nauton, L; Théry, V; Dezhenkova, LG; Kaluzhny, DN; Mazov, VN; Shtil, AA; Anizon, F; Moreau, P Synthesis and biological activity of pyrazole analogues of the staurosporine aglycon K252c. Bioorg Med Chem 24: 3116-24 (2016)
- Tripathy, R; Angeles, TS; Yang, SX; Mallamo, JP TrkA kinase inhibitors from a library of modified and isosteric Staurosporine aglycone. Bioorg Med Chem Lett 18: 3551-5 (2008)
- Walker, EH; Pacold, ME; Perisic, O; Stephens, L; Hawkins, PT; Wymann, MP; Williams, RL Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 6: 909-19 (2000)
- Hirozane, Y; Toyofuku, M; Yogo, T; Tanaka, Y; Sameshima, T; Miyahisa, I; Yoshikawa, M Structure-based rational design of staurosporine-based fluorescent probe with broad-ranging kinase affinity for kinase panel application. Bioorg Med Chem Lett 29: (2019)
- ChEMBL_698124 (CHEMBL1646044) Inhibition of DNase gamma-mediated HMGB1 release expressed in human staurosporine-induced HeLaS3 cells treated 1 hr before staurosporine challenge measured after 24 hrs by Western blotting
- ChEMBL_574191 (CHEMBL1060392) Antagonist activity at human 5HT transporter expressed in staurosporine treated human JAR cells
- ChEMBL_2232555 (CHEMBL5146327) Inhibition of GST-tagged CDK2 (unknown origin) expressed in Escherichia coli in presence of Staurosporine by competitive binding assay
- ChEBML_201366 Affinity for HC Serotonin transporter determined in vitro by incubating compound and [3H]5-HT with human carcinoma (Jar cells), previously treated with staurosporine
- ChEMBL_741337 (CHEMBL1764806) Inhibition of staurosporine induced activation of caspase 3 activation in human Hela cells assessed as hydrolysis of Z-DEVD-R110 substrate by microplate fluorescence assay
- ChEMBL_2369785 Inhibition of 8-((4-chlorophenyl)amino)naphthalene-1-sulfonic acid binding to CDK2 (unknown origin) incubated for 2 hrs in presence of staurosporine by fluorescence based analysis
- In-Vitro Kinase Inhibition Assay Compounds 2-4 and cabozantinib were each tested for binding of c-Met, VEGFR2, TIE2 and the control compound, staurosporine. Specifically, each compound was tested at a 3-fold serial dilution starting at 10 microMolar ( μM ) in a 10-dose IC50 mode into an enzyme/substrate mixture using acoustic technology, and pre-incubated for 20 minutes to ensure compounds were equilibrated and bound to the enzyme. Staurosporine was used as a control and was tested at a 4-fold serial dilution starting at 20 μM. Next, 5 concentrations of ATP were added to initiate the reaction. The activity was monitored every 5-15 min for a time course study.
- Kinase Activity Assay Compounds VKT-007, VKT-034, VKT-036, and VKT-511 were tested in 10-dose IC50 duplicate mode with a 3-fold serial dilution starting at 10 μM. Control compound, Staurosporine, was tested in 10-dose IC50 mode with 4-fold serial dilution starting at 20 μM. Reactions were carried out at 10 μM ATP. Compound VKT-320 was tested in 10-dose IC50 triplicate mode with a 3-fold serial dilution starting at 9.827 μM. Control compound, Staurosporine, was tested in 10-dose IC50 mode with 4-fold serial dilution starting at 20 μM. Reactions were carried out at 10 μM ATP.
- LRRK2 Kinase Assay Procedure: Enzyme was incubated with substrate peptide in reaction buffer in the presence and absence of test compounds or Staurosporine. All additions were done on ice, followed by the addition of ATP mix. Wells were uniformly mixed using an Eppendorff plate shaker and incubated at 30° C. for 20 min, and stopped by the addition of 5 μl of 3% phosphoric acid. Volume was increased to 100 μl by adding 0.8% phosphoric acid which was then transferred to PC filter mats (Millipore), pre-equilibrated with 70% ethanol and water. Plates were washed thrice with 100 μl 0.8% phosphoric acid and dried for an hour at 60° C. 100 μl scintillation fluid was added into each well and reading taken in Perkin Elmer TOPCOUNT beta counter. The data analysis was performed by averaging the duplicate top count readings for each standard, negative, positive control (enzyme control) and samples and subtracting the average negative control from each reading which results in corrected values. A validation EC50 curve was generated by plotting CPM for each Staurosporine concentration on y-axis against the Log concentration of Staurosporine (nM) on the x-axis followed by a best fit curve through the points.
- Z-LYTE biochemical assay To the assay kit, 2.5 µL of different concentrate of the test compounds, Pazopanib or water (control) were added and incubated for 1 h at room temperature followed by the addition of 5 µL development agent. The reaction was stopped by the addition of 5 µL stop reagent. The fluorescence intensity at 445 and 520 nm were monitored and the standard inhibitory reference compound was Staurosporine.
- Inhibition Assay The inhibitory activities of compounds of Formula I were assay at Reaction Biology Corporation, One Great Valley Parkway, Malvern, Pa., USA. Human ALK and cMet enzymes were used and the substrate was a peptide substrate, poly[Glu:Tyr] (4:1) at a concentration of 0.2 mg/ml. The ATP concentration for the assay was 10 uM and Staurosporine was used as a standardwith an IC50 of 2.3 nM, and 75 nM, respectively for ALK and cMet.
- IC50 Kinase Assays Table 8-10: Compounds were tested in 10-dose IC50 mode with 3-fold serial dilution starting at 10 uM, and are relative to DMSO, the negative control. The positive control, Staurosporine, was tested in a 10-dose IC50 mode with 4-fold serial dilution starting at 20 uM. Reactions were carried out at 10 uM ATP. Curve fits were performed to determine IC50 where the enzyme activities at the highest concentration of compounds were less than 65%.
- Inhibition Assay To test the efficacy of caspase-3 inhibitors at the cellular level, the ability of selected compounds to inhibit the proteolytic cleavage of PARP (poly ADP-ribose polymerase) was evaluated in live Hela cells.Briefly, in this assay Hela cells are seeded in 96 well plates and incubated for 4 hours with staurosporine, a well characterized inducer of apoptosis, alone or together with different concentrations of compound (50, 25, 10 and 3 uM). After formaldehyde-based fixation, the cells are stained with a fluorescein-labeled anti-cleaved PARP antibody (Cell signaling, Cat#: 9547) and counterstained with Hoechst33342 (Invitrogen, Cat#: H3570) to mark all nuclei.
- Kinase Inhibition Assay The compound of Example 39, 2-(1H-indol-5-ylamino)-6-(2,4-difluorophenylsulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one, was subjected to a kinase inhibition assay for the kinases. Compounds were tested in 5 dose IC50 mode with 10-fold serial dilution starting at 10 μM. Staurosporine, a known protein kinase inhibitor, was tested in 5-dose IC50 mode with 3-fold serial dilution starting at 20 μM. Reactions were carried out in 10 μM ATP. The results are shown in Table 5. The results show that the compound of Example 39 is a kinase inhibitor highly selective for the kinase Plk2.
- ADP-Glo Kinase Assay The STK3 kinase assay was performed in 5 μL reaction buffer containing 50 ng recombinant human STK3 protein (full length, SignalChem #S24-10G; Richmond, Canada), 250 μg/mL myelin basic protein (Sigma-Aldrich #M1891; St. Louis, MO, USA), and 50 μM ATP (Sigma-Aldrich #A7699). The STK4 kinase assay was performed in 5 μL reaction buffer containing 50 ng recombinant human STK4 protein (full length, SignalChem #S25-10G), 300 μg/mL Axltide (SignalChem #A16-58), and 50 μM ATP. IC50 values were determined with 10 concentrations of compounds serially diluted 3-fold from a starting concentration of 30 μM. Staurosporine, a non-selective protein kinase inhibitor, was included in the assay as a positive control. Three experiments were performed, each in triplicate.
- Biochemical Inhibition of Enzymatic Activities Assay Table D: Corresponding biochemical inhibition of enzymatic activities of FLT3 (wt), FLT3 (D835Y), and FLT3 (ITD) were measured using recombinant protein constructs of kinase domains via activity based FLT3 kinase assay for compound screening and profiling via radiometric HotSpot™ kinase assay (Reaction Biology). Peptide substrate [EAIYAAPFAKKK]. Compounds were dissolved to 10 mM in DMSO. Compounds were tested in 10-dose IC50 mode with a 3-fold serial dilution starting at 0.3 μM. Control compound, Staurosporine, was tested in 10-dose IC50 mode with 4-fold serial dilution starting at 20 μM. Alternate control compounds were tested in 10-dose IC50 mode with 3-fold serial dilution starting at 20 μM. Reactions were carried out at 1 μM ATP.
- IMAP High-Throughput Screening The IMAP Screening Express Kit (Molecular Devices, Sunnyvale, CA) was used for the high-throughput screening experiments. Compounds from the Developmental Therapeutics Program (DTP) Open Repository were solubilized and diluted in DMSO and initially tested at one concentration in the assay. Active compounds were subsequently titrated at 20 2-fold dilutions. Reactions were performed using recombinant Chk2 with the indicated drug concentrations in reaction buffer in 384-well black microplate (Greiner Bio-One, Longwood, FL)] for 60 min at room temperature. IMAP binding reagent was added to each well, plates were incubated for 30 min at room temperature, and fluorescence polarization was measured using a Tecan Ultra plate reader. Each screening plate contained staurosporine as a positive control.
- Inhibition In Vitro Assay Selected compounds disclosed herein were tested in CDK4/cyclinD1, CDK2/CycA and CDK2/cyclinE kinase assays to determine their inhibitory effect on these CDKs. The assays were performed using microfluidic kinase detection technology. The compounds were tested in 12-point dose-response format in singlicate at Km for ATP. Phosphoacceptor substrate peptide concentration used was 1 μM for all assays and Staurosporine was used as the reference compound for all assays. Specifics of each assay are as described below:CDK2/CyclinA: Enzyme concentration: 0.2 nM; ATP concentration: 50 μM; Incubation time: 3 hr.CDK2/CyclinE: Enzyme concentration: 0.28 nM; ATP concentration: 100 μM; Incubation time: 1 hr.CDK4/CyclinD1: Enzyme concentration: 1 nM; ATP concentration: 200 μM; Incubation time: 10 hr.
- Caliper Assay Selected compounds disclosed herein were tested in CDK4/cyclinD1, CDK2/CycA and CDK2/cyclinE kinase assays by Nanosyn (Santa Clara, Calif.) to determine their inhibitory effect on these CDKs. The assays were performed using microfluidic kinase detection technology (Caliper Assay Platform). The compounds were tested in 12-point dose-response format in singlicate at Km for ATP. Phosphoacceptor substrate peptide concentration used was 1 μM for all assays and Staurosporine was used as the reference compound for all assays. Specifics of each assay are as described below:CDK2/CyclinA: Enzyme concentration: 0.2 nM; ATP concentration: 50 μM; Incubation time: 3 hr.CDK2/CyclinE: Enzyme concentration: 0.28 nM; ATP concentration: 100 μM; Incubation time: 1 hr.CDK4/CyclinD1: Enzyme concentration: 1 nM; ATP concentration: 200 μM; Incubation time: 10 hr.
- Inhibition In Vitro Assay Selected compounds disclosed herein were tested in CDK4/cyclinD1, CDK2/CycA and CDK2/cyclinE kinase assays by Nanosyn (Santa Clara, Calif.) to determine their inhibitory effect on these CDKs. The assays were performed using microfluidic kinase detection technology (Caliper Assay Platform). The compounds were tested in 12-point dose-response format in singlicate at Km for ATP. Phosphoacceptor substrate peptide concentration used was 1 μM for all assays and Staurosporine was used as the reference compound for all assays. Specifics of each assay are as described below:CDK2/CyclinA: Enzyme concentration: 0.2 nM; ATP concentration: 50 μM; Incubation time: 3 hr.CDK2/CyclinE: Enzyme concentration: 0.28 nM; ATP concentration: 100 μM; Incubation time: 1 hr.CDK4/CyclinD1: Enzyme concentration: 1 nM; ATP concentration: 200 μM; Incubation time: 10 hr.
- Flashplate Assay The kinase assay is performed as 384-well flashplate assay (for example for Topcount measurement).0.6 nM TANK binding kinase (TBK1), 800 nM biotinylated MELK-derived peptide (Biotin-Ah-Ah-AKPKGNKDYHLQTCCGSLAYRRR) and 10 uM ATP (spiked with 0.25 uCi of 33P-ATP/well) are incubated at 30 C. for 120 min in a total volume of 50 ul (10 mM MOPS, 10 mM Mg acetate, 0.1 mM EGTA, 1 mM DTT, 0.02% of Brij35, 0.1% of BSA, pH 7.5) with or without test compound. The reaction is stopped with 25 ul of 200 mM EDTA. After 30 min at room temperature, the liquid is removed, and each well is washed three times with 100 ul of 0.9% sodium chloride solution. Non-specific reaction is measured in the presence of 100 nM staurosporine. The radioactivity is measured in a Topcount (PerkinElmer).
- In Vitro Inhibitory Activity Assay For the inhibitory activity measurement of each compound, the compound of the present invention or staurosporine was first serially diluted with dimethyl sulfoxide (DMSO). Next, the HER2 protein, the substrate peptide (final concentration: 0.5 uM), manganese chloride (final concentration: 10 mM), ATP (final concentration: 6 uM), and the solution of the compound of the present invention in DMSO (final concentration of DMSO: 5%) were added into a buffer solution for kinase reaction (15 mM Tris (pH 7.5), 2 mM dithiothreitol, and 0.01% Tween 20), and the mixture was incubated at 25° C. for 40 minutes for kinase reaction. The reaction was terminated by adding EDTA (final concentration: 30 mM) thereto. Finally, the unphosphorylated substrate peptide (S) and the phosphorylated peptide (P) were separated and detected by microcapillary electrophoresis using LabChip EZ Reader II (PerkinElmer Inc.).
- Inhibition Assay The experimental batches are carried out in a flashplate system with 384 wells/microtitration plate.In each case, the PDK1 sample His6-PDK1 (1-50)(3.4 nM), the PDK1 substrate biotin-bA-bA-KTFCGTPEYLAPEVRREPRILSEEEQEMFRDFDYIADWC (400 nM), 4 μM ATP (with 0.2 μCi of 33P-ATP/well) and the test substance in 50 μl of conventional experimental solution per well are incubated at 30° C. for 60 min. The test substances are employed in corresponding concentrations (if desired in a dilution series). The control is carried out without test substance. The reaction is stopped using standard methods and washed. The activity of the kinase is measured via the incorporated radioactivity in top count. In order to determine the non-specific kinase reaction (blank value), the experimental batches are carried out in the presence of 100 nM staurosporine.
- Kinase Assay Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1 Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated and phosphorylated forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- Kinase Inhibition Assay 2.5 times of the kinase (VEGFR2 or EGFR or FYN) buffer solution was added to the compound diluted in equal concentration in the 384-well plate and incubated at 22-27° C. for 10 minutes, then 2.5 times of the FAM-labeled peptide substrate and ATP buffer solution were added therein and incubated at 28° C., and then the stop buffer solution was added to stop the reaction. The data was read by Caliper and the following formula was used to calculate the IC50 inhibition: Percent inhibition=(max-conversion)/(max-min)*100. Staurosporine was used as a control. In kinase inhibition assays, some of the compounds of the present invention are effective in inhibiting VEGFR2/FYN/EGFR, so they have the potential for treating or preventing cancer or central nervous system diseases (Table 1).
- RIPK1 HTRF Binding Assay A solution was prepared containing 0.2 nM Anti GST-Tb (Cisbio, 61GSTTLB), 90.6 nM probe and 1 nM His-GST-TVMV-hRIPK1 (1-324) in FRET Buffer (20 mM HEPES, 10 mM MgCl2, 0.015% Brij-35, 4 mM DTT, 0.05 mg/mL BSA). Using Formulatrix Tempest, the detection antibody/enzyme/probe solution (2 mL) was dispensed into wells of a 1536 plate (Black Low Binding Polystyrene 1536 Plate (Corning, 3724)) containing 10 nL of compounds of interest at appropriate concentration in DMSO. The plate was incubated at rt for 1 h. FRET was measured using the EnVision plate reader (Excitation: 340 nM, Emission: 520 nM/495 nM). Total signal (0% inhibition) was calculated from wells containing 10 nL DMSO only. Blank signal (100% inhibition) calculated from wells containing 10 nL of 15 nM staurosporine and internal controls.
- RIPK1 HTRF Binding Assay A solution was prepared containing 0.2 nM Anti GST-Tb (Cisbio, 61GSTTLB), 90.6 nM probe and 1 nM His-GST-TVMV-hRIPK1(1-324) in FRET Buffer (20 mM HEPES, 10 mM MgCl2, 0.015% Brij-35, 4 mM DTT, 0.05 mg/mL BSA). Using Formulatrix Tempest, the detection antibody/enzyme/probe solution (2 mL) was dispensed into wells of a 1536 plate (Black Low Binding Polystyrene 1536 Plate (Corning, 3724)) containing 10 nL of compounds of interest at appropriate concentration in DMSO. The plate was incubated at rt for 1 h. FRET was measured using the EnVision plate reader (Excitation: 340 nM, Emission: 520 nM/495 nM). Total signal (0% inhibition) was calculated from wells containing 10 nL DMSO only. Blank signal (100% inhibition) calculated from wells containing 10 nL of 15 nM staurosporine and internal controls.
- TBK1 Enzyme Assay The kinase assay is performed as 384-well Flashplate assay assay (for e.g. Topcount measurement. 0.6 nM TANK binding kinase (TBK1), 800 nM biotinylated MELK-derived peptide (Biotin-Ah-Ah-AKPKGNKDYHLQTCCGSLAYRRR) SEQ ID NO: 2 and 10 μM ATP (spiked with 0.25 μCi 33P-ATP/well) are incubated in a total volume of 50 μl (10 mM MOPS, 10 mM Mg-acetat, 0.1 mM EGTA, 1 mM DTT, 0.02% Brij35, 0.1% BSA, pH 7.5) with or without test compound for 120 Min at 30° C. The reaction is stopped with 25 μl 200 mM EDTA. After 30 Min at room temperature the liquid is removed and each well washed thrice with 100 μl 0.9% sodium chloride solution. Nonspecific reaction is determined in presence of 100 nM Staurosporine. Radioactivity is measured in a Topcount (PerkinElmer).
- HER2-Phosphorylating Activity For setting the conditions for the method for measuring the in vitro inhibitory activity of a compound against HER2-phosphorylating activity, ProfilerPro Peptide 22 from PerkinElmer Inc. was used as a substrate on the basis of the report (PLoS One, 6 (7), e21487, 2011) on HER2 kinase reaction using, as a substrate, a peptide having the same sequence (5-FAM-EEPLYWSFPAKKK-CONH2) as that of ProfilerPro Peptide 22. The purified recombinant human HER2 protein used in the test was purchased from Carna Biosciences, Inc. Also, staurosporine (Eur. J. Biochem., 234, p. 317-322, 1995; and Nat. Biotechnol., 26 (1), p. 127-132, 2008), which is a multikinase inhibitor having Her2 inhibitory activity, was purchased from Enzo Life Sciences, Inc. (item No.: ALX-380-014) and used as a positive control in this test.For the inhibitory activity measurement of each compound, the compound of the present invention or staurosporine was first serially diluted with dimethyl sulfoxide (DMSO). Next, the HER2 protein, the substrate peptide (final concentration: 0.5 uM), manganese chloride (final concentration: 10 mM), ATP (final concentration: 6 uM), and the solution of the compound of the present invention in DMSO (final concentration of DMSO: 5%) were added into a buffer solution for kinase reaction (15 mM Tris (pH 7.5), 2 mM dithiothreitol, and 0.01% Tween 20), and the mixture was incubated at 25° C. for 40 minutes for kinase reaction. The reaction was terminated by adding EDTA (final concentration: 30 mM) thereto. Finally, the unphosphorylated substrate peptide (S) and the phosphorylated peptide (P) were separated and detected by microcapillary electrophoresis using LabChip EZ Reader II (PerkinElmer Inc.). The amount of phosphorylation reaction was determined from the respective peak heights of S and P. The compound concentration which can suppress the phosphorylation reaction by 50% was defined as an IC50 value (nM).
- LanthaScreen Eu Kinase Binding Assay The reagent was prepared through a series of dilutions of the tracer. The tracer was first diluted to 3000 nM by adding 3.6 μL of 50 μL stock tracer to 56 μL of 1× Kinase Buffer A. 50 μL of 1× Kinase Buffer A was added to 5 wells in each of two columns of a 96-well plate. 50 μL of the 3000 nM tracer was added to well A1 and mixed. 50 μL of solution was removed from A1 and transferred to A2 and mixed. 50 μL of the solution in well A2 was removed and transferred to well B1 and mixed. This protocol was repeated nine times to the desired concentration. The kinase/antibody solution was prepared at 15 nM kinase, 6 nM antibody, and 6 nM Eu-Streptavidin. Both the antibody tube and Eu-Streptavidin tube were centrifuged at approximately 10,000×g for ten minutes, and the desired volume was aspirated from the top. The volume of reagents added to Kinase Buffer A were calculated using the equations provided in the LanthaScreen® Eu Kinase Binding Assay Validation Packet. 30 μM staurosporine (“competitor solution”) was prepared by diluting 30 μL of 1 mM staurosporine (from a stock in DMSO) into 970 μL Kinase Buffer A. A 3% DMSO control solution was prepared by adding 30 μL DMSO to 970 μL Kinase Buffer A.5 μL of each concentration of serially diluted tracer was added to six replicate assay wells in a 384-well plate. 5 μL of competitor solution was added to three wells for each tracer concentration. 5 μL of DMSO control solution was added to the other three wells for each tracer concentration. 5 μL of kinase/antibody solution was added to all wells, and the plate was incubated at room temperature for 60 mins.
- Caliper Assay Selected compounds disclosed herein were tested in CDK4/cyclinD1, CDK6/CycD3 CDK2/CycA and CDK2/cyclinE kinase assays by Nanosyn (Santa Clara, Calif.) to determine their inhibitory effect on these CDKs. The assays were performed using microfluidic kinase detection technology (Caliper Assay Platform). The compounds were tested in 12-point dose-response format in singlicate at Km for ATP. Phosphoacceptor substrate peptide concentration used was 1 μM for all assays and Staurosporine was used as the reference compound for all assays. Specifics of each assay are as described below:CDK2/CyclinA: Enzyme concentration: 0.2 nM; ATP concentration: 50 μM; Incubation time: 3 hr.CDK2/CyclinE: Enzyme concentration: 0.28 nM; ATP concentration: 100 μM; Incubation time: 1 hr.CDK4/CyclinD1: Enzyme concentration: 1 nM; ATP concentration: 200 μM; Incubation time: 10 hr.CDK6/CyclinD3: Enzyme concentration: 1 nM; ATP concentration: 300 μM; Incubation time: 3 hr.
- DYRK1A kinase assay Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1X Kinase buffer to final concentrations of 0.25 μg/mL, 15 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (luM top) was run to serve as a positive compound control. After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- Kinase Assay The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ("0% Control") and phosphorylated ("100% control") forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (luM top) was run to serve as a positive compound control.
- Kinase Assay The kinase assay is performed as 384-well flashplate assay.0.6 nM TANK binding kinase (TBK1), 800 nM biotinylated MELK-derived peptide (biotin-Ah-Ah-AKPKGNKDYHLQTCCGSLAYRRR) and 10 μM ATP (with 0.25 μCi of 33P-ATP/well) are incubated in a total volume of 50 μl (10 mM MOPS, 10 mM magnesium acetate, 0.1 mM EGTA, 1 mM DTT, 0.02% of Brij35, 0.1% of BSA, pH 7.5) with or without test substance at 30° C. for 120 min. The reaction is stopped using 25 μl of 200 mM EDTA solution, filtered off with suction after 30 min at room temperature, and the wells are washed 3 times with 100 μl of 0.9% NaCl solution. The non-specific proportion of the kinase reaction (blank) is determined using 100 nM staurosporine. Radioactivity is measured in the Topcount. IC50 values are calculated using RS1.
- PDK1 Enzymatic Assay The experimental batches are carried out in a flashplate system with 384 wells/microtitration plate.In each case, the PDK1 sample His6-PDK1(Δ1-50)(3.4 nM) (His6 disclosed as SEQ ID NO: 2), the PDK1 substrate biotin-bA-bAKTFCGTPEYLAPEVRREPRILSEEEQEMFRDFDYIADWC (SEQ ID NO: 3) (400 nM), 4 uM ATP (with 0.2 uCi of 33P-ATP/well) and the test substance in 50 ul of conventional experimental solution per well are incubated at 30° C. for 60 min. The test substances are employed in corresponding concentrations (if desired in a dilution series). The control is carried out without test substance. The reaction is stopped using standard methods and washed. The activity of the kinase is measured via the incorporated radioactivity in top count. In order to determine the non-specific kinase reaction (blank value), the experimental batches are carried out in the presence of 100 nM staurosporine.
- Phosphoinositide 3-kinase (PI3KD) Biochemical Assay A solution was prepared containing 0.2 nM Anti-HIS-Terbium (Cisbio, 64CUSTAZU), 40 nM designed-probe and 1.0 nM His-PIK3CD/PIK3R1 (p110 delta/p85 alpha) in FRET Buffer (20 mM HEPES, 10 mM MgCl2, 0.015% Brij-35, 4 mM DTT, 0.05 mg/mL BSA). Using Formulatrix Tempest for liquid handling, the detection antibody/enzyme/probe solution (2 uL per well) was dispensed into wells of a 1536 plate (Black Low Binding Polystyrene 1536 Plate (Corning, 3724) containing 10 nL of compounds of interest at appropriate concentration in DMSO. The plate was incubated at room temperature for 1 h. FRET was measured using the EnVision plate reader (Excitation: 340 nM, Emission: 520 nM/495 nM). Total signal (0% inhibition) was calculated from wells containing 10 nL DMSO only. Blank signal (100% inhibition) calculated from wells containing 10 nL of 15 nM staurosporine and internal controls.
- ROCK1, ROCK2, PRKX, and PKA Inhibition Assay Compounds as a powder were dissolved in dimethyl sulfoxide to make a 10 mM stock. Compounds were tested in 10-dose IC50 triplicate mode with a 3-fold serial dilution starting at 1 μM. The control compound, staurosporine, was tested in 10-dose IC50 mode with 4-fold serial dilution starting at 20 μM. The HotSpot kit employing 33P-ATP was used with reactions conducted at 10 μM ATP for each tested enzyme. The percent activity relative to DMSO controls for each concentration was then fitted to a curve using GraphPad Prism to determine the IC50. Curve fits were performed where the enzyme activities at the highest concentration of compounds were less than 65%. An IC50 value less than 50.8 μM or higher than 1 μM is estimated based on the best curve fitting available.
- The DYRK1A kinase assa Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated (0% Control) and phosphorylated (100% control) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- DYRKIA Kinase Activity Assay The DYRKIA kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRKIA kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.
- Dose-response biochemical assay for inhibitors of protein kinase A (PKA) activity Source (MLSCN Center Name): The Scripps Research Institute Molecular Screening Center Center Affiliation: The Scripps Research Institute, TSRI Assay Provider: Scripps Florida Network: Molecular Library Screening Center Network (MLSCN) Grant Proposal Number: NA PKA is an ubiquitous serine/threonine protein kinase and belongs to the AGC kinase family. It has several functions in the cell, including regulation of immune response [1], transcription [2], cell cycle and apoptosis [3]. PKA is a cAMP dependent enzyme that exists in its native inactive form as a 4 subunit enzyme with two regulatory and two catalytic subunits. Binding of cAMP to the regulatory subunit leads to the disassembly of the complex and release of now active catalytic subunits. This screen is designed to identify inhibitors of PKA. The known PKA inhibitor Staurosporine was used as a positive control. Keywords: PKA, kinase, Protein kinase A, luciferase, luminescence, apoptosis, immune response, cAMP dependant enzyme,
- EMSA For IC50 measurements, a 384-well microtiter plate contained 8-point serial dilutions of inhibitors and 16-point serial dilutions of staurosporine as a reference compound. First, 4.5 μL of 2× kinase solution was added to 0.05 μL of compound (maximum concentration 1.8 mM in 90% DMSO and 10% H2O). To allow for possible slow association, plates with type II inhibitors were incubated for 60 min at 30 °C. The assay started after the addition of 4.5 μL of 2× peptide with ATP and was run for 60 min at 30 °C, after which 16 μL of stop solution was added (100 mM HEPES pH 7.5, 5% DMSO, 0.1% coating reagent (Caliper Lifescience) 10 mM EDTA, pH 8.0, 0.015% BRIJ35). All reactions were performed in 50 mM HEPES, pH 7.5, 1 mM DTT, 0.02% Tween 20, 0.02% BSA, 10 mM betaglycerophosphate, 0.01 mM Na3VO4, 0.6% DMSO, and 2 μM peptide.
- Enzyme Assay The kinase assay is performed as 384-well Flashplate assay (PerkinElmer LAS Germany GmbH). 3.4 nM His6-PDK1(Delta 1-50) (PDK1 that has a His-tag consisting of six histidines and lacks the first fifty amino acids), 400 nM PDKtide (Biotin-bA-bAKTFCGTPEYLAPEVRREPRILSEEEQEMFRDFDYIADWC as the substrate, and 4 μM ATP (spiked with 0.25 μCi 33P-ATP/well) are incubated in a total volume of 50 μl (50 mM TRIS, 10 mM Mg-acetate, 0.1% Mercaptoethanol, 0.02 Brij35, 0.1% BSA, pH 7.5) with or without test compound (5-10 concentrations) for 60 Min at 30° C. The reaction is stopped with 25 μl 200 mM EDTA. After 30 Min at room temperature the liquid is removed and each well washed thrice with 100 ml 0.9% sodium chloride solution. Nonspecific reaction is determined in presence of 100 nM of the high affinity protein kinase inhibitor Staurosporine. Radioactivity is measured in a Topcount (PerkinElmer LAS Germany GmbH).
- Inhibition Assay The object of this assay was to test the inventive compounds for the kinase inhibitory activity in vitro. In this assay, an isotopic labeling method was used to label the gamma phosphate group on ATP. EGFR (including wild type, L858R mutant type and L858R/T790M double mutant type), VEGFR2, ALK, BTK, c-KIT, c-SRC, MET, PDGFRalpha and FLT3 kinases were tested in vitro for the activity inhibition. Staurosporine was used as a reference molecule (or referred to as a positive control). The kinase inhibitory activities of the tested compounds were expressed in the IC50 value (half inhibition concentration) or the kinase activity inhibitory rate by the tested compounds at 10 uM. The IC50 value can be obtained by the calculation of the inhibitory rates at a series of different concentrations of the tested compounds. 1. Materials: 20mM 3-(N-morpholinyl)propylsulfonic acid (MOPS); 1mM Ethylenediaminetetraacetic acid (EDTA); 0.01% Polyethylene glycol lauryl ether (Brij-35).
- Kinase Assay The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.
- Kinase Assay for EGFR The assay is performed in a Black 384-well plate (available from Corning). EGFR kinase (is diluted in TR-FRET Dilution Buffer (PV3189, InVitrogen) at concentration of 0.4 ug/ml as stock solution, and then 2-fold serial diluted. Addition of 1 mM ATP initiated the reaction, and the reaction is incubated for 111 reaction at room temperature. 10 μL of the Tb-antibody (from InVitrogen)+EDTA (from InVitrogen) solution prepared was added to each well of the assay plate and mix briefly, and incubated for 30 min. The signal is monitored by using M5 microplate reader (Ex=332 nm, Em=488 nm and 518 nm). Each compound is tested in duplicate wells. EGFR without compound is used as control. Staurosporine (available from Sigma) is used as positive control compound. Inhibition was calculated as percentage of the EGFR activity (without compound). Each compound in Examples 1 to 7 showed >50% inhibition at 100 nM.
- PI3Kdelta HTRF Binding Assay A solution was prepared containing 0.2 nM Anti GST-Tb (Cisbio, 61GSTTLB), 40 nM probe and 1 nM GST-tagged PIK3C5 in complex with PIK3R1 (Invitrogen #PV5273) in FRET Buffer (20 mM HEPES, 10 mM MgC12, 0.015% Brij-35, 4mM DTT, 0.05 mg/mL BSA). Using Formulatrix Tempest, the detection antibody/enzyme/probe solution (2 mL) was dispensed into wells of a 1536 plate (Black Low Binding Polystyrene 1536 Plate (Corning, 3724)) containing 10 nL of compounds of interest at appropriate concentration in DMSO. The plate was incubated at rt for 1 h. FRET was measured using the EnVision plate reader (Excitation: 340 nM, Emission: 520 nM/495 nM). Total signal (0% inhibition) was calculated from wells containing 10 nL DMSO only. Blank signal (100% inhibition) calculated from wells containing 10 nL of 15 nM staurosporine and internal controls.Preferred compounds have low to no activity against PI3K, preferably compounds have PIK3 activity of 1 pM or greater.
- Phosphoinositide 3-kinase α (PI3Kα) Biochemical Assay A solution was prepared containing 0.2 nM Anti-HIS-Terbium (Cisbio, 64CUSTAZU), 3.7 nM designed-probe and 1.0 nM His-PIK3CA/PIK3R1 (p110 alpha/p85 alpha) in FRET Buffer (20 mM HEPES, 10 mM MgCl2, 0.015% Brij-35, 4 mM DTT, 0.05 mg/mL BSA). Using Formulatrix Tempest for liquid handling, the detection antibody/enzyme/probe solution (2 uL per well) was dispensed into wells of a 1536 plate (Black Low Binding Polystyrene 1536 Plate (Corning, 3724) containing 10 nL of compounds of interest at appropriate concentration in DMSO. The plate was incubated at room temperature for 1 h. FRET was measured using the EnVision plate reader (Excitation: 340 nM, Emission: 520 nM/495 nM). Total signal (0% inhibition) was calculated from wells containing 10 nL DMSO only. Blank signal (100% inhibition) calculated from wells containing 10 nL of 15 nM staurosporine and internal controls.
- IRAK4 Biochemical Assay IRAK4 enzyme (Carna Biosciences, Chuo-ku, Kobe, Japan) activity was measured by detecting phosphorylated peptide substrate formation using an antibody against the phosphorylated peptide substrate. This is a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay, based on the STK1 KinEASE Assay (Cisbio, Bedford, Mass.). The assay was designed as a simple two-step, endpoint assay (a 5 μl enzyme reaction followed by 5 μl stop and detect Solution) performed in ProxiPlate-384 Plus plates (Perkin Elmer, Waltham, Mass.). Staurosporine, a non-selective kinase inhibitor was used as a positive control. Compounds diluted in DMSO were spotted into 384 well plates using a Labcyte Echo 550 Liquid Handling System prior to addition of IRAK4 enzyme and peptide substrate. Reaction solutions were delivered using a Multi-Flo (Bio-Tek Instruments). The enzyme and peptide solution was incubated with compound for 15 minutes at room temp before the reaction was initiated by the addition of ATP. The standard 5 μl reaction mixture contained 500 μM ATP, 2 M peptide (STK1 Peptide), 0.75 nM of IRAK4 in reaction buffer (50 mM HEPES, pH 7.0, 0.02% NaN3, 0.01% BSA, 0.1 mM Orthovanadate, 5 mM MgC2, 0.025% NP-40, 1 mM DTT). After 120 min of incubation at room temperature, 5 μl of Stop and Detect Solution (1:100 Cryptate labeled anti-phosphorylated peptide antibody solution and 125 nM Tracer in a 50 mM HEPES pH 7.0 detection buffer containing sufficient EDTA) was added. The plate was then further incubated for 60 minutes at room temperature and read on Envision 2103 Multilabeled reader (PerkinElmer) with excitation/emission/FRET emission at 340 nm/615 nm665 nm, respectively. Fluorescence intensities at 615 nm and 665 nm emission wavelengths were expressed as aratio (665 nm/615 nm). Percentage of inhibition was calculated as below:% Inhibition=100×(Ratiosample−Ratio0% Inhibition)/(Ratio100% Inhibition−Ratio0% Inhibition)The 0% inhibition value comes from control wells lacking inhibitor. The 100% inhibition value comes from control wells containing a saturating amount of known inhibitor staurosporine.
- In Vitro Inhibition Assay ITK and JAK3 Kinase assay procedures:Enzyme was incubated with substrate peptide in reaction buffer in the presence and absence of test compounds or Staurosporine. All additions were done on ice, followed by the addition of ATP mix. Wells were uniformly mixed using an Eppendorff plate shaker and incubated at 30° C. for 20 min, and stopped by the addition of 5 μL of 3% phosphoric acid. Volume was increased to 100 μL by adding 0.8% phosphoric acid which was then transferred to PC filter mats (Millipore), pre-equilibrated with 70% ethanol and water. Plates were washed thrice with 100 μL 0.8% phosphoric acid and dried for an hour at 60° C. 100 μL scintillation fluid was added into each well and reading taken in Perkin Elmer TOPCOUNT beta counter. The data analysis was performed by averaging the duplicate top count readings for each standard, negative, positive control (enzyme control) and samples and subtracting the average negative control from each reading which results in corrected values.
- Inhibitory Activity of Compounds on TRKA, TRKB, TRKC and ROS1 The compound powder was dissolved in 100% DMSO (Sigma, Cat. D8418-1l) to prepare a 10 mM storage solution. The compounds had an initial test concentration of 100 nM, were 3-fold serially diluted to obtain 10 samples for multiple hole inspection. For TRKA, TRKB and TRKC kinase targets, Loxo-101 (Selleckchem, Cat. S7960) was used as positive reference compound; for ROS1, Staurosporine (Selleckchem, Cat. S1421) was used as positive reference compound. The gradient diluted compounds were mixed with TRKA/TRKB/TRKC/ROS1 kinase (Carna, Cat. 08-186/08-187/08-197/08-163) with final concentration of 2.5 nM/2.55 nM/2.5 nM/0.3 nM in a Optiplate-384F plate (PerkinElmer, Cat. 6007270), and incubated at room temperature for 10 minutes. After that, ATP was added with final concentration of 47.8 μM/71.2 μM/44.4 μM/26.7 μM, 3 μM Kinase Substrate22 (GL Biochem, Cat. 112393) was added. The reaction was carried out at room temperature for 30 min/40 min/20 min/20 min respectively.
- JAK/TYK2 Assay 10 mM test compound stock or 1 mM control compound stock (tofocitinib, ruxolitinib or staurosporine) in DMSO was diluted to 0.4 mM in DMSO. A 3-fold series dilution was then performed in DMSO to generate 10 different compound concentrations. The assay was carried out in 384-well white plate. 0.5 uL of 40× compound DMSO solution at different concentrations was mixed with 10 uL 2× enzyme prepared in reaction buffer (20 mM HEPES, 10 mM MgCl2, 0.01% Tween, 1 mM DTT, pH 7.5). 10 uL 2× substrate mixture prepared in reaction buffer was then added to start the reaction. A short spin was done to settle down all solutions to the bottom of the plate. Final concentrations of test compound in the reaction mixture were 10000, 3333, 1111, 370, 123, 41.2, 13.7, 4.57, 1.52 and 0.51 nM. Concentrations of control compound were ten times less. Enzymatic reaction was conducted at 25° C. for 1-2 hours. 10 uL of Kinase Glo Reagents was added to stop the reaction and generate the luminescent signal which was measured using Envision.
- MYLK/MLCK Assay MYLK dissociation constants (Kd) for compounds were determined using the DiscoverX KdELECT platform. The MYLK kinase (accession number NP_444254.3) was labeled with a DNA tag for subsequent qPCR readout while a known active site binding ligand (staurosporine) was immobilized on a solid support (beads). Test compounds were prepared as 111X stocks in 100% DMSO and Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. The assay plates were incubated at room temperature with shaking for 1 hour to equilibrate. The affinity beads were washed (lx PBS, 0.05% Tween 20) to remove unbound kinase and quantify MYLK captured on solid support by qPCR. The Kd was determined by measuring the amount of MYLK captured on the solid support as a function of the test compound concentration. The Kd values were calculated by fitting dose-response curves to the Hill binding equation using the Levenberg-Marquardt algorithm.
- Z-lyte Assay The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions. This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1x Kinase buffer to final concentrations of 0.19 uMg/mL, 30 uM, and 4 uM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Additionally, an 11-point dose-response curve of Staurosporine (1 M top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- IRAK4 Biochemical Assay IRAK4 enzyme (Carna Biosciences, Chuo-ku, Kobe, Japan) activity was measured by detecting phosphorylated peptide substrate formation using an antibody against the phosphorylated peptide substrate. This is a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay, based on the STK1 KinEASE Assay (Cisbio, Bedford, Mass.). The assay was designed as a simple two-step, endpoint assay (a 5 μl enzyme reaction followed by 5 μl stop and detect Solution) performed in ProxiPlate-384 Plus plates (Perkin Elmer, Waltham, Mass.). Staurosporine, a non-selective kinase inhibitor was used as a positive control. Compounds diluted in DMSO were spotted into 384 well plates using a Labcyte Echo 550 Liquid Handling System prior to addition of IRAK4 enzyme and peptide substrate. Reaction solutions were delivered using a Multi-Flo (Bio-Tek Instruments). The enzyme and peptide solution was incubated with compound for 15 minutes at room temp before the reaction was initiated by the addition of ATP. The standard 5 μl reaction mixture contained 500 μM ATP, 2 μM peptide (STK1 Peptide), 0.75 nM of IRAK4 in reaction buffer (50 mM HEPES, pH 7.0, 0.02% NaN3, 0.01% BSA, 0.1 mM Orthovanadate, 5 mM MgCl2, 0.025% NP-40, 1 mM DTT). After 120 min of incubation at room temperature, 5 μl of Stop and Detect Solution (1:100 Cryptate labeled anti-phosphorylated peptide antibody solution and 125 nM Tracer in a 50 mM HEPES pH 7.0 detection buffer containing sufficient EDTA) was added. The plate was then further incubated for 60 minutes at room temperature and read on Envision 2103 Multilabeled reader (PerkinElmer) with excitation/emission/FRET emission at 340 nm/615 nm/665 nm, respectively. Fluorescence intensities at 615 nm and 665 nm emission wavelengths were expressed as a ratio (665 nm/615 nm). Percentage of inhibition was calculated as below:% Inhibition=100×(RatioSample−Ratio0% Inhibition)/(Ratio100% Inhibition−Ratio0% Inhibition)The 0% inhibition value comes from control wells lacking inhibitor. The 100% inhibition value comes from control wells containing a saturating amount of known inhibitor staurosporine.
- IKKe Biochemical Assay Test compounds were transferred into Labcyte polypropylene 384 well plates (P055-25) and diluted to 3 mM using DMSO. 3 mM test compounds were dispensed using Labcyte ECHO dose response module into Greiner 784075 plates (columns 3-12 and 13-22, 10 point 1:4) so that high concentration was 30 uM final. 100 uM of a reference compound (1 uM final high concentration). Backfilling was performed if necessary so that all wells contain 1% DMSO final:add 75 nl DMSO/well into columns 1, 2 and 24 using Labcyte Echo.add 75 nl 1.0 mM staurosporine/well into column 23 using Labcyte Echo (10 uM final)add 4.5 ul enzyme/well using multidrop dispenseradd 3 ul substrate/well using multidrop dispenserincubate at 25° C. for 90 min.add 7.5 ul 2× stop bufferread on labchip ez reader II using IKKε. jobRaw data files were opened in the Caliper LabChip Reviewer program (Version 3.0.265.0 SP2) and peak assignments were adjusted to reflect substrate first with the software's post-run analysis options. A spline-fit baseline was applied using the software's analysis algorithm.
- TBK Biochemical Assay Test compounds were transferred into Labcyte polypropylene 384 well plates (P055-25) and diluted to 3 mM using DMSO. 3 mM test compounds were dispensed using Labcyte ECHO dose response module into Greiner 784075 plates (columns 3-12 and 13-22, 10 point 1:4) so that high concentration was 30 uM final. 100 uM of a reference compound (1 uM final high concentration). Backfilling was performed if necessary so that all wells contain 1% DMSO final:add 75 nl DMSO/well into columns 1, 2 and 24 using Labcyte Echo.add 75 nl 1.0 mM staurosporine/well into column 23 using Labcyte Echo (10 uM final)add 4.5 ul enzyme/well using multidrop dispenseradd 3 ul substrate/well using multidrop dispenserincubate at 25° C. in Heidolph incubator for 90 min.add 7.5 ul 2× stop buffer using multidrop dispenserread on labchip ez reader II using TBK1.jobRaw data files were opened in the Caliper LabChip Reviewer program (Version 3.0.265.0 SP2) and peak assignments were adjusted to reflect substrate first with the software's post-run analysis options. A spline-fit baseline was applied using the software's analysis algorithm.
- in vitro Kinase Assay Shown are the IC50s (concentrations causing 50% inhibition) of DM and the analogues for the in vitro kinase assays using the following purified human enzymes: the BMP type-I receptor activin receptor-like kinase 2 (ALK2/BMPR-I), the TGFβ type-I receptor activin receptor-like kinase 5 (ALK5/ TGFβR-I), the VEGF type-2 receptor (VEGFR2/KDR), the AMP-activated protein kinase (AMPK), and the platelet-derived growth factor receptor-β (PDGFR β). In in vitro kinase assays, DM was relatively nonspecific, targeting ALK2, AMPK, and KDR with IC50s of <250 nM. LDN-193189 was slightly more selective but still had significant effects against ALK5 and KDR. By comparison, DMH1, DMH2, and DMH3 were much more selective ALK2 inhibitors. In particular, DMH1 had no detectible activity against any of the kinases tested besides ALK2. DMH4 was a selective KDR inhibitor with modest effect on ALK2 (IC50 3.6 uM) and minimal effect on AMPK (IC50 8.0 uM). Nonspecific kinase inhibitor staurosporine was used as a control. All of the reactions were carried out in the presence of 10 uM ATP.
- Biochemical Assay The activity of the inhibitor compounds against DDR1 and DDR2 was tested using KinaseProfiler (Eurofins). Human DDR1/DDR2 kinase was incubated with 8 mM MOPS buffer (pH=7.0), 0.2 mM EDTA, 250 μM IGF 1Rtide protein kinase substrate (e.g., derived from human IRS-1, and is a substrate for TRK1, JAK2, and RET Kinases enzolifesciences.com/BML-P257/igf-1rtide/), 10 mM Magnesium acetate/Manganese chloride, respectively, and [γ-33P]-ATP. The enzymatic reaction processed in the presence of Mg2+ cations and ATP at room temperature for 40 minutes and terminated by addition of phosphoric acid. The reaction mixture (10 μL) was spotted onto a P30 filtermat and washed four times using 0.425% phosphoric acid and once with methanol. All the compounds were prepared in 100% DMSO. Staurosporine was used as a reference inhibitor and was added to each plate at an estimated concentration resulted in complete inhibition. The results for some of the compounds are listed in Table 1, which shows the ability to inhibit DDR1 and DDR2. As such, these compounds be used as inhibitors of the discoidin domain receptor family, such as for DDR1 and DDR2. However, these compounds may also inhibit other DDR family receptors
- DYRK1A Kinase Activity Assay The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturers instructions (Life Technologies- a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission. Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1X Kinase buffer to final concentrations of 0.25 µg/mL, 15 µM, and 4 µM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated (0% Control) and phosphorylated (100% control) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1uM top) was run to serve as a positive compound control.vAfter incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- DYRK1A Kinase Assay The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission. Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1×Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control. After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- JAK/TYK2 Assay 10 mM test compound stock or 1 mM control compound stock (tofocitinib, ruxolitinib or staurosporine) in DMSO was diluted to 0.4 mM in DMSO. A 3-fold series dilution was then performed in DMSO to generate 10 different compound concentrations. The assay was carried out in 384-well white plate. 0.5 uL of 40× compound DMSO solution at different concentrations was mixed with 10 uL 2× enzyme prepared in reaction buffer (20 mM HEPES, 10 mM MgCl2, 0.01% Tween, 1 mM DTT, pH 7.5). 10 uL 2× substrate mixture prepared in reaction buffer was then added to start the reaction. A short spin was done to settle down all solutions to the bottom of the plate. Final concentrations of test compound in the reaction mixture were 10000, 3333, 1111, 370, 123, 41.2, 13.7, 4.57, 1.52and 0.51 nM. Concentrations of control compound were ten times less. Enzymatic reaction was conducted at 25° C. for 1-2 hours. 10 uL of Kinase Glo Reagents was added to stop the reaction and generate the luminescent signal which was measured using Envision. Luminescence signal was inversely related to kinase activity. Reaction mixture which did not contain enzyme served as negative control. The mixture without any compound was the positive control.
- Kinase Assay The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- Kinase Assay The DYRKIA kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies-a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission. Briefly, recombinant DYRKIA kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ("0% Control") and phosphorylated ("100% control") forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control. After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- Kinase Inhibition Assay The assay used was the HotSpot assay (Reaction Biology Corp).Compounds were tested in 10-dose IC50 mode with a 3-fold serial dilution starting at 10 μM. Control Compound, Staurosporine, was tested in 10-dose IC50 mode with 4-fold serial dilution starting at 20 μM. Reactions were carried out at 10 μM ATP.ReagentsBase Reaction buffer; 20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO.*Required cofactors are added individually to each kinase reactionReaction Procedure1. The peptide substrate was freshly prepared in Base Reaction Buffer2. Any required cofactors were delivered to the substrate solution above3. The human recombinant Casein kinase 16 was delivered into the substrate solution and gently mixed4. The compounds in DMSO were delivered into the kinase reaction mixture by Acoustic technology (Echo550; nanoliter range), and incubated for 20 minutes at room temperature5. 33P-ATP (specific activity 10 mCi/mL) together with ATP (10 μM) was delivered into the reaction mixture6. The kinase reaction was incubated for 2 hours at room temperature7. The reaction mixtures were spotted onto P81 ion exchange paper8. The kinase activity was detected by measuring 33P-ATP-labelled product peptide using a filter-binding method.
- HTRF KinEASE Assay ASK1 kinase was from Thermofisher (Catalogue # PV4011), ATP was from Sigma (Catalogue # A7699), HTRF KinEASE Assay System was obtained from Cisbio (Bedford, Mass.). Area plate was from Perkin Elmer (Catalogue #6005560). HTRF KinEASE-STK is a generic method for measuring serine/threonine kinase activities using time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. The IC50 value for each compound was determined in the presence of compound (various concentration from 0 to 10 uM) and a fixed amount of ATP, peptide substrates. Test compound, 1 uM STK3 peptide substrate, 5 nM of ASK1 kinase are incubated with kinase reaction buffer, containing 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA for 30 minutes, then 100 uM ATP is added to start kinase reaction and incubated for 3 hours. The STK3-antibody labeled with Eu3+-Cryptate and 125 nM streptavidin-XL665 are mixed in a single addition with stop reagents provided by the Cisbio kit used to stop the kinase reaction. Fluorescence is detected using Envision Multilabeled 2014 reader from PerkinElmer. The Fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well. The resulting TR-FRET is proportional to the phosphorylation level. Staurosporine was used as the positive control. IC50 was determined by Xlfit 5.3.
- HTRF KinEASE Assay ASK1 was purchased from Thermofisher (Catalogue # PV4011), ATP was purchased from Sigma (Catalogue # A7699), HTRF KinEASE Assay System was obtained from Cisbio (Bedford, Mass.). Area plate was purchased from Perkin Elmer (Catalogue # #6005560). HTRF KinEASE -STK is a generic method for measuring serine/threonine kinase activities using a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. The IC50 value for each compound was determined in the presence of compound (various concentration from 0 to 10 μM) and a fixed amount of ATP and peptide substrates. The test compound, 1 uM STK3 peptide substrate, and 5 nM of ASK1 kinase are incubated with kinase reaction buffer containing 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA for 30 minutes. 100 uM ATP is added to start kinase reaction and incubated for 3 hours. The STK3-antibody labeled with Eu3+-Cryptate and 125 nM streptavidin-XL665 are mixed in a single addition with stop reagents provided by the Cisbio kit used to stop the kinase reaction. Fluorescence is detected using an Envision Multilabeled 2014 reader from PerkinElmer. The Fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well. The resulting TR-FRET is proportional to the phosphorylation level. Staurosporine was used as the positive control.
- HTRF KinEASE Assay ASK1 was purchased from Thermofisher (Catalogue #PV4011), ATP was purchased from Sigma (Catalogue #A7699), HTRF KinEASE Assay System was obtained from Cisbio (Bedford, Mass.). Area plate was purchased from Perkin Elmer (Catalogue # #6005560). HTRF KinEASE -STK is a generic method for measuring serine/threonine kinase activities using a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. The IC50 value for each compound was determined in the presence of compound (various concentration from 0 to 10 μM) and a fixed amount of ATP and peptide substrates. The test compound, 1 uM STK3 peptide substrate, and 5 nM of ASK1 kinase are incubated with kinase reaction buffer containing 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA for 30 minutes. 100 uM ATP is added to start kinase reaction and incubated for 3 hours. The STK3-antibody labeled with Eu3+-Cryptate and 125 nM streptavidin-XL665 are mixed in a single addition with stop reagents provided by the Cisbio kit used to stop the kinase reaction. Fluorescence is detected using an Envision Multilabeled 2014 reader from PerkinElmer. The Fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well. The resulting TR-FRET is proportional to the phosphorylation level. Staurosporine was used as the positive control.
- HTRF KinEASE Assay ASK1 was purchased from Thermofisher (Catalogue #PV4011), ATP was purchased from Sigma (Catalogue #A7699), HTRF KinEASE Assay System was obtained from Cisbio (Bedford, Mass.). Area plate was purchased from Perkin Elmer (Catalogue ##6005560). HTRF KinEASE -STK is a generic method for measuring serine/threonine kinase activities using a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. The IC50 value for each compound was determined in the presence of compound (various concentration from 0 to 10 μM) and a fixed amount of ATP and peptide substrates. The test compound, 1 uM STK3 peptide substrate, and 5 nM of ASK1 kinase are incubated with kinase reaction buffer containing 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA for 30 minutes. 100 uM ATP is added to start kinase reaction and incubated for 3 hours. The STK3-antibody labeled with Eu3+-Cryptate and 125 nM streptavidin-XL665 are mixed in a single addition with stop reagents provided by the Cisbio kit used to stop the kinase reaction. Fluorescence is detected using an Envision Multilabeled 2014 reader from PerkinElmer. The Fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well. The resulting TR-FRET is proportional to the phosphorylation level. Staurosporine was used as the positive control.
- p-Cl-ANS Fluorescent Binding Assay Compounds in DMSO (1% final) were added to 384-well black clear bottom plates (Greiner 781091) in 8-point dose response in duplicate using the Echo 550 (Beckman). SU9516 and DMSO were used as controls representing 100% and 0% inhibition of p-Cl-ANS binding, respectively. 20 μL of assay buffer (150 mM NaCl, 50 mM HEPES, pH 7.5 containing 0.01% Triton and 2 mM DTT) was added to each well, the plate was shaken for 1 minute, centrifuged at 2000 rpm for 5 min intervals until air bubbles were eliminated and then read on a CLARIOstar plate reader (BMG; Ex 388 nm, Em 455 nm) to obtain background fluorescence measurements for correction of intrinsic compound fluorescence. Absorbance was then measured at both the excitation and emission wavelengths for correction of the inner filter effect. Next, 5 μL p-Cl-ANS (Wang, N., et al., ACS Omega 2019, 4 (19), 18472-7; 15 μM final) was added to each well in assay buffer. Where present, staurosporine (Enzo Life Sciences; final 5 μM) was added as a premixed solution with p-Cl-ANS. Lastly, 5 uL of 3 μM CDK2 (final 0.5 μM) in buffer was added (final volume of 30 μL) and the plate was shaken, centrifuged, and read as described above.
- HTRF KinEASE Assay ASK1 was purchased from Thermofisher (Catalogue # PV4011), ATP was purchased from Sigma (Catalogue # A7699), HTRF KinEASE Assay System was obtained from Cisbio (Bedford, Mass.). Area plate was purchased from Perkin Elmer (Catalogue # #6005560). HTRF KinEASE -STK is a generic method for measuring serine/threonine kinase activities using a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. The IC50 value for each compound was determined in the presence of compound (various concentration from 0 to 10 μM) and a fixed amount of ATP and peptide substrates. The test compound, 1 uM STK3 peptide substrate, and 5 nM of ASK1 kinase are incubated with kinase reaction buffer containing 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA for 30 minutes. 100 uM ATP is added to start kinase reaction and incubated for 3 hours. The STK3-antibody labeled with Eu3+-Cryptate and 125 nM streptavidin-XL665 are mixed in a single addition with stop reagents provided by the Cisbio kit used to stop the kinase reaction. Fluorescence is detected using an Envision Multilabeled 2014 reader from PerkinElmer. The Fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well. The resulting TR-FRET is proportional to the phosphorylation level. Staurosporine was used as the positive control. IC50 was determined by XLfit 5.3.
- HTRF KinEASE Assay ASK1 was purchased from Thermofisher (Catalogue #PV4011), ATP was purchased from Sigma (Catalogue #A7699), HTRF KinEASE Assay System was obtained from Cisbio (Bedford, Mass.). Area plate was purchased from Perkin Elmer (Catalogue ##6005560). HTRF KinEASE-STK is a generic method for measuring serine/threonine kinase activities using a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. The IC50 value for each compound was determined in the presence of compound (various concentration from 0 to 10 μM) and a fixed amount of ATP and peptide substrates. The test compound, 1 μM STK3 peptide substrate, and 5 μM of ASK1 kinase are incubated with kinase reaction buffer containing 50 mM HEPES Ph 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA for 30 minutes. 100 μM ATP is added to start kinase reaction and incubated for 3 hours. The STK3-antibody labeled with Eu3+-Cryptate and 125 nM streptavidin-XL665 are mixed in a single addition with stop reagents provided by the Cisbio kit used to stop the kinase reaction. Fluorescence is detected using an Envision Multilabeled 2014 reader from PerkinElmer. The Fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well. The resulting TR-FRET is proportional to the phosphorylation level. Staurosporine was used as the positive control. IC50 was determined by Xlfit 5.3.
- HTRF KinEASE Assay ASK1 was purchased from Thermofisher (Catalogue # PV4011), ATP was purchased from Sigma (Catalogue # A7699), HTRF KinEASE Assay System was obtained from Cisbio (Bedford, Mass.). A Area plate was purchased from Perkin Elmer (Catalogue # #6005560). HTRF KinEASE -STK is a generic method for measuring serine/threonine kinase activities using a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. The IC50 value for each compound was determined in the presence of compound (various concentration from 0 to 10 μM) and a fixed amount of ATP and peptide substrates. The test compound, 1 uM STK3 peptide substrate, and 5 nM of ASK1 kinase are incubated with kinase reaction buffer containing 50 mM HEPES pH 7.5, 0.01% BRU-35, 10 mM MgCl2, and 1 mM EGTA for 30 minutes. 100 uM ATP is added to start kinase reaction and incubated for 3 hours. The STK3-antibody labeled with Eu3+-Cryptate and 125 nM streptavidin-XL665 are mixed in a single addition with stop reagents provided by the Cisbio kit used to stop the kinase reaction. Fluorescence is detected using an Envision Multilabeled 2014 reader from PerkinElmer. The Fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well. The resulting TR-FRET is proportional to the phosphorylation level. Staurosporine was used as the positive control. IC50 was determined by XLfit 5.3.
- HTRF KinEASE Assay ASK1 was purchased from Thermofisher (Catalogue #PV4011), ATP was purchased from Sigma (Catalogue #A7699), HTRF KinEASE Assay System was obtained from Cisbio (Bedford, Mass.). Area plate was purchased from Perkin Elmer (Catalogue ##6005560). HTRF KinEASE -STK is a generic method for measuring serine/threonine kinase activities using a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. The IC50 value for each compound was determined in the presence of compound (various concentration from 0 to 10 μM) and a fixed amount of ATP and peptide substrates. The test compound, 1 μM STK3 peptide substrate, and 5 μM of ASK1 kinase are incubated with kinase reaction buffer containing 50 mM HEPES Ph 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA for 30 minutes. 100 μM ATP is added to start kinase reaction and incubated for 3 hours. The STK3-antibody labeled with Eu3+-Cryptate and 125 nM streptavidin-XL665 are mixed in a single addition with stop reagents provided by the Cisbio kit used to stop the kinase reaction. Fluorescence is detected using an Envision Multilabeled 2014 reader from PerkinElmer. The Fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well. The resulting TR-FRET is proportional to the phosphorylation level. Staurosporine was used as the positive control. IC50 was determined by Xlfit 5.3.
- HotSpot Assay In vitro profiling protein kinases was performed using the HotSpot assay platform. Briefly, specific kinase/substrate pairs along with required cofactors were prepared in reaction buffer. Compounds were delivered into the reaction, followed 15-20 minutes later by addition of a mixture of ATP (Sigma, St. Louis Mo.) and 33P ATP (Perkin Elmer, Waltham Mass.) to a final concentration of 10 μM. Reactions were carried out at room temperature for 120 min, followed by spotting of the reaction mixtures onto P81 ion exchange filter paper (Whatman Inc., Piscataway, N.J.). Unbound phosphate was removed by extensive washing of filters in 0.75% phosphoric acid. After subtraction of background derived from control reactions containing inactive enzyme, kinase activity data was expressed as the percent of remaining kinase activity in test samples compared to vehicle (dimethyl sulfoxide) reactions. IC50 values and curve fits were obtained using Prism (GraphPad Software).Reaction Conditions: Buffer Conditions: 20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, and 1% DMSO. ATP concentration: 10 μM. Reaction Time: 2 hours. Compound 3 was tested at 10 concentrations as follows with a comparison to the common kinase inhibitor reference compound staurosporine. Compounds were tested in a 10-point IC50 mode with 3-fold serial dilution starting at 10 uM. Control Compound was tested in a 10-point IC50 with 3-fold serial dilution starting at 20 uM.
- Inhibition Assay JAK1 (IC50): Recombinant human JAK1 catalytic domain (amino acids 850-1154; catalog number 08-144) is purchased from Carna Biosciences. 10 ng of JAK1 is incubated with 12.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (15 mM Tris-HCl pH 7.5, 1 mM DTT, 0.01% Tween-20, 10 mM MgCl2, 2 μM non-radioactive ATP, 0.25 μCi33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 pt, in a polypropylene 96-well plate (Greiner, V-bottom). After 45 min at 30° C., reactions are stopped by adding of 25 μL /well of 150 mM phosphoric acid. All of the terminated kinase reaction is transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates are washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates is sealed. 40 μL/well of Microscint-20 is added, the top of the plates is sealed and readout is performed using the Topcount (Perkin Elmer). Kinase activity is calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle.
- Inhibition Assay JAK2 (IC50): Recombinant human JAK2 catalytic domain (amino acids 808-1132; catalog number PV4210) is purchased from Invitrogen. 0.025 mU of JAK2 is incubated with 2.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (5 mM MOPS pH 7.5, 9 mM MgAc, 0.3 mM EDTA, 0.06% Brij and 0.6 mM DTT, 1 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 90 min at 30° C., reactions are stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction is transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates are washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates is sealed. 40 μL/well of Microscint-20 is added, the top of the plates is sealed and readout is performed using the Topcount (Perkin Elmer). Kinase activity is calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle.
- Inhibition Assay JAK3 (IC50): Recombinant human JAK3 catalytic domain (amino acids 781-1124; catalog number PV3855) is purchased from Invitrogen. 0.025 mU of JAK3 is incubated with 2.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (25 mM Tris pH 7.5, 0.5 mM EGTA, 0.5 mM Na3VO4, 5 mM b-glycerolphosphate, 0.01% Triton X-100, 1 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 105 mM at 30° C., reactions are stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction is transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates are washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates is sealed. 40 μL/well of Microscint-20 is added, the top of the plates is sealed and readout is performed using the Topcount (Perkin Elmer). Kinase activity is calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle.
- Kinase Assay The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1 Kinase buffer to final concentrations of 0.25 ug/mL, 15 uM, and 4 uM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer). The Emission ratio (Em) was calculated as a ratio of the coumarin (C) emission signal (at 445 nm)/Fluorescein (F) emission signal (at 520 nm). The percent phosphorylation was then calculated using the following formula: [1+-((Em ratio F100%)+-C100%)/((C0%+-C100%)+(Em ratio (F100%+-F0%)))].
- hJAK2 Biological Assay Enzymatic activity of hJAK2 was measured using a LANCE homogeneous time resolved fluorescence resonance energy transfer (TR-FRET) assay that monitors enzyme dependent phosphorylation of a biotinylated serine/threonine peptide substrate (PTK). An increase in the amount of phosphorylated peptide results in an increase in TR-FRET signal. hJAK2 was expressed and purified as full length recombinant proteins. Detection reagents for the assay were purchased from PerkinElmer. hJAK2 enzyme was assayed under initial rate conditions in the presence of 2×Km ATP (30 μM) and 1 μM peptide, 50 mM Tris-Cl (pH 7.5), 10 mM MgCl2, 1 mM dithiothreitol, 0.01% BSA, 0.05% (v/v) DMSO at the following concentration of enzyme: hJAK2 at 0.3 nM. After an assay reaction time of 40 minutes at 25° C., reactions were terminated with EDTA and LANCE Detection Buffer.Amount of phosphorylated peptide was determined by the addition of 20 nM SA-APC and 1 nM europium cryptate labeled anti-phospho PTK monoclonal antibody PY66 and the resulting TR-FRET signal was recorded on an Envision plate reader (Ex: 340 nm; Em: 615/665 nm; 100 μs delay and 200 μs read window). Data was normalized based on a positive (1 μM Staurosporine) and negative (DMSO) controls and IC50 values calculated from the fit of the dose-response curves to a four-parameter equation. All IC50 values represent geometric mean values of a minimum of four determinations.
- Biological Assay Compounds of the present disclosure were tested in a 10-dose IC50 mode with a 3-fold serial dilution starting from 0.5 μM, and the control compound Staurosporine was tested in a 10-dose IC50 mode with a 4-fold serial dilution starting from 20 μM. Reactions were carried out in the presence of 10 μM ATP. Conditions and Protocol: Procedures:1. Compounds were prepared in freshly prepared reaction buffers containing 20 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.01% Brij35, 0.02 mg/ml, BSA, 0.1 mM Na3VO4, 2 mM DTT, and 1% DMSO.2. Required cofactor such as 1 μg (1.5 μM) of recombinant retinoblastoma protein in the case of CDK subtypes was added to the substrate solutions as mentioned above.3. Kinase such as 10 ng of recombinant CDK4/cyclin D1 (Life Technologies PV4204) diluted in a kinase buffer (20 mM Tris pH7.5, 10 mM MgCl2, 0.01% NP-40, 2 mM DTT), and incubated at room temperature for 30 minutes together with indicated concentration of inhibitors.4. Compounds in DMSO were added into the kinase reaction mixture utilizing acoustic technology (Echo550).5. 33P-ATP (specific activity 0.01 μCi/μl final) was added into the reaction mixture to initiate the reaction.6. The reaction mixture was incubated for 120 minutes at room temperature.7. Reactions are spotted onto P81 ion exchange paper (Whatman #3698-915).8. Filters were extensively washed with 0.75% phosphoric acid.9. The radioactive phosphorylated substrate remaining on the filter paper was measured.
- Biological Assay for TBK1 and IKKs Enzymatic activity of IKKε and TBK1 was measured using a homogeneous time resolved fluorescence resonance energy transfer (TR-FRET) assay that monitors enzyme dependent phosphorylation of a biotinylated serine/threonine peptide substrate. An increase in the amount of phopshorylated peptide results in an increase in TR-FRET signal. TBK1 and IKKs were expressed and purified as full length recombinant proteins. Detection reagents for the assay were purchased from Cisbio. TBK1 and IKKε enzymes were assayed under initial rate conditions in the presence of 2× Km ATP (40-80 μM) and 1 μM peptide, hepes (pH 7), 0.1 mM orthovanadate, 0.02% NaN3, 0.01% BSA, 10 mM MgCl2, 0.01% (v/v) tritonX, 1 mM dithiothreitol, 0.5% (v/v) DMSO at the following concentrations for each enzyme: TBK1 at 2.5 nM and IKKε at 0.3 nM. After an assay reaction time of 240 minutes at 25° C., reactions were terminated with EDTA.Amount of phosphorylated peptide was determined by the addition of 125 nM streptavidin XL665 and europium cryptate labeled anti-phospho monoclonal antibody and the resulting TR-FRET signal was recorded on an Envision plate reader (Ex: 340 nm; Em: 615/665 nm; 100 μs delay and 200 μs read window). Data was normalized based on a positive (1 μM Staurosporine) and negative (DMSO) controls and IC50 values calculated from the fit of the dose-response curves to a four-parameter equation. All IC50 values represent geometric mean values of a minimum of four determinations.
- Biological for hJAK2 Enzymatic activity of hJAK2 was measured using a LANCE homogeneous time resolved fluorescence resonance energy transfer (TR-FRET) assay that monitors enzyme dependent phosphorylation of a biotinylated serine/threonine peptide substrate (PTK). An increase in the amount of phopshorylated peptide results in an increase in TR-FRET signal. hJAK2 was expressed and purified as full length recombinant proteins. Detection reagents for the assay were purchased from PerkinElmer. hJAK2 enzyme was assayed under initial rate conditions in the presence of 2×Km ATP (30 μM) and 1 μM peptide, 50 mM Tris-Cl (pH 7.5), 10 mM MgCl2, 1mM dithiothreitol, 0.01% BSA, 0.05% (v/v) DMSO at the following concentration of enzyme: hJAK2 at 0.3 nM. After an assay reaction time of 40 minutes at 25° C., reactions were terminated with EDTA and LANCE Detection Buffer.Amount of phosphorylated peptide was determined by the addition of 20 nM SA-APC and 1 nM europium cryptate labeled anti-phospho PTK monoclonal antibody PY66 and the resulting TR-FRET signal was recorded on an Envision plate reader (Ex: 340 nm; Em: 615/665 nm; 100 μs delay and 200 μs read window). Data was normalized based on a positive (1 μM Staurosporine) and negative (DMSO) controls and IC50 values calculated from the fit of the dose response curves to a four-parameter equation. All IC50 values represent geometric mean values of a minimum of four determinations. These assays generally produced results within 3-fold of the reported mean.
- DYRK1A Kinase Assay Each compound was dissolved in DMSO as a 10 mM stock and used to prepare compound source plates. Serial dilution (1:3, 11-point dose-response curves from 10 μM to 0.00016 μM) and compound transfer was performed using the ECHO 550 (Labcyte, Sunnyvale, CA) into 1536-well black-walled round bottom plates (Corning).The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies—a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.25 μg/mL, 15 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated (“0% Control”) and phosphorylated (“100% control”) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- Enzymatic Assay Briefly, specific kinase/substrate pairs along with required cofactors were prepared in reaction buffer. Compounds were delivered into the reaction, followed 15-20 minutes later by addition of a mixture of ATP (Sigma, St. Louis Mo.) and 33P ATP (Perkin Elmer, Waltham Mass.) to a final concentration of 10 μM. Reactions were carried out at room temperature for 120 min, followed by spotting of the reaction mixtures onto P81 ion exchange filter paper (Whatman Inc., Piscataway, N.J.). Unbound phosphate was removed by extensive washing of filters in 0.75% phosphoric acid. After subtraction of background derived from control reactions containing inactive enzyme, kinase activity data was expressed as the percent of remaining kinase activity in test samples compared to vehicle (dimethyl sulfoxide) reactions. IC50 values and curve fits were obtained using Prism (GraphPad Software). Reaction Conditions: Buffer Conditions: 20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, and 1% DMSO. ATP concentration: 10 μM. Reaction Time: 2 hours. Compound 3 was tested at 10 concentrations as follows with a comparison to the common kinase inhibitor reference compound staurosporine. Compounds were tested in a 10-point IC50 mode with 3-fold serial dilution starting at 10 uM. Control Compound was tested in a 10-point IC50 with 3-fold serial dilution starting at 20 uM. All kinase reactions were performed at 10 uM ATP. Test concentrations are provided below in molar units (M).
- Inhibition Assay The assay is a capture of radioactive 33P phosphorylated product through filtration. The interactions of Btk, biotinylated SH2 peptide substrate (Src homology), and ATP lead to phosphorylation of the peptide substrate. Biotinylated product is bound streptavidin sepharose beads. All bound, radiolabeled products are detected by scintillation counter.Plates assayed are 96-well polypropylene (Greiner) and 96-well 1.2 μm hydrophilic PVDF filter plates (Millipore). Concentrations reported here are final assay concentrations: 10-100 μM compounds in DMSO (Burdick and Jackson), 5-10 nM Btk enzyme (His-tagged, full-length), 30 μM peptide substrate (Biotin-Aca-AAAEEIYGEI-NH2), 100 μM ATP (Sigma), 8 mM imidazole (Sigma, pH 7.2), 8 mM glycerol-2-phosphate (Sigma), 200 μM EGTA (Roche Diagnostics), 1 mM MnCl2 (Sigma), 20 mM MgCl2 (Sigma), 0.1 mg/ml BSA (Sigma), 2 mM DTT (Sigma), 1 μCi 33P ATP (Amersham), 20% streptavidin sepharose beads (Amersham), 50 mM EDTA (Gibco), 2 M NaCl (Gibco), 2 M NaCl w/1% phosphoric acid (Gibco), microscint-20 (Perkin Elmer).IC50 determinations are calculated from 10 data points per compound utilizing data produced from a standard 96-well plate assay template. One control compound and seven unknown inhibitors were tested on each plate and each plate was run twice. Typically, compounds were diluted in half-log starting at 100 μM and ending at 3 nM. The control compound was staurosporine. Background was counted in the absence of peptide substrate. Total activity was determined in the presence of peptide substrate. The following protocol was used to determine Btk inhibition.
- Inhibition Assay The assay is a capture of radioactive 33P phosphorylated product through filtration. The interactions of Btk, biotinylated SH2 peptide substrate (Src homology), and ATP lead to phosphorylation of the peptide substrate. Biotinylated product is bound streptavidin sepharose beads. All bound, radiolabeled products are detected by scintillation counter.Plates assayed are 96-well polypropylene (Greiner) and 96-well 1.2 μm hydrophilic PVDF filter plates (Millipore). Concentrations reported here are final assay concentrations: 10-100 μM compounds in DMSO (Burdick and Jackson), 5-10 nM Btk enzyme (His-tagged, full-length), 30 μM peptide substrate (Biotin-Aca-AAAEEIYGEI-NH2), 100 μM ATP (Sigma), 8 mM imidazole (Sigma, pH 7.2), 8 mM glycerol-2-phosphate (Sigma), 200 μM EGTA (Roche Diagnostics), 1 mM MnCl2 (Sigma), 20 mM mgCl2 (Sigma), 0.1 mg/mL BSA (Sigma), 2 mM DTT (Sigma), 1 μCi 33P ATP (Amersham), 20% streptavidin sepharose beads (Amersham), 50 mM EDTA (Gibco), 2 M NaCl (Gibco), 2 M NaCl w/1% phosphoric acid (Gibco), microscint-20 (Perkin Elmer).IC50 determinations are calculated from 10 data points per compound utilizing data produced from a standard 96-well plate assay template. One control compound and seven unknown inhibitors were tested on each plate and each plate was run twice. Typically, compounds were diluted in half-log starting at 100 μM and ending at 3 nM. The control compound was staurosporine. Background was counted in the absence of peptide substrate. Total activity was determined in the presence of peptide substrate. The following protocol was used to determine Btk inhibition.
- Kinase Assay DYRK1A: Each compound was dissolved in DMSO as a 10 mM stock and used to prepare compound source plates. Serial dilution (1:3, 11-point dose-response curves from 10 M to 0.00016 M) and compound transfer was performed using the ECHO 550 (Labcyte, Sunnyvale, Calif.) into 1536-well black-walled round bottom plates (Corning).The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- Potency of IRAK4 Inhibitor Compounds in cKit Enzyme Assay Potency of IRAK4 Inhibitor Compounds in cKit Enzyme Assay. Evaluation of the effects of the IRAK4 inhibitor compounds on the activity of the human cKit kinase quantified by measuring the phosphorylation of the substrate Ulight-TK peptide using a human recombinant enzyme and the LANCE detection method at Eurofins CEREP, catalog item 3070, SOP no 1C768: The test compound, reference compound or water (control) are mixed with the enzyme (0.38 ng) in a buffer containing 40 mM Hepes/Tris (pH 7.4), 0.8 mM EGTA/Tris, 8 mM MgCl2, 1.6 mM DTT and 0.008% Tween 20. Thereafter, the reaction is initiated by adding 100 nM of the substrate Ulight-TK peptide and 50 μM ATP, and the mixture is incubated for 30 min at room temperature. For control basal measurements, the enzyme is omitted from the reaction mixture. Following incubation, the reaction is stopped by adding 13 mM EDTA. After 5 min, the anti-phopho-PT66 antibody labeled with europium chelate is added. After 60 more min, the fluorescence transfer is measured at lex=337 nm, lem=620 nm and lem=665 nm using a microplate reader (Envision, Perkin Elmer). The enzyme activity is determined by dividing the signal measured at 665 nm by that measured at 620 nm (ratio). The results are expressed as a percent inhibition of the control enzyme activity. The standard inhibitory reference compound is staurosporine, which is tested in each experiment at several concentrations to obtain an inhibition curve from which its IC50 value is calculated.
- TBK1 and IKKE Biological Assays Enzymatic activity of IKKε and TBK1 was measured using a homogeneous time resolved fluorescence resonance energy transfer (TR-FRET) assay that monitors enzyme dependent phosphorylation of a biotinylated serine/threonine peptide substrate. An increase in the amount of phosphorylated peptide results in an increase in TR-FRET signal. TBK1 and IKKε were expressed and purified as full length recombinant proteins. Detection reagents for the assay were purchased from Cisbio. TBK1 and IKKε enzymes were assayed under initial rate conditions in the presence of 2×Km ATP (40-80 μM) and 1 μM peptide, hepes (pH 7), 0.1 mM orthovanadate, 0.02% NaN3, 0.01% BSA, 10 mM MgCl2, 0.01% (v/v) tritonX, 1 mM dithiothreitol, 0.5% (v/v) DMSO at the following concentrations for each enzyme: TBK1 at 2.5 nM and IKKε at 0.3 nM. After an assay reaction time of 240 minutes at 25° C., reactions were terminated with EDTA.Amount of phosphorylated peptide was determined by the addition of 125 nM streptavidin XL665 and europium cryptate labeled anti-phospho monoclonal antibody and the resulting TR-FRET signal was recorded on an Envision plate reader (Ex: 340 nm; Em: 615/665 nm; 100 μs delay and 200 μs read window). Data was normalized based on a positive (1 μM Staurosporine) and negative (DMSO) controls and IC50 values calculated from the fit of the dose-response curves to a four-parameter equation. All IC50 values represent geometric mean values of a minimum of four determinations.
- TR-FRET ASK1 kinase assay The assay measures the phosphorylation level of a biotinylated peptide substrate by the ASK1 kinase using HTRF detection (6.1). This is a competitive, time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay, based on HTRF KinEASE -STK manual from Cisbio (6.1). Test compound, 1 μM STK3 peptide substrate, 4 nM of ASK1 kinase are incubated with 10 mM MOP buffer, pH. 7.0 containing 10 mM Mg-acetate, 0.025% NP-40, 1 mM DTT, 0.05% BSA and 1.5% glycerol for 30 minutes then 100 μM ATP is added to start the kinase reaction and incubated for 3 hr. Peptide antibody labeled with 1×Eu3+ Cryptate buffer containing 10 mM EDTA and 125 nM Streptavidin XL665 are added to stop the reaction and phosphorylated peptide substrate is detected using Envision 2103 Multilabeled reader from PerkinElmer. The fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well. The resulting TR-FRET level (a ratio of 665 nm/615 nm) is proportional to the phosphorylation level. Under these assay conditions, the degree of phosphorylation of peptide substrate was linear with time and concentration for the enzyme. The assay system yielded consistent results with regard to Km and specific activities for the enzyme. For inhibition experiments (IC50 values), activities were performed with constant concentrations of ATP, peptide and several fixed concentrations of inhibitors. Staurosporine, the nonselective kinase inhibitor, was used as the positive control. All enzyme activity data are reported as an average of quadruplicate determination.
- Biological Assay for TBK1 and IKKepsilon Enzymatic activity of IKKε and TBK1 was measured using a homogeneous time resolved fluorescence resonance energy transfer (TR-FRET) assay that monitors enzyme dependent phosphorylation of a biotinylated serine/threonine peptide substrate. An increase in the amount of phopshorylated peptide results in an increase in TR-FRET signal. TBK1 and IKKε were expressed and purified as full length recombinant proteins. Detection reagents for the assay were purchased from Cisbio. TBK1 and IKKε enzymes were assayed under initial rate conditions in the presence of 2×Km ATP (40-80 μM) and 1 μM peptide, hepes (pH 7), 0.1 mM orthovanadate, 0.02% NaN3, 0.01% BSA, 10 mM MgCl2, 0.01% (v/v) tritonX, 1 mM dithiothreitol, 0.5% (v/v) DMSO at the following concentrations for each enzyme: TBK1 at 2.5 mM and IKKε at 0.3 nM. After an assay reaction time of 240 minutes at 25° C., reactions were terminated with EDTA.Amount of phosphorylated peptide was determined by the addition of 125 nM streptavidin XL665 and europium cryptate labeled anti-phospho monoclonal antibody and the resulting TR-FRET signal was recorded on an Envision plate reader (Ex: 340 nm; Em: 615/665 nm; 100 μs delay and 200 μs read window). Data was normalized based on a positive (1 μM Staurosporine) and negative (DMSO) controls and IC50 values calculated from the fit of the dose-response curves to a four-parameter equation. All IC50 values represent geometric mean values of a minimum of four determinations. These assays generally produced results within 3-fold of the reported mean.
- Biological Assays for TBK1 and IKKe Enzymatic activity of IKKε and TBK1 was measured using a homogeneous time resolved fluorescence resonance energy transfer (TR-FRET) assay that monitors enzyme dependent phosphorylation of a biotinylated serine/threonine peptide substrate. An increase in the amount of phopshorylated peptide results in an increase in TR-FRET signal. TBK1 and IKKε were expressed and purified as full length recombinant proteins. Detection reagents for the assay were purchased from Cisbio. TBK1 and IKKε enzymes were assayed under initial rate conditions in the presence of 2× Km ATP (40-80 μM) and 1 μM peptide, hepes (pH 7), 0.1 mM orthovanadate, 0.02% NaN3, 0.01% BSA, 10 mM MgCl2, 0.01% (v/v) tritonX, 1 mM dithiothreitol, 0.5% (v/v) DMSO at the following concentrations for each enzyme: TBK1 at 2.5 nM and IKKε at 0.3 nM. After an assay reaction time of 240 minutes at 25° C., reactions were terminated with EDTA.Amount of phosphorylated peptide was determined by the addition of 125 nM streptavidin XL665 and europium cryptate labeled anti-phospho monoclonal antibody and the resulting TR-FRET signal was recorded on an Envision plate reader (Ex: 340 nm; Em: 615/665 nm; 100 μs delay and 200 μs read window). Data was normalized based on a positive (1 μM Staurosporine) and negative (DMSO) controls and IC50 values calculated from the fit of the dose response curves to a four-parameter equation. All IC50 values represent geometric mean values of a minimum of four determinations. These assays generally produced results within 3-fold of the reported mean.
- Chip Based Microfluidic Mobility Shift Assay All assays were performed in 384 well microtiter plates. Each assay plate contained 8-point serial dilutions for 40 test compounds, as well as four 8-point serial dilutions of staurosporine as reference compound, plus 16 high- and 16 low controls. Liquid handling and incubation steps were done on a Thermo CatX workstation equipped with a Innovadyne Nanodrop Express. Between pipetting steps, tips were cleaned in wash cycles using wash buffer. The assay plates were prepared by addition of 50 nl per well of compound solution in 90% DMSO. The kinase reactions were started by stepwise addition of 4.5 ul per well of peptide/ATP-solution (50 mM HEPES, pH 7.5, 1 mM DTT, 0.02% Tween20, 0.02% BSA, 0.6% DMSO, 10 mM beta-glycerophosphate, and 10 uM sodium orthovanadate, 1 mM MgCl2, 3 mM MnCl2, 4 uM ATP, 4 uM peptide (5-Fluo-Ahx-GAPDYENLQELNKK-Amid) (purchased from Biosyntan, Berlin, Germany) and 4.5 ul per well of enzyme solution (50 mM HEPES, pH 7.5, 1 mM DTT, 0.02% Tween20, 0.02% BSA, 0.6% DMSO, 10 mM beta-glycerophosphate, and 10 uM sodium orthovanadate, 1 mM MgCl2, 3 mM MnCl2, 4 nM Syk (Syk(2-635) (UniProtKB/Swiss-Prot: KSYK_HUMAN, P43405), produced in-house from insect cells). Kinase reactions were incubated at 30° C. for 60 minutes and subsequently terminated by addition of 16 ul per well of stop solution (100 mM HEPES pH 7.5, 5% DMSO, 0.1% Caliper coating reagent, 10 mM EDTA, and 0.015% Brij35). Plates with terminated kinase reactions were transferred to the Caliper LC3000 workstations for reading. Phosphorylated and unphosphorylated peptides were separated using the Caliper microfluidic mobility shift technology.
- DYRK1A Kinase Activity Assay Each compound was dissolved in DMSO as a 10 mM stock and used to prepare compound source plates. Serial dilution (1:3, 11-point dose-response curves from 10 μM to 0.00016 μM) and compound transfer was performed using the ECHO 550 (Labcyte, Sunnyvale, Calif.) into 1536-well black-walled round bottom plates (Corning). The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission. Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control. After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- DYRK1A Kinase Activity Assay Each compound was dissolved in DMSO as a 10 mM stock and used to prepare compound source plates. Serial dilution (1:3, 11-point dose-response curves from 10 μM to 0.00016 μM) and compound transfer was performed using the ECHO 550 (Labcyte, Sunnyvale, Calif.) into 1536-well black-walled round bottom plates (Corning).The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission. Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1×Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control. After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).
- JAK/TYK2 Assay 10 mM test compound stock or 1 mM control compound stock (tofocitinib, ruxolitinib or staurosporine) in DMSO was diluted to 0.4 mM in DMSO. A 3-fold series dilution was then performed in DMSO to generate 10 different compound concentrations. The assay was carried out in 384-well white plate. 0.5 uL of 40× compound DMSO solution at different concentrations was mixed with 10 uL 2× enzyme prepared in reaction buffer (20 mM HEPES, 10 mM MgCl2, 0.01% Tween, 1 mM DTT, pH 7.5). 10 uL 2× substrate mixture prepared in reaction buffer was then added to start the reaction. A short spin was done to settle down all solutions to the bottom of the plate. Final concentrations of test compound in the reaction mixture were 10000, 3333, 1111, 370, 123, 41.2, 13.7, 4.57, 1.52 and 0.51 nM. Concentrations of control compound were ten times less. Enzymatic reaction was conducted at 25° C. for 1-2 hours. 10 uL of Kinase Glo Reagents was added to stop the reaction and generate the luminescent signal which was measured using Envision. Luminescence signal was inversely related to kinase activity. Reaction mixture which did not contain enzyme served as negative control. The mixture without any compound was the positive control. Final concentration of enzymes and substrates and incubation time are summarized in the table below.[enz] [ATP] [sub] timeJAK1 7.5 nM 2 uM 30 uM (IRS-1) 1 hrJAK2 0.8 nM 2 uM 4 uM (pEY) 1 hrJAK3 1.5 nM 2 uM 4 uM (pEY) 1 hrTYK2 9 nM 2 uM 30 uM (IRS-1) 1 hr
- Recombinant CDK8 Protein-Cyclin C Complex In Vitro Capability of compounds of the present invention to bind to CDK8 protein was detected using LanthaScreen (ThermoFisher) assay. We detected FRET signal proportional to the amount of CDK8-bound fluorescently-labeled ligand (Tracer 236) that competes with inhibitor for ATP binding site. We carried out the measurements in a 15 μl reaction volume using a 384-well plate (Corning, #CLS4513). Enzyme CDK8/Cyclin C (ThermoFisher, #PR7261B) was mixed with antibodies Anti-His-tag-Biotin (ThermoFisher, #PV6090), Streptavidin-Eu (ThermoFisher, #PV6025), the resulting mixture was added to plate wells (5 MKπ/well). Final concentrations of substances were as follows: CDK8/Cyclin C 5 nM, Streptavidin-Eu 3 nM, Anti-His-tag-Biotin 3 nM. Staurosporine (S4400, Sigma) was used as a reference inhibitor, and 0.1% solution of dimethyl sulphoxide (DMSO) in reaction buffer was used as a blank, the reaction buffer comprised 250 mM HEPES (pH 7.5), 50 mM MgCl2, 5 mM EGTA, and 0.05% Brij-35.The inhibitors and controls in question were added to corresponding wells (5 μl/well). The plate was incubated at room temperature for 20 minutes. After incubating, 5 μl/well of tracer solution Alexa Fluor-647 (Kinase Tracer 236, ThermoFisher, #PV5592)) was added to the wells. Final concentration of tracer was 10 nM. Reaction buffer, instead of tracer solution, was used as a negative control. The plate was incubated for 40 minutes at 25 C., TR-FRET signal was then measured according to the manufacturer's guidelines on SPARK20 plate reader (Tecan, Switzerland), and the value was converted to the amount of kinase-bound tracer.
- Biological Assay IRAK4 enzyme (Carna Biosciences, Chuo-ku, Kobe, Japan) activity was measured by detecting phosphorylated peptide substrate formation using an antibody against the phosphorylated peptide substrate. This is a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay, based on the STK1 KinEASE Assay (Cisbio, Bedford, Mass.). The assay was designed as a simple two-step, endpoint assay (a 5 μl enzyme reaction followed by 5 μl stop and detect Solution) performed in ProxiPlate-384 Plus plates (Perkin Elmer, Waltham, Mass.). Staurosporine, a non-selective kinase inhibitor was used as a positive control. Compounds diluted in DMSO were spotted into 384 well plates using a Labcyte Echo 550 Liquid Handling System prior to addition of IRAK4 enzyme and peptide substrate. Reaction solutions were delivered using a Multi-Flo (Bio-Tek Instruments). The enzyme and peptide solution was incubated with compound for 15 minutes at room temp before the reaction was initiated by the addition of ATP. The standard 5 μl reaction mixture contained 500 μM ATP, 2 μM peptide (STK1 Peptide), 0.75 nM of IRAK4 in reaction buffer (50 mM HEPES, pH 7.0, 0.02% NaN3, 0.01% BSA, 0.1 mM Orthovanadate, 5 mM MgCl2, 0.025% NP-40, 1 mM DTT). After 120 min of incubation at room temperature, 5 μl of Stop and Detect Solution (1:100 Cryptate labeled anti-phosphorylated peptide antibody solution and 125 nM Tracer in a 50 mM HEPES pH 7.0 detection buffer containing sufficient EDTA) was added. The plate was then further incubated for 60 minutes at room temperature and read on Envision 2103 Multilabeled reader (PerkinElmer) with excitation/emission/FRET emission at 340 nm/615 nm/665 nm, respectively. Fluorescence intensities at 615 nm and 665 nm emission wavelengths were expressed as a ratio (665 nm/615 nm).
- Enzyme-linked immunosorbent assays (ELISA) Table B4: Enzyme-linked immunosorbent assays (ELISA) were performed to measure phosphorylated EGFR levels using A431 cells (10% FBS). A431 (1.0*104cells/40 μl/well) cells were seeded in 384 well. Compounds were dissolved in DMSO, serially diluted in DMSO and then were added, mixed, and incubated for 4 hours at 37° C., 500 CO2. Following the 4-hours incubation, cells were stimulated for 10 minutes with EGF (Invitrogen, cat #PHG0311) at a final concentration of 30 ng/mL in the incubator. The media was aspirated and cells were lysed in 10 μL lysis buffer with protease and phosphatase inhibitors (PerkinElmer, cat #ALSU-PEGFR-A50K). The plates were placed on a shaker for 5 minutes and then incubated for 30 min at 4° C. for complete lysis. The lysate was transferred to an Optiplate (Perkin Elmer, cat #6007290). Acceptor mix (PerkinElmer, cat #ALSU-PEGFR-A50K) was prepared just before use and 5 μL was dispensed to all the wells, followed by a 1.5-2 h incubation at room temperature in dark. The donor mix (PerkinElmer, cat #ALSU-PEGFR-A50K) was prepared under low light conditions prior to use and 5 μl of donor mix was added to all the wells under subdued lighting or green filters. The plates were placed on a shaker for 5 min, sealed, and incubated overnight at room temperature in dark. Plates were read on the Envision (PerkinElmer) using standard AlphaLISA settings. The percentage of inhibition on EGFR phosphorylation was calculated following equation: % Inhibition=100×(LumHC−LumSample)/(LumHC−LumLC). The low and high controls (LC/HC) are generated from lysate from wells with cells treated with DMSO or 10 mM Staurosporine (BioAustralis, cat #BIA-S1086), respectively.
- In Vitro c-FMS Activity In a 384 well polypropylene plate, c-FMS (0.14 nM, Carna 08-155) was pre-incubated in a 100 mM Hepes-NaOH pH 7.5 buffer containing 0.01% Triton X-100, 10 mM MgCl2, 0.1% BSA, 1 mM DTT, 10 uM sodium orthovanadate and 10 μM beta-glycerophosphate and compound with a concentration of 2.5% DMSO for 15 minutes at room temperature. The reaction was initiated with an equal volume of peptide substrate (Caliper Life Sciences catalog no. 760430) and ATP in the aforementioned buffer. The final concentrations in the reaction were 70 pM c-FMS, 1.5 μM peptide substrate and 500 μM ATP (ATP Km). The reaction was incubated at room temperature for 120 minutes and terminated with a buffer containing excess EDTA (100 mM Hepes-NaOH pH 7.5, 0.02% Brij, 0.1% CR-3, 0.36% DMSO and 100 mM EDTA). The plate was run for one cycle on a LabChip 3000 (Caliper Life Sciences, Hopkinton, Mass.) in an off-chip mobility shift type assay with an upstream voltage of −2250 volts, a downstream voltage of −500 volts and a vacuum pressure of −1.6 psi. The LabChip 3000 separates and measures the fluorescent signal of fluorescein labeled peptide substrate and fluorescein labeled peptide product present in each well. Results are expressed as percent conversion by measuring peak height for both the substrate and product and dividing the product peak height by the sum of peak heights for both substrate and product. On every plate 100% inhibition (with a saturating concentration of staurosporine) and 0% inhibition (substrate with enzyme and DMSO) controls were used to calculate percent inhibition of tested compounds and a Z′ prime value.
- In Vitro TrkA Activity In a 384 well polypropylene plate, TrkA (0.4 nM, Carna 08-186) was pre-incubated in a 100 mM Hepes-NaOH pH 7.5 buffer containing 0.01% Triton X-100, 10 mM MgCl2, 0.1% BSA, 1 mM DTT, 10 μM sodium orthovanadate and 10 μM beta-glycerophosphate and compound with a concentration of 2.5% DMSO for 15 minutes at room temperature. The reaction was initiated with an equal volume of peptide substrate (Caliper Life Sciences catalog no. 760430) and ATP in the aforementioned buffer. The final concentrations in the reaction were 200 pM TrkA, 1.5 μM peptide substrate and 55 μM ATP (ATP Km). The reaction was incubated at room temperature for 180 minutes and terminated with a buffer containing excess EDTA (100 mM Hepes-NaOH pH 7.5, 0.02% Brij, 0.1% CR-3, 0.36% DMSO and 100 mM EDTA). The plate was run for one cycle on a LabChip 3000 (Caliper Life Sciences, Hopkinton, Mass.) in an off-chip mobility shift type assay with an upstream voltage of −2250 volts, a downstream voltage of −500 volts and a vacuum pressure of −1.6 psi. The LabChip 3000 separates and measures the fluorescent signal of fluorescein labeled peptide substrate and fluorescein labeled peptide product present in each well. Results are expressed as percent conversion by measuring peak height for both the substrate and product and dividing the product peak height by the sum of peak heights for both substrate and product. On every plate 100% inhibition (with a saturating concentration of staurosporine) and 0% inhibition (substrate with enzyme and DMSO) controls were used to calculate percent inhibition of tested compounds and a Z′ prime value.
- In Vitro TrkB Activity In a 384 well polypropylene plate, TrkB (0.6 nM, Carna 08-187) was pre-incubated in a 100 mM Hepes-NaOH pH 7.5 buffer containing 0.01% Triton X-100, 10 mM MgCl2, 0.1% BSA, 1 mM DTT, 10 uM sodium orthovanadate and 10 μM beta-glycerophosphate and compound with a concentration of 2.5% DMSO for 15 minutes at room temperature. The reaction was initiated with an equal volume of peptide substrate (Caliper Life Sciences catalog no. 760430) and ATP in the aforementioned buffer. The final concentrations in the reaction were 300 pM TrkB, 1.5 μM peptide substrate and 70 μM ATP (ATP Km). The reaction was incubated at room temperature for 180 minutes and terminated with a buffer containing excess EDTA (100 mM Hepes-NaOH pH 7.5, 0.02% Brij, 0.1% CR-3, 0.36% DMSO and 100 mM EDTA). The plate was run for one cycle on a LabChip 3000 (Caliper Life Sciences, Hopkinton, Mass.) in an off-chip mobility shift type assay with an upstream voltage of −2250 volts, a downstream voltage of −500 volts and a vacuum pressure of −1.6 psi. The LabChip 3000 separates and measures the fluorescent signal of fluorescein labeled peptide substrate and fluorescein labeled peptide product present in each well. Results are expressed as percent conversion by measuring peak height for both the substrate and product and dividing the product peak height by the sum of peak heights for both substrate and product. On every plate 100% inhibition (with a saturating concentration of staurosporine) and 0% inhibition (substrate with enzyme and DMSO) controls were used to calculate percent inhibition of tested compounds and a Z′ prime value.
- JAK1 Inhibition Assay Recombinant human JAK1 catalytic domain (amino acids 850-1154; catalog number 08-144) was purchased from Carna Biosciences. 10 ng of JAK1 was incubated with 12.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (15 mM Tris-HCl pH 7.5, 1 mM DTT, 0.01% Tween-20, 10 mM MgCl2, 2 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 45 min at 30° C., reactions were stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 μL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100%.
- JAK2 Inhibition Assay Recombinant human JAK2 catalytic domain (amino acids 808-1132; catalog number PV4210) was purchased from Invitrogen. 0.025 mU of JAK2 was incubated with 2.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (5 mM MOPS pH 7.5, 9 mM MgAc, 0.3 mM EDTA, 0.06% Brij and 0.6 mM DTT, 1 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 90 min at 30° C., reactions were stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 μL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100%.
- JAK3 Inhibition Assay Recombinant human JAK3 catalytic domain (amino acids 781-1124; catalog number PV3855) was purchased from Invitrogen. 0.025 mU of JAK3 was incubated with 2.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (25 mM Tris pH 7.5, 0.5 mM EGTA, 0.5 mM Na3VO4, 5 mM b-glycerophosphate, 0.01% Triton X-100, 1 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 105 min at 30° C., reactions were stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 μL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100%.
- Measurement of Human IRAK-4 Inhibitory Activity For the measurement of the activity of the human IRAK-4 (Invitrogen, Cat. PV3362), phosphorylation of the IRAK-4 peptide substrate (biotin-KKKKRFSFKKSFKC) by the enzyme in the presence of 10 µM ATP (Sigma-Aldrich, Cat. A7699) was measured by the TR-FRET method. The enzymatic reaction was performed in a reaction buffer containing 50 mM HEPES (pH 7.2), 1 mM DTT, 0.1 mM Na3VO4, 5 mM MgCl2, 1 mM MnCl2, and 0.1% bovine serum albumin. For the measurement of the IRAK-4 inhibitory activity, a test compound was added to the reaction buffer containing 1 nM IRAK-4, 0.5 µM peptide substrate, and 10 µM ATP, and the mixture was incubated at 23° C. for 30 minutes. Then, a detection solution containing an antibody labeled with europium cryptate (0.3 µg/mL, the antibody was prepared by using the IRAK-4 peptide substrate as the antigen), streptavidin-XL665 (2 µg/mL, CisBio, Cat. 610SAXLB), 50 mM HEPES (pH 7.2), 0.1% BSA, 120 mM KF, and 66.7 mM EDTA (all the concentrations of the reagents are final concentrations) was added to terminate the reaction, and then the mixture was further incubated at 23° C. for 60 minutes. Fluorescence intensity was measured at wavelengths of 665 nm and 620 nm with a microplate reader, and the enzymatic activity was calculated as the ratio of fluorescence intensities at 665 nm and 620 nm (665 nm/620 nm). The IRAK-4 suppression ratio observed with addition of 12.5 µM staurosporine (LC Laboratories, Cat. S-9300) was defined to be 100%, the IRAK-4 suppression ratio observed with no addition of test compound was defined to be 0%, and IC50 of the test compound was calculated by using the 4-parameter logistic model of the data analysis software XLfit (ID Business Solutions Ltd.).
- TYK2 Inhibition Assay Recombinant human TYK2 catalytic domain (amino acids 871-1187; catalog number 08-147) was purchased from Carna biosciences. 5 ng of TYK2 was incubated with 12.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (25 mM Hepes pH 7.5, 100 mM NaCl, 0.2 mM Na3VO4, 0.1% NP-40, 0.1 μM non-radioactive ATP, 0.125 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 90 min at 30° C., reactions were stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 μL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100%.
- The DYRKIA kinase assay The DYRKIA kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRKIA kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).The Emission ratio (Em) was calculated as a ratio of the coumarin (C) emission signal (at 445 nm)/Fluorescein (F) emission signal (at 520 nm). The percent phosphorylation was then calculated using the following formula: [1−((Em ratio×F100%)−C100%)/((C0%−C100%)+(Em ratio×(F100%−F0%)))]. Dose-response curves were generated and inhibitory concentration (IC50) values were calculated using non-linear regression curve fit in the Dotmatics' Studies Software (Bishops Stortford, UK).
- TrkB Activity Reagents and consumables were purchased from Sigma Aldrich, Carna Biosciences, or Caliper Life Sciences. All assay reaction conditions for IC50 determinations were within the linear range with respect to time and enzyme concentration. In a 384 well polypropylene plate, TrkB (0.6 nM, Carna 08-187) was pre-incubated in a 100 mM Hepes-NaOH pH 7.5 buffer containing 0.01% Triton X-100, 10 mM MgCl2, 0.1% BSA, 1 mM DTT, 10 uM sodium orthovanadate and 10 μM beta-glycerophosphate and compound with a concentration of 2.5% DMSO for 15 minutes at room temperature. The reaction was initiated with an equal volume of peptide substrate (Caliper Life Sciences catalog no. 760430) and ATP in the aforementioned buffer. The final concentrations in the reaction were 300 pM TrkB, 1.5 μM peptide substrate and 70 μM ATP (ATP Km). The reaction was incubated at room temperature for 180 minutes and terminated with a buffer containing excess EDTA (100 mM Hepes-NaOH pH 7.5, 0.02% Brij, 0.1% CR-3, 0.36% DMSO and 100 mM EDTA). The plate was run for one cycle on a LabChip 3000 (Caliper Life Sciences, Hopkinton, Mass.) in an off-chip mobility shift type assay with an upstream voltage of −2250 volts, a downstream voltage of −500 volts and a vacuum pressure of −1.6 psi. The LabChip 3000 separates and measures the fluorescent signal of fluorescein labeled peptide substrate and fluorescein labeled peptide product present in each well. Results are expressed as percent conversion by measuring peak height for both the substrate and product and dividing the product peak height by the sum of peak heights for both substrate and product. On every plate 100% inhibition (with a saturating concentration of staurosporine) and 0% inhibition (substrate with enzyme and DMSO) controls were used to calculate percent inhibition of tested compounds and a Z′ prime value.
- TrkB Activity Reagents and consumables were purchased from Sigma Aldrich, Carna Biosciences, or Caliper Life Sciences. All assay reaction conditions for IC50 determinations were within the linear range with respect to time and enzyme concentration. In a 384 well polypropylene plate, TrkB (0.6 nM, Carna 08-187) was pre-incubated in a 100 mM Hepes-NaOH pH 7.5 buffer containing 0.01% Triton X-100, 10 mM MgCl2, 0.1% BSA, 1 mM DTT, 10 uM sodium orthovanadate and 10 μM beta-glycerophosphate and compound with a concentration of 2.5% DMSO for 15 minutes at room temperature. The reaction was initiated with an equal volume of peptide substrate (Caliper Life Sciences catalog no. 760430) and ATP in the aforementioned buffer. The final concentrations in the reaction were 300 pM TrkB, 1.5 μM peptide substrate and 70 μM ATP (ATP Km). The reaction was incubated at room temperature for 180 minutes and terminated with a buffer containing excess EDTA (100 mM Hepes-NaOH pH 7.5, 0.02% Brij, 0.1% CR-3, 0.36% DMSO and 100 mM EDTA). The plate was run for one cycle on a LabChip 3000 (Caliper Life Sciences, Hopkinton, Mass.) in an off-chip mobility shift type assay with an upstream voltage of −2250 volts, a downstream voltage of −500 volts and a vacuum pressure of −1.6 psi. The LabChip 3000 separates and measures the fluorescent signal of fluorescein labeled peptide substrate and fluorescein labeled peptide product present in each well. Results are expressed as percent conversion by measuring peak height for both the substrate and product and dividing the product peak height by the sum of peak heights for both substrate and product. On every plate 100% inhibition (with a saturating concentration of staurosporine) and 0% inhibition (substrate with enzyme and DMSO) controls were used to calculate percent inhibition of tested compounds and a Z′prime value.
- Caliper Assay All assays were performed in 384-well microtiter plates. Each assay plate contained 8-point serial dilutions for 40 test compounds, as well as four 8-point serial dilutions of staurosporine as a reference compound, plus 16 high and 16 low controls. Liquid handling and incubation steps were done on a Thermo CatX workstation equipped with Innovadyne Nanodrop Express. Between pipetting steps, tips were cleaned in wash cycles using wash buffer. The assay plates were prepared by addition of 50 nL per well of compound solution in 90% DMSO. The kinase reactions were started by stepwise addition of 4.5 μL per well of peptide/ATP-solution (50 mM HEPES, pH 7.5, 1 mM DTT, 0.02% BSA, 0.6% DMSO, 10 mM beta-glycerophosphate, and 10 μM sodium orthovanadate, 20 mM MgCl2, 2 mM MnCl2, 4 μM ATP, 4 μM peptide (FITC-Ahx-EAIYAAPFAKKK-NH2)) and 4.5 μL per well of enzyme solution (50 mM HEPES, pH 7.5, 1 mM DTT, 0.02% BSA, 0.6% DMSO, 10 mM beta-glycerophosphate, and 10 μM sodium orthovanadate, 20 mM MgCl2, 2 mM MnCl2, 3.5 nM ABL (ABL(64-515), produced in-house from E. coli)). Kinase reactions were incubated at 30 °C. for 60 minutes and subsequently terminated by addition of 16 μL per well of stop solution (100 mM HEPES pH 7.5, 5% DMSO, 0.1% Caliper coating reagent, 10 mM EDTA, and 0.015% Brij35). Plates with terminated kinase reactions were transferred to the Caliper LC3000 workstations for reading. Phosphorylated and unphosphorylated peptides were separated using the Caliper microfluidic mobility shift technology. Briefly, samples from terminated kinase reactions were applied to the chip. Analytes are transported through the chip by constant buffer flow and the migration of the substrate peptide is monitored by the fluorescence signal of its label. Phosphorylated peptide (product) and unphosphorylated peptide (substrate) are separated in an electric field by their charge/mass ratio.
- Chip Based Microfluidic Mobility Shift Assay All assays were performed in 384 well microtiter plates. Each assay plate contained 8-point serial dilutions for 40 test compounds, as well as four 8-point serial dilutions of staurosporine as reference compound, plus 16 high- and 16 low controls. Liquid handling and incubation steps were done on a Thermo CatX workstation equipped with a Innovadyne Nanodrop Express. Between pipetting steps, tips were cleaned in wash cycles using wash buffer. The assay plates were prepared by addition of 50 nl per well of compound solution in 90% DMSO. The kinase reactions were started by stepwise addition of 4.5 μl per well of peptide/ATP-solution (50 mM HEPES, pH 7.5, 1 mM DTT, 0.02% Tween20, 0.02% BSA, 0.6% DMSO, 10 mM beta-glycerophosphate, and 10 μM sodium orthovanadate, 1 mM MgCl2, 3 mM MnCl2, 4 μM ATP, 4 μM peptide (5-Fluo-Ahx-GAPDYENLQELNKK-Amid) and 4.5 μl per well of enzyme solution (50 mM HEPES, pH 7.5, 1 mM DTT, 0.02% Tween20, 0.02% BSA, 0.6% DMSO, 10 mM beta-glycerophosphate, and 10 μM sodium orthovanadate, 1 mM MgCl2, 3 mM MnCl2, 4 nM SYK (SYK(2-635), produced in-house from insect cells). Kinase reactions were incubated at 30 °C. for 60 minutes and subsequently terminated by addition of 16 μl per well of stop solution (100 mM HEPES pH 7.5, 5% DMSO, 0.1% Caliper coating reagent, 10 mM EDTA, and 0.015% Brij35). Plates with terminated kinase reactions were transferred to the Caliper LC3000 workstations for reading. Phosphorylated and unphosphorylated peptides were separated using the Caliper microfluidic mobility shift technology. Briefly, samples from terminated kinase reactions were applied to the chip. Analytes are transported through the chip by constant buffer flow and the migration of the substrate peptide is monitored by the fluorescence signal of its label. Phosphorylated peptide (product) and unphosphorylated peptide (substrate) are separated in an electric field by their charge/mass ratio. Kinase activities were calculated from the amounts of formed phospho-peptide.
- In Vitro Enzymatic Inhibitory Activity Assay The test was performed in a U-shaped bottom 384-well plate (coming, 4512 #), and the reaction temperature was 27° C. CDK7/CyclinH was diluted in a test buffer solution (20 mM MES PH6.75, 0.01% Tween20, 50 ug/mL BSA, 6 mM MgCl2) to obtain a corresponding enzyme solution with a 2.4× concentration. CDK2/CyclinE1 was diluted in a test buffer solution (20 mM MES PH6.75, 0.01% Tween20, 50 ug/mL BSA, 6 mM MgCl2) to obtain a corresponding enzyme solution with a 2.4× concentration. CDK9/CyclinT1 was diluted in a test buffer solution (20 mM MES PH6.75, 0.01% Tween20, 50 ug/mL BSA, 10 mM MgCl2) to obtain a corresponding enzyme solution with a 2.4× concentration. CDK12/CyclinK was diluted in a test buffer solution (80 mM MES PH6.5, 0.01% Tween20, 50 ug/mL BSA, 10 mM MgCl2) to obtain a corresponding enzyme solution with a 2.4× concentration. The compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM. When in use, the compounds were diluted with DMSO into 10 concentration gradients ranging from 25 nM to 500 uM, which were respectively diluted by 8.3 times in the test buffer solution to obtain compound solutions with a 6× concentration. A polypeptide substrate and ATP were diluted in the test buffer solution to obtain a mixed solution of the polypeptide substrate and ATP with a 2.4× concentration. 2 ul of a test compound solution was mixed with 5 ul of enzyme solution. After incubating for 10 min, 5 ul of the mixed solution of the polypeptide substrate and ATP was added. After incubating at 27° C. for 180 min, 4 uL of EDTA with a 120 mM concentration was added to each sample to stop the reaction. The test buffer solution containing 20 uM of staurosporine replaced the compound solution as 100% inhibitory control, the DMSO replaced the compound solution as 0% inhibitory control, and each test at least contained 2 parallel controls.
- JAK1 Inhibition Assay Recombinant human JAK1 (catalytic domain, amino acids 866-1154; catalog number PV4774) was purchased from Invitrogen. 1 ng of JAK1 was incubated with 20 nM Ulight-JAK1(tyr1023) peptide (Perkin Elmer catalog number TRF0121) in kinase reaction buffer (25 mM MOPS pH6.8, 0.016% Brij-35, 8.33 mM MgCl2, 3.33 mM DTT, 7 uM ATP) with or without 4 uL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 20 uL, in a white 384 Luminotrac 200 plate (Greiner, catalog number 781075). After 60 min at room temperature, reactions were stopped by adding 20 uL/well of detection mixture (1x detection buffer (Perkin Elmer, catalog number CR97-100C), 0.5 nM Europium-anti-phosphotyrosine (PT66) (Perkin Elmer, catalog number AD0068), 10 mM EDTA). Readout is performed using the Envision with excitation at 320 nm and measuring emission at 615 nm (Perkin Elmer). Kinase activity was calculated by subtracting relative fluorescence units (RFU) obtained in the presence of a positive control inhibitor (10 uM staurosporine) from RFU obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as: Percentage inhibition=((RFU determined for sample with test compound present-RFU determined for sample with positive control inhibitor) divided by (RFU determined in the presence of vehicle-RFU determined for sample with positive control inhibitor))*100. Dose dilution series were prepared for the compounds enabling the testing of dose-response effects in the JAK1 assay and the calculation of the IC50 for the compound. Each compound is routinely tested at concentration of 20 uM followed by a 1/5 serial dilution, 8 points (20 uM-4 uM-800 nM-160 nM-32 nM-6.4 nM-1.28 nM-0.26 nM) in a final concentration of 1% DMSO. When potency of compound series increases, more dilutions are prepared and/or the top concentration are lowered (e.g. 5 uM, 1 uM). The data are expressed as the average IC50 from the assays+standard error of the mean.
- JAK2 Inhibition Assay Recombinant human JAK2 (catalytic domain, amino acids 866-1154; catalog number PV4210) was purchased from Invitrogen. 0.0125 mU of JAK2 was incubated with 25 nM Ulight-JAK1(tyr1023) peptide (Perkin Elmer catalog number TRF0121) in kinase reaction buffer (41.66 mM HEPES pH7.0, 0.016% Triton X-100, 12.5 mM MgCl2, 3.33 mM DTT, 7.5M ATP) with or without 4L containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 20L, in a white 384 Luminotrac 200 plate (Greiner, catalog number 781075). After 60 min at room temperature, reactions were stopped by adding 20L/well of detection mixture (1x detection buffer (Perkin Elmer, catalog number CR97-100C), 0.5 nM Europium-anti-phosphotyrosine (PT66) (Perkin Elmer, catalog number AD0068), 10 mM EDTA). Readout is performed using the Envision with excitation at 320 nm and measuring emission at 615 nm (Perkin Elmer). Kinase activity was calculated by subtracting relative fluorescence units (RFU) obtained in the presence of a positive control inhibitor (10M staurosporine) from RFU obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as: Percentage inhibition=((RFU determined for sample with test compound present-RFU determined for sample with positive control inhibitor) divided by (RFU determined in the presence of vehicle-RFU determined for sample with positive control inhibitor))*100. Dose dilution series are prepared for compound enabling the testing of dose-response effects in the JAK2 assay and the calculation of the IC50 for the compound. Each compound is routinely tested at concentration of 20M followed by a 1/5 serial dilution, 8 points (20M-4M-800 nM-160 nM-32 nM-6.4 nM-1.28 nM-0.26 nM) in a final concentration of 1% DMSO. When potency of compound series increases, more dilutions are prepared and/or the top concentration are lowered (e.g. 5M, 1M). The data are expressed as the average IC50 from the assays standard error of the mean.
- KIT Kinase Biochemical Assay i. Enzyme The assay has been performed using KIT cytoplasmic domain product and purified in house as GST fused protein. The KIT protein (4.5 microM) was pre activated with 300 microM ATP for 1 hour at 28° C. in order to obtain a linear kinetic.ii. KIT Kinase Buffer (KB) Kinase buffer was composed of 50 mM HEPES pH 7.9 containing 5 mM MgCl2, 1 mM MnCl2, 10 mM DTT, 3 microM Na3VO4, and 0.2 mg/mL BSAiii. Assay Conditions The KIT kinase assay was run with a final pre activated enzyme concentration of 4 nM, in the presence of 4.4 microM ATP (residual ATP from KIT pre activation step is negligible), 3.9 nM 33P-γ-ATP and 2.5 microM of substrate BioDB n*138 (Aminoacidic sequence: KVVEEINGNNYVYIDPTQLPYDHKWEFPRNR SEQ ID NO: 2). The peptide was purchased from American Peptide Company (Sunnyvale, Calif.).Compound Testingi. Compound Dilution For IC50 determination, test compounds were received as a 1 mM solution in 100% DMSO, distributed into 96 well plates: compounds were then plated into the first column of a microtiter plate (A1 to G1), 100 microL/well. An automated station for serial dilutions (Biomek FX, Beckman) was used for producing 1:3 dilutions in 100% DMSO, from line A1 to A10, and for all the compounds in the column. Moreover, 4-5 copies of daughter plates were prepared by reformatting 5 microL of this first set of 100% DMSO dilution plates into 384 deep well-plates: one of these plates with the serial dilutions of test compounds was thawed the day of the experiments, reconstituted at a 3× concentration with water and used in the IC50 determination assays. In a standard experiment, the highest concentration (3×) of all compounds was 30 microM, while the lowest one was 1.5 nM.Each 384 well-plate contained at least one curve of the standard inhibitor staurosporine and reference wells (total enzyme activity vs. no enzymatic activity) for the Z and signal to background evaluation.
- The screening assay for DYRK1A kinase activity Each compound was dissolved in DMSO as a 10 mM stock and used to prepare compound source plates. Serial dilution (1:3, 11-point dose-response curves from 10 μM to 0.00016 μM) and compound transfer was performed using the ECHO 550 (Labcyte, Sunnyvale, Calif.) into 1536-well black-walled round bottom plates (Corning).The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).The Emission ratio (Em) was calculated as a ratio of the coumarin (C) emission signal (at 445 nm)/Fluorescein (F) emission signal (at 520 nm). The percent phosphorylation was then calculated using the following formula: [1−((Em ratio×F100%)−C100%)/((C0%−C100%)+(Em ratio×(F100%−F0%)))]. Dose-response curves were generated and inhibitory concentration (IC50) values were calculated using non-linear regression curve fit in the Dotmatics' Studies Software (Bishops Stortford, UK).
- The screening assay for DYRK1A kinase activity Each compound was dissolved in DMSO as a 10 mM stock and used to prepare compound source plates. Serial dilution (1:3, 11-point dose-response curves from 10 μM to 0.00016 μM) and compound transfer was performed using the ECHO 550 (Labcyte, Sunnyvale, Calif.) into 1536-well black-walled round bottom plates (Corning).The DYRK1A kinase assay was run using the Ser/Thr 18 peptide Z-lyte assay kit according to manufacturer's instructions (Life Technologies a Division of Thermo-Fisher). This is a non-radioactive assay using fluorescence resonance energy transfer (FRET) between coumarin and fluorescein to detect kinase activity which is represented as a ratio of coumarin emission/fluorescein emission.Briefly, recombinant DYRK1A kinase, ATP and Ser/Thr peptide 18 were prepared in 1× Kinase buffer to final concentrations of 0.19 μg/mL, 30 μM, and 4 μM respectively. The mixture was allowed to incubate with the representative compounds for one hour at room temperature. All reactions were performed in duplicate. Unphosphorylated ( 0% Control ) and phosphorylated ( 100% control ) forms of Ser/Thr 18 served as control reactions. Additionally, an 11-point dose-response curve of Staurosporine (1 uM top) was run to serve as a positive compound control.After incubation, Development Reagent A was diluted in Development Buffer then added to the reaction and allowed to further incubate for one hour at room temperature. The plate was read at Ex 400 Em 455 to detect the coumarin signal and Ex 400 Em 520 to measure the signal (EnVision Multilabel Plate Reader, PerkinElmer).The Emission ratio (Em) was calculated as a ratio of the coumarin (C) emission signal (at 445 nm)/Fluorescein (F) emission signal (at 520 nm). The percent phosphorylation was then calculated using the following formula: [1−((Em ratio×F100%)−C100%)/((C0%−C100%)+(Em ratio×(F100%−F0%)))]. Dose-response curves were generated and inhibitory concentration (IC50) values were calculated using non-linear regression curve fit in the Dotmatics' Studies Software (Bishops Stortford, UK).
- TrkA Activity Reagents and consumables were purchased from Sigma Aldrich, Carna Biosciences, or Caliper Life Sciences. All assay reaction conditions for IC50 determinations were within the linear range with respect to time and enzyme concentration. In a 384 well polypropylene plate, TrkA (0.4 nM, Carna 08-186) was pre-incubated in a 100 mM Hepes-NaOH pH 7.5 buffer containing 0.01% Triton X-100, 10 mM MgCl2, 0.1% BSA, 1 mM DTT, 10 μM sodium orthovanadate and 10 μM beta-glycerophosphate and compound with a concentration of 2.5% DMSO for 15 minutes at room temperature. The reaction was initiated with an equal volume of peptide substrate (Caliper Life Sciences catalog no. 760430) and ATP in the aforementioned buffer. The final concentrations in the reaction were 200 pM TrkA, 1.5 μM peptide substrate and 55 μM ATP (ATP Km). The reaction was incubated at room temperature for 180 minutes and terminated with a buffer containing excess EDTA (100 mM Hepes-NaOH pH 7.5, 0.02% Brij, 0.1% CR-3, 0.36% DMSO and 100 mM EDTA). The plate was run for one cycle on a LabChip 3000 (Caliper Life Sciences, Hopkinton, Mass.) in an off-chip mobility shift type assay with an upstream voltage of −2250 volts, a downstream voltage of −500 volts and a vacuum pressure of −1.6 psi. The LabChip 3000 separates and measures the fluorescent signal of fluorescein labeled peptide substrate and fluorescein labeled peptide product present in each well. Results are expressed as percent conversion by measuring peak height for both the substrate and product and dividing the product peak height by the sum of peak heights for both substrate and product. On every plate 100% inhibition (with a saturating concentration of staurosporine) and 0% inhibition (substrate with enzyme and DMSO) controls were used to calculate percent inhibition of tested compounds and a Z′ prime value.
- c-FMS Activity Reagents and consumables were purchased from Sigma Aldrich, Carna Biosciences, or Caliper Life Sciences. All assay reaction conditions for IC50 determinations were within the linear range with respect to time and enzyme concentration. In a 384 well polypropylene plate, c-FMS (0.14 nM, Carna 08-155) was pre-incubated in a 100 mM Hepes-NaOH pH 7.5 buffer containing 0.01% Triton X-100, 10 mM MgCl2, 0.1% BSA, 1 mM DTT, 10 uM sodium orthovanadate and 10 μM beta-glycerophosphate and compound with a concentration of 2.5% DMSO for 15 minutes at room temperature. The reaction was initiated with an equal volume of peptide substrate (Caliper Life Sciences catalog no. 760430) and ATP in the aforementioned buffer. The final concentrations in the reaction were 70 pM c-FMS, 1.5 μM peptide substrate and 500 μM ATP (ATP Km). The reaction was incubated at room temperature for 120 minutes and terminated with a buffer containing excess EDTA (100 mM Hepes-NaOH pH 7.5, 0.02% Brij, 0.1% CR-3, 0.36% DMSO and 100 mM EDTA). The plate was run for one cycle on a LabChip 3000 (Caliper Life Sciences, Hopkinton, Mass.) in an off-chip mobility shift type assay with an upstream voltage of −2250 volts, a downstream voltage of −500 volts and a vacuum pressure of −1.6 psi. The LabChip 3000 separates and measures the fluorescent signal of fluorescein labeled peptide substrate and fluorescein labeled peptide product present in each well. Results are expressed as percent conversion by measuring peak height for both the substrate and product and dividing the product peak height by the sum of peak heights for both substrate and product. On every plate 100% inhibition (with a saturating concentration of staurosporine) and 0% inhibition (substrate with enzyme and DMSO) controls were used to calculate percent inhibition of tested compounds and a Z′ prime value.
- c-FMS Activity Reagents and consumables were purchased from Sigma Aldrich, Carna Biosciences, or Caliper Life Sciences. All assay reaction conditions for IC50 determinations were within the linear range with respect to time and enzyme concentration. In a 384 well polypropylene plate, c-FMS (0.14 nM, Carna 08-155) was pre-incubated in a 100 mM Hepes-NaOH pH 7.5 buffer containing 0.01% Triton X-100, 10 mM MgCl2, 0.1% BSA, 1 mM DTT, 10 uM sodium orthovanadate and 10 μM beta-glycerophosphate and compound with a concentration of 2.5% DMSO for 15 minutes at room temperature. The reaction was initiated with an equal volume of peptide substrate (Caliper Life Sciences catalog no. 760430) and ATP in the aforementioned buffer. The final concentrations in the reaction were 70 pM c-FMS, 1.5 μM peptide substrate and 500 μM ATP (ATP Km). The reaction was incubated at room temperature for 120 minutes and terminated with a buffer containing excess EDTA (100 mM Hepes-NaOH pH 7.5, 0.02% Brij, 0.1% CR-3, 0.36% DMSO and 100 mM EDTA). The plate was run for one cycle on a LabChip 3000 (Caliper Life Sciences, Hopkinton, Mass.) in an off-chip mobility shift type assay with an upstream voltage of −2250 volts, a downstream voltage of −500 volts and a vacuum pressure of −1.6 psi. The LabChip 3000 separates and measures the fluorescent signal of fluorescein labeled peptide substrate and fluorescein labeled peptide product present in each well. Results are expressed as percent conversion by measuring peak height for both the substrate and product and dividing the product peak height by the sum of peak heights for both substrate and product. On every plate 100% inhibition (with a saturating concentration of staurosporine) and 0% inhibition (substrate with enzyme and DMSO) controls were used to calculate percent inhibition of tested compounds and a Z′prime value.
- High Throughput EGFR Biochemical Assay EGFR activity was measured using KinEASE (Cisbio), a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. In this assay, EGFR-catalyzes the phosphorylation of a universal Tyrosine kinase peptide substrate labeled with XL665. Europium conjugated phosphor-tyrosine specific antibody binds the resulting phosphorylated peptide. Formation of phosphorylated peptide is quantified by TR-FRET with Europium as the donor and XL665 the acceptor. The assay was performed in two main steps. The first step is the kinase reaction step and the second step is the detection step with TR-FRET reagents. In brief, test compounds 1:3 serially diluted in DMSO were delivered into Corning white, low volume, non-binding 384 well plates using the Echo 550 acoustic liquid dispenser (Labcyte ). EGFR enzyme (Human EGFR, cytoplasmic domain [669-1210] from Carna Biosciences Cat. No. 08-115) and substrates TK substrate-biotin (included in Cisbio HTRF KinEASE-TK kit Cat. No. 62TK0PEJ) were dispensed into assay plates using a Multi-Flo (Bio-Tek Instruments). The standard 10 μL reaction mixture contained 6 μM ATP (1×Km) or 12 μM ATP (2×Km), 1 μM biotinylated peptide, 0.3 nM EGFR (for 1×Km ATP) or 0.1 nM EGFR (for 2×Km ATP) in reaction buffer (10 mM MOPS, pH 7.0, 1.5% Glycerol, 0.5 mg/ml BSA, 10 mM Mg-Acetate, 1 mM DTT, 0.025% NP-40). After 60 min of incubation at room temperature, 10 μL of Stop and Detect Solution (1:400 Europium Cryptate labeled anti-phosphorylated peptide antibody solution and 125 nM strepavidin-XL665 Tracer in a 50 mM Hepes pH 7.0 detection buffer containing sufficient EDTA) was added. The plate was then further incubated for over 60 minutes at room temperature and read using an Envision 2103 Multilabeled reader (PerkinElmer) with excitation/emission/FRET emission at 340 nm/615 nm/665 nm, respectively. Fluorescence intensities at 615 nm and 665 nm emission wavelengths were expressed as a ratio (665 nm/615 nm). Percent inhibition was calculated as follows: % Inhibition=100x(RatioSample−Ratio0% Inhibition)/(Ratio100% Inhibition−Ratio0% Inhibition) where 0.05% DMSO (0% inhibition) was the negative control and 100 μM Staurosporine and Gefitinib (100% inhibition) was used as the positive control.
- High Throughput EGFR Biochemical Assay EGFR activity was measured using KinEASE (Cisbio), a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. In this assay, EGFR-catalyzes the phosphorylation of a universal Tyrosine kinase peptide substrate labeled with XL665. Europium conjugated phosphor-tyrosine specific antibody binds the resulting phosphorylated peptide. Formation of phosphorylated peptide is quantified by TR-FRET with Europium as the donor and XL665 the acceptor. The assay was performed in two main steps. The first step is the kinase reaction step and the second step is the detection step with TR-FRET reagents. In brief, test compounds 1:3 serially diluted in DMSO were delivered into Corning white, low volume, non-binding 384 well plates using the Echo 550 acoustic liquid dispenser (Labcyte ). EGFR enzyme (Human EGFR, cytoplasmic domain [669-1210] from Carna Biosciences Cat. No. 08-115) and substrates TK substrate-biotin (included in Cisbio HTRF KinEASE-TK kit Cat. No. 62TK0PEJ) were dispensed into assay plates using a Multi-Flo (Bio-Tek Instruments). The standard 10 μL reaction mixture contained 6 μM ATP (1×Km) or 12 μM ATP (2×Km), 1 μM biotinylated peptide, 0.3 nM EGFR (for 1×Km ATP) or 0.1 nM EGFR (for 2×Km ATP) in reaction buffer (10 mM MOPS, pH 7.0, 1.5% Glycerol, 0.5 mg/ml BSA, 10 mM Mg-Acetate, 1 mM DTT, 0.025% NP-40). After 60 min of incubation at room temperature, 10 μL of Stop and Detect Solution (1:400 Europium Cryptate labeled anti-phosphorylated peptide antibody solution and 125 nM strepavidin-XL665 Tracer in a 50 mM Hepes pH 7.0 detection buffer containing sufficient EDTA) was added. The plate was then further incubated for over 60 minutes at room temperature and read using an Envision 2103 Multilabeled reader (PerkinElmer) with excitation/emission/FRET emission at 340 nm/615 nm/665 nm, respectively. Fluorescence intensities at 615 nm and 665 nm emission wavelengths were expressed as a ratio (665 nm/615 nm). Percent inhibition was calculated as follows:% Inhibition=100×(RatioSample−Ratio0% Inhibition)/(Ratio100% Inhibition−Ratio0% Inhibition)where 0.05% DMSO (0% inhibition) was the negative control and 100 μM Staurosporine and Gefitinib (100% inhibition) was used as the positive control.
- TYK2 Inhibition Assay Recombinant human TYK2 catalytic domain (amino acids 871-1187; catalog number 08-147) was purchased from Carna biosciences. 5 ng of TYK2 was incubated with 12.5 ug polyGT substrate (Sigma catalog number Pb0275) in kinase reaction buffer (25 mM Hepes pH 7.5, 100 mM NaCl, 0.2 mM Na3VO4, 0.1% NP-40, 0.1 uM non-radioactive ATP, 0.125 uCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 uL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 uL, in a polypropylene 96-well plate (Greiner, V-bottom). After 90 min at 30° C., reactions were stopped by adding of 25 uL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 uL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 uL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 uM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as: Percentage inhibition=((cpm determined for sample with test compound present-cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle-cpm determined for sample with positive control inhibitor))*100. Dose dilution series were prepared for the compounds enabling the testing of dose-response effects in the TYK2 assay and the calculation of the IC50 for the compound. Each compound was routinely tested at concentration of 20 uM followed by a 1/3 serial dilution, 8 points (20 uM-6.67 uM-2.22 uM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of compound series increased, more dilutions were prepared and/or the top concentration was lowered (e.g. 5 uM, 1 uM).
- JAK1 Inhibition Assay Recombinant human JAK1 catalytic domain (amino acids 850-1154; catalog number 08-144) is purchased from Carna Biosciences. 10 ng of JAK1 is incubated with 12.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (15 mM Tris-HCl pH 7.5, 1 mM DTT, 0.01% Tween-20, 10 mM MgCl2, 2 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 45 min at 30° C., reactions are stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction is transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates are washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates is sealed. 40 μL/well of Microscint-20 is added, the top of the plates is sealed and readout is performed using the Topcount (Perkin Elmer). Kinase activity is calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity is determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100.Dose dilution series are prepared for the compounds enabling the testing of dose-response effects in the JAK1 assay and the calculation of the IC50 for each compound. Each compound is routinely tested at concentration of 20 μM followed by a 1/3 serial dilution, 8 points (20 μM-6.67 μM-2.22 μM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of compound series increased, more dilutions are prepared and/or the top concentration is lowered (e.g. 5 μM, 1 μM).
- JAK1 Inhibition Assay Recombinant human JAK1 catalytic domain (amino acids 850-1154; catalog number 08-144) was purchased from Carna Biosciences. 10 ng of JAK1 was incubated with 12.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (15 mM Tris-HCl pH 7.5, 1 mM DTT, 0.01% Tween-20, 10 mM MgCl2, 2 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 45 min at 30° C., reactions were stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 μL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100. Dose dilution series were prepared for the compounds enabling the testing of dose-response effects in the JAK1 assay and the calculation of the IC50 for each compound. Each compound was routinely tested at concentration of 20 μM followed by a ⅓ serial dilution, 8 points (20 μM-6.67 μM-2.22 μM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of compound series increased, more dilutions were prepared and/or the top concentration was lowered (e.g. 5 μM, 1 μM).
- JAK2 Inhibition Assay Recombinant human JAK2 catalytic domain (amino acids 808-1132; catalog number PV4210) is purchased from Invitrogen. 0.025 mU of JAK2 is incubated with 2.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (5 mM MOPS pH 7.5, 9 mM MgAc, 0.3 mM EDTA, 0.06% Brij and 0.6 mM DTT, 1 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 90 min at 30° C., reactions are stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction is transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates are washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates is sealed. 40 μL/well of Microscint-20 is added, the top of the plates is sealed and readout is performed using the Topcount (Perkin Elmer). Kinase activity is calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity is determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100.Dose dilution series are prepared for the compounds enabling the testing of dose-response effects in the JAK2 assay and the calculation of the IC50 for each compound. Each compound is routinely tested at concentration of 20 μM followed by a 1/3 serial dilution, 8 points (20 μM-6.67 μM-2.22 μM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of compound series increased, more dilutions are prepared and/or the top concentration is lowered (e.g. 5 μM, 1 μM).
- JAK2 Inhibition Assay Recombinant human JAK2 catalytic domain (amino acids 808-1132; catalog number PV4210) was purchased from Invitrogen. 0.025 mU of JAK2 was incubated with 2.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (5 mM MOPS pH 7.5, 9 mM MgAc, 0.3 mM EDTA, 0.06% Brij and 0.6 mM DTT, 1 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 90 min at 30° C., reactions were stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prcwashcd (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 μL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100.Dose dilution series were prepared for the compounds enabling the testing of dose-response effects in the JAK2 assay and the calculation of the IC50 for each compound. Each compound was routinely tested at concentration of 20 μM followed by a ⅓ serial dilution, 8 points (20 μM-6.67 μM-2.22 μM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of compound series increased, more dilutions were prepared and/or the top concentration was lowered (e.g. 5 μM, 1 μM).
- JAK3 Inhibition Assay Recombinant human JAK3 catalytic domain (amino acids 781-1124; catalog number PV3855) is purchased from Invitrogen. 0.025 mU of JAK3 is incubated with 2.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (25 mM Tris pH 7.5, 0.5 mM EGTA, 0.5 mM Na3VO4, 5 mM b-glycerolphosphate, 0.01% Triton X-100, 1 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 105 min at 30° C., reactions are stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction is transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates are washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates is sealed. 40 μL/well of Microscint-20 is added, the top of the plates is sealed and readout is performed using the Topcount (Perkin Elmer). Kinase activity is calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity is determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100.Dose dilution series are prepared for the compounds enabling the testing of dose-response effects in the JAK3 assay and the calculation of the IC50 for each compound. Each compound is routinely tested at concentration of 20 μM followed by a 1/3 serial dilution, 8 points (20 μM-6.67 μM-2.22 μM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of compound series increased, more dilutions are prepared and/or the top concentration is lowered (e.g. 5 μM, 1 μM).
- JAK3 Inhibition Assay Recombinant human JAK3 catalytic domain (amino acids 781-1124; catalog number PV3855) was purchased from Invitrogen. 0.025 mU of JAK3 was incubated with 2.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (25 mM Tris pH 7.5, 0.5 mM EGTA, 0.5 mM Na3VO4, 5 mM b-glycerolphosphate, 0.01% Triton X-100, 1 μM non-radioactive ATP, 0.25 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 105 min at 30° C., reactions were stopped by adding of 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 μL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100.Dose dilution series were prepared for the compounds enabling the testing of dose-response effects in the JAK3 assay and the calculation of the IC50 for each compound. Each compound was routinely tested at concentration of 20 μM followed by a ⅓ serial dilution, 8 points (20 μM-6.67 μM-2.22 μM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of compound series increased, more dilutions were prepared and/or the top concentration was lowered (e.g. 5 μM, 1 μM).
- JAK3 Inhibition Assay Recombinant human JAK3 catalytic domain (amino acids 781-1124; catalog number PV3855) was purchased from Invitrogen. 0.025 mU of JAK3 was incubated with 2.5 ug polyGT substrate (Sigma catalog number Pb0275) in kinase reaction buffer (25 mM Tris pH 7.5, 0.5 mM EGTA, 0.5 mM Na3VO4, 5 mM b-glycerolphosphate, 0.01% Triton X-100, 1 uM non-radioactive ATP, 0.25 uCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 uL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 uL, in a polypropylene 96-well plate (Greiner, V-bottom). After 105 min at 30° C., reactions were stopped by adding of 25 uL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 uL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 uL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 uM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as: Percentage inhibition=((cpm determined for sample with test compound present-cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle-cpm determined for sample with positive control inhibitor))*100. Dose dilution series were prepared for the compound enabling the testing of dose-response effects in the JAK3 assay and the calculation of the IC50 for the compound. Each compound was routinely tested at concentration of 20 uM followed by a 1/3 serial dilution, 8 points (20 uM-6.67 uM-2.22 uM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of the compound series increased, more dilutions were prepared and/or the top concentration was lowered (e.g. 5 uM, 1 uM). The data are expressed as the average IC50 from the assays standard error of the mean.
- TYK2 Inhibition Assay Recombinant human TYK2 catalytic domain (amino acids 871-1187; catalog number 08-147) is purchased from Carna biosciences. 5 ng of TYK2 is incubated with 12.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (25 mM Hepes pH 7.5, 100 mM NaCl, 0.2 mM Na3VO4, 0.1% NP-40, 0.1 μM non-radioactive ATP, 0.125 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 90 min at 30° C., reactions are stopped by adding 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction is transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates are washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates is sealed. 40 μL/well of Microscint-20 is added, the top of the plates is sealed and readout is performed using the Topcount (Perkin Elmer). Kinase activity is calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity is determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100.Dose dilution series are prepared for the compounds enabling the testing of dose-response effects in the TYK2 assay and the calculation of the IC50 for each compound. Each compound is routinely tested at concentration of 20 μM followed by a 1/3 serial dilution, 8 points (20 μM-6.67 μM-2.22 μM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of compound series increased, more dilutions are prepared and/or the top concentration is lowered (e.g. 5 μM, 1 μM).
- TYK2 Inhibition Assay Recombinant human TYK2 catalytic domain (amino acids 871-1187; catalog number 08-147) was purchased from Carna biosciences. 5 ng of TYK2 was incubated with 12.5 μg polyGT substrate (Sigma catalog number P0275) in kinase reaction buffer (25 mM Hepes pH 7.5, 100 mM NaCl, 0.2 mM Na3VO4, 0.1% NP-40, 0.1 μM non-radioactive ATP, 0.125 μCi 33P-gamma-ATP (GE Healthcare, catalog number AH9968) final concentrations) with or without 5 μL containing test compound or vehicle (DMSO, 1% final concentration), in a total volume of 25 μL, in a polypropylene 96-well plate (Greiner, V-bottom). After 90 min at 30° C., reactions were stopped by adding 25 μL/well of 150 mM phosphoric acid. All of the terminated kinase reaction was transferred to prewashed (75 mM phosphoric acid) 96 well filter plates (Perkin Elmer catalog number 6005177) using a cell harvester (Perkin Elmer). Plates were washed 6 times with 300 μL per well of a 75 mM phosphoric acid solution and the bottom of the plates was sealed. 40 μL/well of Microscint-20 was added, the top of the plates was sealed and readout was performed using the Topcount (Perkin Elmer). Kinase activity was calculated by subtracting counts per minute (cpm) obtained in the presence of a positive control inhibitor (10 μM staurosporine) from cpm obtained in the presence of vehicle. The ability of a test compound to inhibit this activity was determined as:Percentage inhibition=((cpm determined for sample with test compound present−cpm determined for sample with positive control inhibitor) divided by (cpm determined in the presence of vehicle−cpm determined for sample with positive control inhibitor))*100.Dose dilution series were prepared for the compounds enabling the testing of dose-response effects in the TYK2 assay and the calculation of the IC50 for each compound. Each compound was routinely tested at concentration of 20 μM followed by a ⅓ serial dilution, 8 points (20 μM-6.67 μM-2.22 μM-740 nM-247 nM-82 nM-27 nM-9 nM) in a final concentration of 1% DMSO. When potency of compound series increased, more dilutions were prepared and/or the top concentration was lowered (e.g. 5 μM, 1 μM).
- Kinase Assay Materials used include HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), Brij-35 (dodecyl polyglycol ether), DTT (dithiothreitol), EDTA (ethylenediamine tetraacetic acid), EGFR (human epidermal growth factor receptor), HER2 (human epidermal growth factor receptor 2), EGFR T790M (human epidermal growth factor receptor T790M mutation), Peptide FAM-P22 (fluorescein-labeled peptide 22), ATP (adenosine triphosphate), DMSO (dimethyl sulfoxide), 96-well plate, 384-well plate, staurosporine, Coating Reagent #3 and so on, all of which are commercially available.1. Preparation of Kinase Base Buffer and Stop Buffer1× Kinase buffer without MnCl2 was prepared from 50 mM HEPES, pH 7.5, 0.0015% Brij-35, 10 mM MgCl2, and 2 mM DTT. Stop buffer was prepared from 100 mM HEPES, pH 7.5, 0.015% Brij-35, 0.2% Coating Reagent #3, and 50 mM EDTA.2. Preparation of the Compounds for Testing KinasesPreparation of the compounds was performed according to the following procedures: (1) the compound to be tested was diluted to the concentration of 500 μM with 100% DMSO which is 50× of the final desired highest inhibitor concentration, and 100 μL of the diluted compound solution was transferred to a well in a 96-well plate; (2) the compound was gradiently diluted by transferring 20 μL original solution to 60 μL of 100% DMSO in the next well and so forth for a total of 10 concentrations; (3) DMSO (100 μL, 100%) was added to two empty wells as a no-compound control and a no-enzyme control in the same 96-well plate, and the plate was marked as source plate; (4) intermediate plate was prepared by transferring 10 μL of each compound from source plate to a new 96-well plate as the intermediate plate, and to each well of the intermediate plate was added 90 μL of 1× Kinase base buffer, then the intermediate plate was mixed for 10 min on shaker; and (5) assay plate was prepared by transferring 5 μL of each well from the 96-well intermediate plate to a 384-well plate in duplicates.3. Kinase ReactionKinase reaction was performed according to the following procedures: (1) 2.5× kinase solution was prepared by adding kinase into 1× kinase base buffer; (2) 2.5× peptide solution was prepared by adding FAM-labeled peptide and ATP into 1× kinase base buffer; (3) 2.5× kinase solution (10 μL) was added to each well of the 384-well assay plate containing 5 μL of compound in 10% DMSO and then the assay plate was incubated at room temperature for 10 minutes; (4) 2.5× peptide solution (10 μL) was added to each well of the 384-well assay plate; and (5) stop buffer (25 μL) was added to stop the kinase reaction after incubation at 28° C. for a specified period of time.4. Data MeasurementMultifunctional microplate reader (BMG LABTECH, PHER Astar FS) with a 320 nM excitation wavelength was used to read and collected the data of by absorbing light at 665 nM and 620 nM, which was converted from the rate of 665 signal/620 signal.
- Biochemical Assay for Inhibitors of FLT3 Kinase Activity i. Enzyme The assay was performed using FLT3 cytoplasmic domain product and purified in house as GST fused protein. The FLT3 protein (1 microM) was pre activated with 800 microM ATP for 1 hour at 28° C. in order to obtain a linear kinetic.ii. FLT3 Kinase Buffer (KB) Kinase buffer was composed of 50 mM HEPES pH 7.9 containing 4 mM MgCl2, 1 mM DTT, 10 microM Na3VO4, and 0.2 mg/mL BSAiii. Assay conditions The FLT3 kinase assay was run with a final pre activated enzyme concentration of 2 nM, in the presence of 254 microM ATP (residual ATP from KIT pre activation step is negligible), 8 nM 33P-γ-ATP and 55 microM of substrate BioDB n*24 (Aminoacidic sequence: GGKKKVSRSGLYRSPSMPENLNRPR SEQ ID NO: 1). The peptide was purchased from American Peptide Company (Sunnyvale, Calif.).Compound Testingi. Compound Dilution For IC50 determination, test compounds were received as a 1 mM solution in 100% DMSO, distributed into 96 well plates: compounds were then plated into the first column of a microtiter plate (A1 to G1), 100 microL/well. An automated station for serial dilutions (Biomek FX, Beckman) was used for producing 1:3 dilutions in 100% DMSO, from line A1 to A10, and for all the compounds in the column. Moreover, 4-5 copies of daughter plates were prepared by reformatting 5 microL of this first set of 100% DMSO dilution plates into 384 deep well-plates: one of these plates with the serial dilutions of test compounds was thawed the day of the experiments, reconstituted at a 3× concentration with water and used in the IC50 determination assays. In a standard experiment, the highest concentration (3×) of all compounds was 30 microM, while the lowest one was 1.5 nM.Each 384 well-plate contained at least one curve of the standard inhibitor staurosporine and reference wells (total enzyme activity vs. no enzymatic activity) for the Z′ and signal to background evaluation,ii. Assay Scheme 384-well plates, V bottom (test plates) were prepared with 5 microL of the compound dilution (3×) and then placed onto a PlateTrak 12 robotized station (Perkin Elmer; the robot had one 384-tip pipetting head for starting the assay plus one 96-tip head for dispensing the resin) together with one reservoir for the Enzyme mix (3×) and one for the ATP mix (3×). At the start of the run, the robot aspirated 5 μl of ATP mix, made an air gap inside the tips (3 microL) and aspirated 5 microL of Enzyme mix. The following dispensation into the plates plus 3 cycles of mixing, done by the robot itself, started the kinase reaction. At this point, the correct concentrations were restored for all the reagents. The robot incubated the plates for 60 minutes at r.t., and then stopped the reaction by pipetting 60 microL of dowex resin suspension into the reaction mix. In order to avoid tip clogging, wide bore tips were used to dispense the resin suspension. Three cycles of mixing were done immediately after the addition of the resin. Another mixing cycle was performed after all the plates were stopped, this time using normal tips: the plates were then allowed to rest for about one hour in order to allow resin sedimentation. At this point, 27 microL of the supernatant were transferred into 384-Optiplates (Perkin-Elmer), with 50 microL of Microscipt 40 (Perkin-Elmer); after 5 min of orbital shaking the plates were read on a Perkin-Elmer Top Count radioactivity counter.iii. Data Fitting Data were analyzed by an internally customized version of the SW package Assay Explorer that provided sigmoidal fitting of the ten-dilutions curves for IC50 determination in the secondary assays/hit confirmation routines.
- Biochemical Assay for Inhibitors of KIT Kinase Activity i. Enzyme The assay has been performed using KIT cytoplasmic domain product and purified in house as GST fused protein. The KIT protein (4.5 microM) was pre activated with 300 microM ATP for 1 hour at 28° C. in order to obtain a linear kinetic.ii. KIT kinase Buffer (KB) Kinase buffer was composed of 50 mM HEPES pH 7.9 containing 5 mM MgCl2, 1 mM MnCl2, 10 mM DTT, 3 microM Na3VO4, and 0.2 mg/mL BSAiii. Assay conditions The KIT kinase assay was run with a final pre activated enzyme concentration of 4 nM, in the presence of 4.4 microM ATP (residual ATP from KIT pre activation step is negligible), 3.9 nM 33P-γ-ATP and 2.5 microM of substrate BioDB n*138 (Aminoacidic sequence: KVVEEINGNNYVYIDPTQLPYDHKWEFPRNR SEQ ID NO: 2). The peptide was purchased from American Peptide Company (Sunnyvale, Calif.).Compound Testingi. Compound Dilution For IC50 determination, test compounds were received as a 1 mM solution in 100% DMSO, distributed into 96 well plates: compounds were then plated into the first column of a microtiter plate (A1 to G1), 100 microL/well. An automated station for serial dilutions (Biomek FX, Beckman) was used for producing 1:3 dilutions in 100% DMSO, from line A1 to A10, and for all the compounds in the column. Moreover, 4-5 copies of daughter plates were prepared by reformatting 5 microL of this first set of 100% DMSO dilution plates into 384 deep well-plates: one of these plates with the serial dilutions of test compounds was thawed the day of the experiments, reconstituted at a 3× concentration with water and used in the IC50 determination assays. In a standard experiment, the highest concentration (3×) of all compounds was 30 microM, while the lowest one was 1.5 nM.Each 384 well-plate contained at least one curve of the standard inhibitor staurosporine and reference wells (total enzyme activity vs. no enzymatic activity) for the Z′ and signal to background evaluation,ii. Assay Scheme 384-well plates, V bottom (test plates) were prepared with 5 microL of the compound dilution (3×) and then placed onto a PlateTrak 12 robotized station (Perkin Elmer; the robot had one 384-tip pipetting head for starting the assay plus one 96-tip head for dispensing the resin) together with one reservoir for the Enzyme mix (3×) and one for the ATP mix (3×). At the start of the run, the robot aspirated 5 μl of ATP mix, made an air gap inside the tips (3 microL) and aspirated 5 microL of Enzyme mix. The following dispensation into the plates plus 3 cycles of mixing, done by the robot itself, started the kinase reaction. At this point, the correct concentrations were restored for all the reagents. The robot incubated the plates for 60 minutes at r.t., and then stopped the reaction by pipetting 60 microL of dowex resin suspension into the reaction mix. In order to avoid tip clogging, wide bore tips were used to dispense the resin suspension. Three cycles of mixing were done immediately after the addition of the resin. Another mixing cycle was performed after all the plates were stopped, this time using normal tips: the plates were then allowed to rest for about one hour in order to allow resin sedimentation. At this point, 27 microL of the supernatant were transferred into 384-Optiplates (Perkin-Elmer), with 50 microL of Microscipt 40 (Perkin-Elmer); after 5 min of orbital shaking the plates were read on a Perkin-Elmer Top Count radioactivity counter.iii. Data Fitting Data were analyzed by an internally customized version of the SW package Assay Explorer that provided sigmoidal fitting of the ten-dilutions curves for IC50 determination in the secondary assays/hit confirmation routines.
- IRAK4 Monocyte TNFalpha Cell Based Assay Cryopreserved human monocytes (Stem Cell Technologies) were thawed, diluted in RPMI with GlutaMAX (Gibco 200 mM L-alanyl-L-glutamine) (10 mM HEPES, 1× Pen-Strep, 55 μM -mercaptoethanol, 1 mM Sodium pyruvate) media containing 10% FBS to 0.125×106 cells/ml and recovered at 37° C. for 2 hours. The cell suspension was then plated at a density of 5,000 cells/well onto black 384 well Greiner clear bottom plates. Plates were pre-spotted with test compounds and serially diluted in DMSO where 200 nL/well were delivered using the Echo 550 acoustic liquid dispenser (Labcyte ) for a final DMSO concentration of 0.5%. Plated cells were treated with compound for 1 hour at 37° C. Cells were then stimulated with 50 pg/ml of LPS (Sigma) excluding outside columns of plate used for unstimulated cell control wells. Cells were incubated for an additional 4 hours at 37° C. Cells were then spun out of the media and 5 μl of sample were taken and analyzed for total TNFα content using the TR-FRET Human TNFα detection system (CisBio). This system utilizes two labeled antibodies (cryptate and XL665) that bind to two different epitopes of the TNFα molecule and produce FRET signal proportional to the concentration of TNFα in the sample. Detection antibodies are mixed 50:50 and 5 μL were dispensed into each well. Plates were covered with clear seals and incubated at room temp overnight. The following morning plates were read using an Envision 2103 Multilabeled reader (PerkinElmer) with excitation/emission/FRET emission at 340 nm/615 nm/665 nm, respectively. Fluorescence intensities at 615 nm and 665 nm emission wavelengths were expressed as a ratio (665 nm/615 nm). Percent of control was calculated as follows:% Control=100×(RatioSample−Ratio0% stimulation)/(Ratio100% Stimulation−Ratio0% Stimulation)where unstimulated cells (0% stimulation) were the negative control and stimulated cells (100% stimulation) were used as the positive control.IRAK4 Biochemical Assay Procedure:IRAK4 enzyme (Carna Biosciences, Chuo-ku, Kobe, Japan) activity was measured by detecting phosphorylated peptide substrate formation using an antibody against the phosphorylated peptide substrate. This is a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay, based on the STK1 KinEASE Assay (Cisbio, Bedford, Mass.). The assay was designed as a simple two-step, endpoint assay (a 5 μl enzyme reaction followed by 5 μl stop and detect Solution) performed in ProxiPlate-384 Plus plates (Perkin Elmer, Waltham, Mass.). Staurosporine, a non-selective kinase inhibitor was used as a positive control. Compounds diluted in DMSO were spotted into 384 well plates using a Labcyte Echo 550 Liquid Handling System prior to addition of IRAK4 enzyme and peptide substrate. Reaction solutions were delivered using a Multi-Flo (Bio-Tek Instruments). The enzyme and peptide solution was incubated with compound for 15 minutes at room temp before the reaction was initiated by the addition of ATP. The standard 5 μl reaction mixture contained 500 □M ATP, 2 □M peptide (STK1 Peptide), 0.75 nM of IRAK4 in reaction buffer (50 mM HEPES, pH 7.0, 0.02% NaN3, 0.01% BSA, 0.1 mM Orthovanadate, 5 mM MgCl2, 0.025% NP-40, 1 mM DTT). After 120 min of incubation at room temperature, 5 □l of Stop and Detect Solution (1:100 Cryptate labeled anti-phosphorylated peptide antibody solution and 125 nM Tracer in a 50 mM HEPES pH 7.0 detection buffer containing sufficient EDTA) was added. The plate was then further incubated for 60 minutes at room temperature and read on Envision 2103 Multilabeled reader (PerkinElmer) with excitation/emission/FRET emission at 340 nm/615 nm/665 nm, respectively. Fluorescence intensities at 615 nm and 665 nm emission wavelengths were expressed as a ratio (665 nm/615 nm).
- In-Vitro Inhibition of CDK Kinase Activity Assay This experiment was used for determining the inhibition of the activities of CDK2, CDK7, CDK9 and CDK12 kinases by the compound. The kinase reaction carried out by the present invention was measured in a 384-well plate, the final measured volume was 16 μl, and the reaction temperature was 27° C. The concentrations of the kinases were determined by optimization experiment. The specific experimental process was as follows:1) preparation of kinase solutions:kinase solution (CDK2/Cyclin E1): the kinase was diluted in a assay buffer solution ((20 mM MES pH 6.75, 0.01% Tween 20, 0.05 mg/mL BSA, 2 mM MgCl2) to obtain an enzyme solution with the corresponding 2.4× concentration;kinase solution (CDK7/Cyclin H/MAT1): the kinase was diluted in the assay buffer solution (20 mM MES pH 6.75, 0.01% Tween 20, 0.05 mg/mL BSA, 6 mM MgCl2) to obtain an enzyme solution with the corresponding 2.4× concentration;kinase solution (CDK9/Cyclin T1): the kinase was diluted in the assay buffer solution (20 mM MES pH 6.75, 0.01% Tween 20, 0.05 mg/mL BSA, 10 mM MgCl2) to obtain an enzyme solution with the corresponding 2.4× concentration; andkinase solution (CDK12/Cyclin K): the kinase was diluted in a assay buffer solution (80 mM MES pH 6.5, 0.01% Tween 20, 0.05 mg/mL BSA, 10 mM MgCl2) to obtain an enzyme solution with the corresponding 2.4× concentration.2) Preparation of compound solutions: the compound was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM, the compound was diluted to 10 concentration gradients of 25 nM to 500 μM with DMSO during use, diluted in ultrapure water by 8.3 times respectively to obtain compound solutions with 6× concentration.3) Preparation of a polypeptide substrate and ATP solution: the polypeptide substrate and ATP were diluted in the assay buffer solution to obtain a polypeptide substrate and ATP mixed solution with 2.4× concentration.4) Kinase reaction process:2 ul of the test compound solution, 5 ul of the polypeptide substrate and ATP mixed solution and 5 ul of an enzyme solution were mixed and incubated at 27° C. (CDK2 was incubated for 60 min, CDK7 was incubated for 70 min, CDK9 was incubated for 70 min, and CDK12 was incubated for 280 min), and then 4 ul of EDTA with a concentration of 150 mM was added into each sample to terminate the reaction. The assay buffer solution containing 20 μM of staurosporine was used for replacing the compound solution as 100% inhibition, and DMSO was used for replacing the compound solution as 0% inhibition. Each test had at least two parallel controls. Final concentration of a reagent in CDK2 assay: ATP was 100 μM, the polypeptide substrate (5-FAM-YSPTSPSYSPTSPSYSPT SPSKKKK) was 2 μM, and CDK2/Cyclin E1 was 0.5 nM; final concentration of a reagent in CDK7 assay: ATP was 50 μM, the polypeptide substrate (5-FAM-YSPTSPSYSPTSPSYSPT SPSKKKK) was 2 μM, and CDK7/Cyclin H/MAT1 was 3 nM; final concentration of a reagent in CDK9 assay: ATP was 50 μM, the polypeptide substrate (FITC-Ahx-GSRTPMY-NH2) was 2 μM, and CDK9/Cyclin T1 was 8 nM; and final concentration of a reagent in CDK12 assay: ATP was 30 μM, the polypeptide substrate (FITC-Ahx-GSRTPMY-NH2) was 2 μM, and CDK12/Cyclin K was 50 nM.5) Data calculation and analysis: electrophoretic separation was carried out on a fluorescent substrate and a phosphorylated product on a Caliper EZ Reader II to analyze a reaction mixture. The data was calculated with GraphPad Prism version 9.0, and an IC50 value was obtained by adjusting a nonlinear regression model using a dose reaction curve. The calculation formula was Y=Bottom+(Top-Bottom)/(1+10{circumflex over ( )}((LogIC50−X)*HillSlope)). X represents a log value (log of dose or concentration); Y represents an inhibition rate (% inhibition, increasing as x increases) which was increased along with the increase of X; Top represents maximum response; Bottom represents baseline response; and HillSlope represents a curve slope. Due to the limitation of the detection lower limit of a CDK7
- RIPK1 HTRF Binding Assay A solution was prepared containing 0.2 nM Anti GST-Tb (Cisbio, 61GSTTLB), 90.6 nM probe and 1 nM His-GST-TVMV-hRIPK1(1-324) in FRET Buffer (20 mM HEPES, 10 mM MgCl2, 0.015% Brij-35, 4 mM DTT, 0.05 mg/mL BSA). Using Formulatrix Tempest, the detection antibody/enzyme/probe solution (2 mL) was dispensed into wells of a 1536 plate (Black Low Binding Polystyrene 1536 Plate (Corning, 3724)) containing 10 nL of compounds of interest at appropriate concentration in DMSO. The plate was incubated at rt for 1 h. FRET was measured using the EnVision plate reader (Excitation: 340 nM, Emission: 520 nM/495 nM). Total signal (0% inhibition) was calculated from wells containing 10 nL DMSO only. Blank signal (100% inhibition) calculated from wells containing 10 nL of 15 nM staurosporine and internal controls.Cloning and Baculovirus Expression of RIPK1 ConstructThe coding region of human RIPK1(1-324) flanked by NdeI site at 5′ end and stop codon TGA and XhoI site at 3′ end was codon optimized and gene synthesized at GenScript USA Inc. (Piscataway, N.J.) and subcloned into a modified pFastBacl vector (Invitrogen, Carlsbad, Calif.) with N-terminal His-GST-TVMV tag, to generate His-GST-TVMV-hRIPK1(1-324)-pFB. The fidelity of the synthetic fragment was confirmed by sequencing.Baculovirus was generated for the construct using the Bac-to-Bac baculovirus expression system (Invitrogen) according to the manufacturer's protocol. Briefly, recombinant bacmid was isolated from transformed DH10Bac E. coli competent cells (Invitrogen) and used to transfect Spodoptera frugiperda (Sf9) insect cells (Invitrogen). Baculovirus was harvested 72 hours post transfection and a virus stock was prepared by infecting fresh Sf9 cells at a 1/1000 (v/v) ratio for 66 hours.For large scale protein production, Sf9 cells (Expression System, Davis, Calif.) grown in ESF921 insect medium (Expression System) at 2×106 cells/ml were infected with virus stock at a 1/100 (v/v) ratio for 66 hours. The production was carried out either at a 10 L scale in a 22 L cellbag (GE Healthcare Bioscience, Pittsburgh, Pa.) or at a 20 L scale in a 50 L cellbag using WAVE-Bioreactor System 20/50 (GE Healthcare Bioscience). The infected cells were harvested by centrifugation at 2000 rpm for 20 min at 4° C. in a SORVALL RC12BP centrifuge. The cell pellets was stored at −70° C. before protein was purified.Purification of His-GST-TVMV-hRIPK1(1-324)RIPK1 containing cell paste was resuspended in 50 mM Tris pH 7.5, 150 mM NaCl, 10 mM imidazole, 5% glycerol, 5 mM MgSO4, 1 mM TCEP, 25 U/ml Benzonase, and Complete Protease Inhibitor tablets (1/50 ml, Roche Diagnostics, Indianapolis, Ind.). The cells were lysed by nitrogen cavitation using an unstirred pressure vessel @ 525 PSI (Parr Instrument Company, Moline, Ill.). The suspension was clarified by centrifugation at 136,000×g for 40 min, at 4° C. The lysate was decanted from the pellet and passed through a 5 ml NiNTA Superflow cartridge (Qiagen, Valencia, Calif.) using an AKTA Pure (GE Healthcare). Column was eluted with 10 CV linear gradient into 50 mM Tris 7.5, 150 mM NaCl, 500 mM imidazole, 5% glycerol, 1 mM TCEP. Peak fractions were pooled and loaded directly onto 5 ml GSTrap 4B column (GE Healthcare). Column was washed with 50 mM Tris 7.0, 150 mM NaCl, 5% glycerol, 1 mM DTT and eluted in 10 CV linear gradient into 50 mM Tris 8.0, 150 mM NaCl, 20 mM reduced glutathione, 5% glycerol, 1 mM DTT. Fractions identified by SDS-PAGE as containing RIPK1 were pooled and concentrated using 30 kDa MWCO spin concentrators (Amicon Ultra-15, Millipore, Billerica, Mass.) and loaded onto a HiLoad 26/600 Superdex 200 column (GE Healthcare) equilibrated in 25 mM Tris 7.5, 150 mM NaCl, 2 mM TCEP, 5% glycerol. The RIPK1 protein eluted as a dimer off the SEC column.The yield was 8 mg/L with a purity >95% as determined by Coomassie staind SDS-PAGE gel analysis.
- RIPK1 HTRF Binding Assay A solution was prepared containing 0.2 nM Anti GST-Tb (Cisbio, 61GSTTLB), 90.6 nM probe and 1 nM His-GST-TVMV-hRIPKl (1-324) in FRET Buffer (20 mM HEPES, 10 mM MgC12, 0.015% Brij-35, 4mM DTT, 0.05 mg/mL BSA). Using Formulatrix Tempest, the detection antibody/enzyme/probe solution (2 mL) was dispensed into wells of a 1536 plate (Black Low Binding Polystyrene 1536 Plate (Corning, 3724)) containing 10 nL of compounds of interest at appropriate concentration in DMSO. The plate was incubated at rt for 1 h. FRET was measured using the EnVision plate reader (Excitation: 340 nM, Emission: 520 nM/495 nM). Total signal (0% inhibition) was calculated from wells containing 10 nL DMSO only. Blank signal (100% inhibition) calculated from wells containing 10 nL of 15 nM staurosporine and internal controls.Cloning and Baculovirus Expression of RIPK1 ConstructThe coding region of human RIPKl(l-324) flanked by Ndel site at 5' end and stop codon TGA and Xhol site at 3' end was codon optimized and gene synthesized at GenScript USA Inc. (Piscataway, NJ) and subcloned into a modified pFastBacl vector (Invitrogen, Carlsbad, CA) with N-terminal His-GST-TVMV tag, to generate His-GST-TVMV-hRIPKl (1-324)-pFB. The fidelity of the synthetic fragment was confirmed by sequencing.Baculovirus was generated for the construct using the Bac-to-Bac baculovirus expression system (Invitrogen) according to the manufacturer's protocol. Briefly, recombinant bacmid was isolated from transformed DHIOBac E.coli competent cells (Invitrogen) and used to transfect Spodoptera frugiperda (Sf9) insect cells (Invitrogen). Baculovirus was harvested 72 hours post transfection and a virus stock was prepared by infecting fresh Sf9 cells at a 1/1000 (v/v) ratio for 66 hours.For large scale protein production, Sf9 cells (Expression System, Davis, CA) grown in ESF921 insect medium (Expression System) at 2 x 106 cells/ml were infected with virus stock at a 1/100(v/v) ratio for 66 hours. The production was carried out either at a 10 L scale in a 22 L cellbag (GE Healthcare Bioscience, Pittsburgh, PA) or at a 20 L scale in a 50 L cellbag using WAVE-Bioreactor System 20/50 (GE Healthcare Bioscience). The infected cells were harvested by centrifugation at 2000 rpm for 20 min at 4 °C in a SORVALL RC12BP centrifuge. The cell pellets was stored at -70 °C before protein was purified.Purification of His-GST-TVMV-hRIPKl(l-324)RIPK1 containing cell paste was resuspended in 50 mM Tris pH 7.5, 150 mM NaCl, 10 mM imidazole, 5% glycerol, 5 mM MgSCE, 1 mM TCEP, 25 U/ml Benzonase, and Complete Protease Inhibitor tablets (1/50 ml, Roche Diagnostics, Indianapolis, IN). The cells were lysed by nitrogen cavitation using an unstirred pressure vessel @ 525 PSI (Parr Instrument Company, Moline, IL). The suspension was clarified by centrifugation at 136,000 x g for 40 min, at 4 °C. The lysate was decanted from the pellet and passed through a 5 ml NiNTA Superflow cartridge (Qiagen, Valencia, CA) using an AKTA Pure (GE Healthcare). Column was eluted with 10 CV linear gradient into 50 mM Tris 7.5, 150 mM NaCl, 500 mM imidazole, 5% glycerol, 1 mM TCEP. Peak fractions were pooled and loaded directly onto 5 ml GSTrap 4B column (GE Healthcare). Column was washed with 50 mM Tris 7.0, 150 mM NaCl, 5% glycerol, 1 mM DTT and eluted in 10 CV linear gradient into 50 mM Tris 8.0, 150 mM NaCl, 20 mM reduced glutathione, 5% glycerol, 1 mM DTT. Fractions identified by SDS-PAGE as containing RIPK1 were pooled and concentrated using 30 kDa MWCO spin concentrators (Amicon Ultra-15, Millipore, Billerica, MA) and loaded onto a HiLoad 26/600 Superdex 200 column (GE Healthcare) equilibrated in 25 mM Tris 7.5, 150 mM NaCl, 2 mM TCEP, 5% glycerol. The RIPK1 protein eluted as a dimer off the SEC column.The yield was ~8 mg/L with a purity >95% as determined by Coomassie stain SDS-PAGE gel analysis.