Target (22)
Compound (13)
Article Title (98)
Assay (448)
Narjes, F; Edfeldt, F; Petersen, J; Öster, L; Hamblet, C; Bird, J; Bold, P; Rae, R; Bäck, E; Stomilovic, S; Zlatoidsky, P; Svensson, T; Hidestål, L; Kunalingam, L; Shamovsky, I; De Maria, L; Gordon, E; Lewis, RJ; Watcham, S; van Rietschoten, K; Mudd, GE; Harrison, H; Chen, L; Skynner, MJ Discovery and Characterization of a Bicyclic Peptide (Bicycle) Binder to Thymic Stromal Lymphopoietin. J Med Chem 67: 2220 -2235 Kim, D; Sheen, YY; Jin, C; Park, C; Domalapally, S; Kota, SR; Maddeboina, K; Vura Bala, S Methods of treating fibrosis, cancer and vascular injuries US Patent USRE47141 (2018) Nakamura, K; Taguchi, E; Miura, T; Yamamoto, A; Takahashi, K; Bichat, F; Guilbaud, N; Hasegawa, K; Kubo, K; Fujiwara, Y; Suzuki, R; Kubo, K; Shibuya, M; Isae, T KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res 66: 9134 -42 Chang, CY; Chuang, HY; Lee, HY; Yeh, TK; Kuo, CC; Chang, CY; Chang, JY; Liou, JP Antimitotic and vascular disrupting agents: 2-hydroxy-3,4,5-trimethoxybenzophenones. Eur J Med Chem 77: 306 -14 (2014) Lin, WH; Wu, SY; Yeh, TK; Chen, CT; Song, JS; Shiao, HY; Kuo, CC; Hsu, T; Lu, CT; Wang, PC; Wu, TS; Peng, YH; Lin, HY; Chen, CP; Weng, YL; Kung, FC; Wu, MH; Su, YC; Huang, KW; Chou, LH; Hsueh, CC; Yen, KJ; Kuo, PC; Huang, CL; Chen, LT; Shih, C; Tsai, HJ; Jiaang, WT Identification of a Multitargeted Tyrosine Kinase Inhibitor for the Treatment of Gastrointestinal Stromal Tumors and Acute Myeloid Leukemia. J Med Chem 62: 11135 -11150 (2019) Musumeci, F; Radi, M; Brullo, C; Schenone, S Vascular endothelial growth factor (VEGF) receptors: drugs and new inhibitors. J Med Chem 55: 10797 -822 (2012) Zhao, H; Caflisch, A Current kinase inhibitors cover a tiny fraction of fragment space. Bioorg Med Chem Lett 25: 2372 -6 (2015) Ingkaninan, K; von Frijtag Drabbe Künzel, JK; IJzerman, AP; Verpoorte, R Interference of linoleic acid fraction in some receptor binding assays. J Nat Prod 62: 912 -4 (1999) Blum, A; Dorsch, D; Linde, N; Brandstetter, S; Buchstaller, HP; Busch, M; Glaser, N; Grädler, U; Ruff, A; Petersson, C; Schieferstein, H; Sherbetjian, E; Esdar, C Identification of M4205─A Highly Selective Inhibitor of KIT Mutations for Treatment of Unresectable Metastatic or Recurrent Gastrointestinal Stromal Tumors. J Med Chem 66: 2386 -2395 (2023) Cherney, RJ; Wang, Z Heterocyclic compounds as S1P1 agonists for the treatment of autoimmune and vascular diseases US Patent US8629282 (2014) Driowya, M; Leclercq, J; Verones, V; Barczyk, A; Lecoeur, M; Renault, N; Flouquet, N; Ghinet, A; Berthelot, P; Lebegue, N Synthesis of triazoloquinazolinone based compounds as tubulin polymerization inhibitors and vascular disrupting agents. Eur J Med Chem 115: 393 -405 (2016) Wang, Y; Gratzke, C; Tamalunas, A; Wiemer, N; Ciotkowska, A; Rutz, B; Waidelich, R; Strittmatter, F; Liu, C; Stief, CG; Hennenberg, M P21-Activated Kinase Inhibitors FRAX486 and IPA3: Inhibition of Prostate Stromal Cell Growth and Effects on Smooth Muscle Contraction in the Human Prostate. PLoS ONE 11: Moloney, GP; Robertson, AD; Martin, GR; MacLennan, S; Mathews, N; Dodsworth, S; Sang, PY; Knight, C; Glen, R A novel series of 2,5-substituted tryptamine derivatives as vascular 5HT1B/1D receptor antagonists. J Med Chem 40: 2347 -62 (1997) Bockman, CS; Jeffries, WB; Abel, PW Binding and functional characterization of alpha-2 adrenergic receptor subtypes on pig vascular endothelium. J Pharmacol Exp Ther 267: 1126 -33 (1993) Marivet, MC; Bourguignon, JJ; Lugnier, C; Mann, A; Stoclet, JC; Wermuth, CG Inhibition of cyclic adenosine-3',5'-monophosphate phosphodiesterase from vascular smooth muscle by rolipram analogues. J Med Chem 32: 1450 -7 (1989) Bligt-Lindén, E; Pihlavisto, M; Szatmári, I; Otwinowski, Z; Smith, DJ; Lázár, L; Fülöp, F; Salminen, TA Novel pyridazinone inhibitors for vascular adhesion protein-1 (VAP-1): old target-new inhibition mode. J Med Chem 56: 9837 -48 (2013) Nagasawa, HT; Kawle, SP; Elberling, JA; DeMaster, EG; Fukuto, JM Prodrugs of nitroxyl as potential aldehyde dehydrogenase inhibitors vis-a-vis vascular smooth muscle relaxants. J Med Chem 38: 1865 -71 (1995) Kaur, N; Fang, YC; Lee, HY; Singh, A; Nepali, K; Lin, MH; Yeh, TK; Lai, MJ; Chan, L; Tu, YK; Banerjee, S; Hu, CJ; Liou, JP Protective effects of 10,11-dihydro-5H-dibenzo[b,f]azepine hydroxamates on vascular cognitive impairment. Eur J Med Chem 187: (2020) Goncalves, V; Gautier, B; Coric, P; Bouaziz, S; Lenoir, C; Garbay, C; Vidal, M; Inguimbert, N Rational design, structure, and biological evaluation of cyclic peptides mimicking the vascular endothelial growth factor. J Med Chem 50: 5135 -46 (2007) Jarvis, A; Allerston, CK; Jia, H; Herzog, B; Garza-Garcia, A; Winfield, N; Ellard, K; Aqil, R; Lynch, R; Chapman, C; Hartzoulakis, B; Nally, J; Stewart, M; Cheng, L; Menon, M; Tickner, M; Djordjevic, S; Driscoll, PC; Zachary, I; Selwood, DL Small molecule inhibitors of the neuropilin-1 vascular endothelial growth factor A (VEGF-A) interaction. J Med Chem 53: 2215 -26 (2010) Yamaki, S; Yamada, H; Nagashima, A; Kondo, M; Shimada, Y; Kadono, K; Yoshihara, K Synthesis and structure activity relationships of carbamimidoylcarbamate derivatives as novel vascular adhesion protein-1 inhibitors. Bioorg Med Chem 25: 6024 -6038 (2017) Cuong, NM; Khanh, PN; Huyen, PT; Duc, HV; Huong, TT; Ha, VT; Durante, M; Sgaragli, G; Fusi, F Vascular L-type Ca²¿ channel blocking activity of sulfur-containing indole alkaloids from Glycosmis petelotii. J Nat Prod 77: 1586 -93 (2014) Zhu, H; Tan, Y; He, C; Liu, Y; Duan, Y; Zhu, W; Zheng, T; Li, D; Xu, J; Yang, DH; Chen, ZS; Xu, S Discovery of a Novel Vascular Disrupting Agent Inhibiting Tubulin Polymerization and HDACs with Potent Antitumor Effects. J Med Chem 65: 11187 -11213 (2022) Du, J; Lei, B; Qin, J; Liu, H; Yao, X Molecular modeling studies of vascular endothelial growth factor receptor tyrosine kinase inhibitors using QSAR and docking. J Mol Graph Model 27: 642 -54 (2008) Wurster, JA; Malone, TC; Hull, III, CE; Rao, S; Yang, R; Yee, R Piperidylpyrimidine derivatives as modulators of protein kinase inhibitors and of vascular endothelial growth factor receptor 2 US Patent US9296747 (2016) Novoa, A; Pellegrini-Moïse, N; Bechet, D; Barberi-Heyob, M; Chapleur, Y Sugar-based peptidomimetics as potential inhibitors of the vascular endothelium growth factor binding to neuropilin-1. Bioorg Med Chem 18: 3285 -98 (2010) Yamaki, S; Suzuki, D; Fujiyasu, J; Neya, M; Nagashima, A; Kondo, M; Akabane, T; Kadono, K; Moritomo, A; Yoshihara, K Synthesis and structure activity relationships of glycine amide derivatives as novel Vascular Adhesion Protein-1 inhibitors. Bioorg Med Chem 25: 187 -201 (2017) Gautier, B; Goncalves, V; Diana, D; Di Stasi, R; Teillet, F; Lenoir, C; Huguenot, F; Garbay, C; Fattorusso, R; D'Andrea, LD; Vidal, M; Inguimbert, N Biochemical and structural analysis of the binding determinants of a vascular endothelial growth factor receptor peptidic antagonist. J Med Chem 53: 4428 -40 (2010) Ye, X; Gaucher, JF; Hu, H; Wang, L; Broussy, S Dimer Peptide Ligands of Vascular Endothelial Growth Factor: Optimizing Linker Length for High Affinity and Antiangiogenic Activity. J Med Chem 66: 9753 -9765 (2023) Liang, J; Huang, YY; Zhou, Q; Gao, Y; Li, Z; Wu, D; Yu, S; Guo, L; Chen, Z; Huang, L; Liang, SH; He, X; Wu, R; Luo, HB Discovery and Optimization of α-Mangostin Derivatives as Novel PDE4 Inhibitors for the Treatment of Vascular Dementia. J Med Chem 63: 3370 -3380 (2020) Murza, A; Sainsily, X; Coquerel, D; Côté, J; Marx, P; Besserer-Offroy, É; Longpré, JM; Lainé, J; Reversade, B; Salvail, D; Leduc, R; Dumaine, R; Lesur, O; Auger-Messier, M; Sarret, P; Marsault, É Discovery and Structure-Activity Relationship of a Bioactive Fragment of ELABELA that Modulates Vascular and Cardiac Functions. J Med Chem 59: 2962 -72 (2016) Harmange, JC; Weiss, MM; Germain, J; Polverino, AJ; Borg, G; Bready, J; Chen, D; Choquette, D; Coxon, A; DeMelfi, T; DiPietro, L; Doerr, N; Estrada, J; Flynn, J; Graceffa, RF; Harriman, SP; Kaufman, S; La, DS; Long, A; Martin, MW; Neervannan, S; Patel, VF; Potashman, M; Regal, K; Roveto, PM; Schrag, ML; Starnes, C; Tasker, A; Teffera, Y; Wang, L; White, RD; Whittington, DA; Zanon, R Naphthamides as novel and potent vascular endothelial growth factor receptor tyrosine kinase inhibitors: design, synthesis, and evaluation. J Med Chem 51: 1649 -67 (2008) Falcon, BL; Barr, S; Gokhale, PC; Chou, J; Fogarty, J; Depeille, P; Miglarese, M; Epstein, DM; McDonald, DM Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res 71: 1573 -83 Ding, X; Stasi, LP; Dai, X; Long, K; Peng, C; Zhao, B; Wang, H; Sun, C; Hu, H; Wan, Z; Jandu, KS; Philps, OJ; Chen, Y; Wang, L; Liu, Q; Edge, C; Li, Y; Dong, K; Guan, X; Tattersall, FD; Reith, AD; Ren, F 5-Substituted-N-pyridazinylbenzamides as potent and selective LRRK2 inhibitors: Improved brain unbound fraction enables efficacy. Bioorg Med Chem Lett 29: 212 -215 (2019) Lu, Y; Mao, F; Li, X; Zheng, X; Wang, M; Xu, Q; Zhu, J; Li, J Discovery of Potent, Selective Stem Cell Factor Receptor/Platelet Derived Growth Factor Receptor Alpha (c-KIT/PDGFRa) Dual Inhibitor for the Treatment of Imatinib-Resistant Gastrointestinal Stromal Tumors (GISTs). J Med Chem 60: 5099 -5119 (2017) Kiselyov, AS; Piatnitski, E; Milligan, D; Ouyang, X 1H-1,2,4-triazol-3-yl-anilines: novel potent inhibitors of vascular endothelial growth factor receptors 1 and 2. Chem Biol Drug Des 69: 331 -7 (2007) Starzec, A; Miteva, MA; Ladam, P; Villoutreix, BO; Perret, GY Discovery of novel inhibitors of vascular endothelial growth factor-A-Neuropilin-1 interaction by structure-based virtual screening. Bioorg Med Chem 22: 4042 -8 (2014) Abdel-Magid, AF Plasma Kallikrein Inhibitors for the Treatment of Retinal Vascular Permeability Associated with Diabetic Retinopathy and Diabetic Macular Edema. ACS Med Chem Lett 8: 776 -777 (2017) Weiss, MM; Harmange, JC; Polverino, AJ; Bauer, D; Berry, L; Berry, V; Borg, G; Bready, J; Chen, D; Choquette, D; Coxon, A; DeMelfi, T; Doerr, N; Estrada, J; Flynn, J; Graceffa, RF; Harriman, SP; Kaufman, S; La, DS; Long, A; Neervannan, S; Patel, VF; Potashman, M; Regal, K; Roveto, PM; Schrag, ML; Starnes, C; Tasker, A; Teffera, Y; Whittington, DA; Zanon, R Evaluation of a series of naphthamides as potent, orally active vascular endothelial growth factor receptor-2 tyrosine kinase inhibitors. J Med Chem 51: 1668 -80 (2008) Wang, L; Zhou, L; Reille-Seroussi, M; Gagey-Eilstein, N; Broussy, S; Zhang, T; Ji, L; Vidal, M; Liu, WQ Identification of Peptidic Antagonists of Vascular Endothelial Growth Factor Receptor 1 by Scanning the Binding Epitopes of Its Ligands. J Med Chem 60: 6598 -6606 (2017) Yu, H; Wang, Z; Zhang, L; Zhang, J; Huang, Q The discovery of novel vascular endothelial growth factor receptor tyrosine kinases inhibitors: pharmacophore modeling, virtual screening and docking studies. Chem Biol Drug Des 69: 204 -11 (2007) Falck, JR; Kodela, R; Manne, R; Atcha, KR; Puli, N; Dubasi, N; Manthati, VL; Capdevila, JH; Yi, XY; Goldman, DH; Morisseau, C; Hammock, BD; Campbell, WB 14,15-Epoxyeicosa-5,8,11-trienoic acid (14,15-EET) surrogates containing epoxide bioisosteres: influence upon vascular relaxation and soluble epoxide hydrolase inhibition. J Med Chem 52: 5069 -75 (2010) Potashman, MH; Bready, J; Coxon, A; Demelfi, TM; Dipietro, L; Doerr, N; Elbaum, D; Estrada, J; Gallant, P; Germain, J; Gu, Y; Harmange, JC; Kaufman, SA; Kendall, R; Kim, JL; Kumar, GN; Long, AM; Neervannan, S; Patel, VF; Polverino, A; Rose, P; Plas, SV; Whittington, D; Zanon, R; Zhao, H Design, Synthesis, and Evaluation of Orally Active Benzimidazoles and Benzoxazoles as Vascular Endothelial Growth Factor-2 Receptor Tyrosine Kinase Inhibitors. J Med Chem 50: 4351 -4373 (2007) Wu, Y; Zhou, Q; Zhang, T; Li, Z; Chen, YP; Zhang, P; Yu, YF; Geng, H; Tian, YJ; Zhang, C; Wang, Y; Chen, JW; Chen, Y; Luo, HB Discovery of Potent, Selective, and Orally Bioavailable Inhibitors against Phosphodiesterase-9, a Novel Target for the Treatment of Vascular Dementia. J Med Chem 62: 4218 -4224 (2019) Meng, CQ; Somers, PK; Hoong, LK; Zheng, XS; Ye, Z; Worsencroft, KJ; Simpson, JE; Hotema, MR; Weingarten, MD; MacDOnald, ML; Hill, RR; Marino, EM; Suen, KL; Luchoomun, J; Kunsch, C; Landers, LK; Stefanopoulos, D; Howard, RB; Sundell, CL; Saxena, U; Wasserman, MA; Sikorski, JA Discovery of novel phenolic antioxidants as inhibitors of vascular cell adhesion molecule-1 expression for use in chronic inflammatory diseases. J Med Chem 47: 6420 -32 (2004) Nakao, Y; Narazaki, G; Hoshino, T; Maeda, S; Yoshida, M; Maejima, H; Yamashita, JK Evaluation of antiangiogenic activity of azumamides by the in vitro vascular organization model using mouse induced pluripotent stem (iPS) cells. Bioorg Med Chem Lett 18: 2982 -4 (2008) Peng, FW; Wu, TT; Ren, ZW; Xue, JY; Shi, L Hybrids from 4-anilinoquinazoline and hydroxamic acid as dual inhibitors of vascular endothelial growth factor receptor-2 and histone deacetylase. Bioorg Med Chem Lett 25: 5137 -41 (2015) Salerno, S; Marini, AM; Fornaciari, G; Simorini, F; La Motta, C; Taliani, S; Sartini, S; Da Settimo, F; García-Argáez, AN; Gia, O; Cosconati, S; Novellino, E; D'Ocon, P; Fioravanti, A; Orlandi, P; Bocci, G; Dalla Via, L Investigation of new 2-aryl substituted Benzothiopyrano[4,3-d]pyrimidines as kinase inhibitors targeting vascular endothelial growth factor receptor 2. Eur J Med Chem 103: 29 -43 (2015) Kuo, GH; Wang, A; Emanuel, S; Deangelis, A; Zhang, R; Connolly, PJ; Murray, WV; Gruninger, RH; Sechler, J; Fuentes-Pesquera, A; Johnson, D; Middleton, SA; Jolliffe, L; Chen, X Synthesis and discovery of pyrazine-pyridine biheteroaryl as a novel series of potent vascular endothelial growth factor receptor-2 inhibitors. J Med Chem 48: 1886 -900 (2005) Yamaki, S; Koga, Y; Nagashima, A; Kondo, M; Shimada, Y; Kadono, K; Moritomo, A; Yoshihara, K Synthesis and pharmacological evaluation of glycine amide derivatives as novel vascular adhesion protein-1 inhibitors without CYP3A4 and CYP2C19 inhibition. Bioorg Med Chem 25: 4110 -4122 (2017) Tymecka, D; Puszko, AK; Lipiński, PFJ; Fedorczyk, B; Wilenska, B; Sura, K; Perret, GY; Misicka, A Branched pentapeptides as potent inhibitors of the vascular endothelial growth factor 165 binding to Neuropilin-1: Design, synthesis and biological activity. Eur J Med Chem 158: 453 -462 (2018) Shan, Y; Si, R; Wang, J; Zhang, Q; Li, J; Ma, Y; Zhang, J Discovery of novel anti-angiogenesis agents. Part 11: Development of PROTACs based on active molecules with potency of promoting vascular normalization. Eur J Med Chem 205: (2020) Nurminen, EM; Pihlavisto, M; Lázár, L; Pentikäinen, U; Fülöp, F; Pentikäinen, OT Novel hydrazine molecules as tools to understand the flexibility of vascular adhesion protein-1 ligand-binding site: toward more selective inhibitors. J Med Chem 54: 2143 -54 (2011) Garofalo, A; Farce, A; Ravez, S; Lemoine, A; Six, P; Chavatte, P; Goossens, L; Depreux, P Synthesis and structure-activity relationships of (aryloxy)quinazoline ureas as novel, potent, and selective vascular endothelial growth factor receptor-2 inhibitors. J Med Chem 55: 1189 -204 (2012) Kuo, GH; Prouty, C; Wang, A; Emanuel, S; Deangelis, A; Zhang, Y; Song, F; Beall, L; Connolly, PJ; Karnachi, P; Chen, X; Gruninger, RH; Sechler, J; Fuentes-Pesquera, A; Middleton, SA; Jolliffe, L; Murray, WV Synthesis and structure-activity relationships of pyrazine-pyridine biheteroaryls as novel, potent, and selective vascular endothelial growth factor receptor-2 inhibitors. J Med Chem 48: 4892 -909 (2005) Nurminen, EM; Pihlavisto, M; Lázár, L; Szakonyi, Z; Pentikäinen, U; Fülöp, F; Pentikäinen, OT Synthesis, in vitro activity, and three-dimensional quantitative structure-activity relationship of novel hydrazine inhibitors of human vascular adhesion protein-1. J Med Chem 53: 6301 -15 (2010) Wissner, A; Floyd, MB; Johnson, BD; Fraser, H; Ingalls, C; Nittoli, T; Dushin, RG; Discafani, C; Nilakantan, R; Marini, J; Ravi, M; Cheung, K; Tan, X; Musto, S; Annable, T; Siegel, MM; Loganzo, F 2-(Quinazolin-4-ylamino)-[1,4]benzoquinones as covalent-binding, irreversible inhibitors of the kinase domain of vascular endothelial growth factor receptor-2. J Med Chem 48: 7560 -81 (2005) Cho, EH; Shin, HJ; Ki, MH; Kwon, HS; Lee, JW; Joo, JH; Lee, KK; Kim, JM; Park, YB; Kang, SH; Cho, HM; Kim, HT; Ahn, SK; Hong, SP; Kim, SH Pyridine derivative inhibiting RAF kinase and vascular endothelial growth factor receptor, method for preparing same, pharmaceutical composition containing same, and use thereof US Patent US10844062 (2020) Inghardt, T; Antonsson, T; Ericsson, C; Hovdal, D; Johannesson, P; Johansson, C; Jurva, U; Kajanus, J; Kull, B; Michaëlsson, E; Pettersen, A; Sjögren, T; Sörensen, H; Westerlund, K; Lindstedt, EL Discovery of AZD4831, a Mechanism-Based Irreversible Inhibitor of Myeloperoxidase, As a Potential Treatment for Heart Failure with Preserved Ejection Fraction. J Med Chem 65: 11485 -11496 (2022) Wang, Q; Liu, F; Wang, B; Zou, F; Chen, C; Liu, X; Wang, A; Qi, S; Wang, W; Qi, Z; Zhao, Z; Hu, Z; Wang, W; Wang, L; Zhang, S; Wang, Y; Liu, J; Liu, Q Discovery of N-(3-((1-Isonicotinoylpiperidin-4-yl)oxy)-4-methylphenyl)-3-(trifluoromethyl)benzamide (CHMFL-KIT-110) as a Selective, Potent, and Orally Available Type II c-KIT Kinase Inhibitor for Gastrointestinal Stromal Tumors (GISTs). J Med Chem 59: 3964 -79 (2016) Mátyus, P; Magyar, K; Pihlavisto, M; Gyires, K; Haider, N; Wang, Y; Woda, P; Dunkel, P; Tóth-Sarudy, P; Túrós, G Compounds for inhibiting semicarbazide-sensitive amine oxidase (SSAO)/vascular adhesion protein-1 (VAP-1) and uses thereof for treatment and prevention of diseases US Patent US8536210 (2013) Grabowska, K; Puszko, AK; Lipinski, PFJ; Laskowska, AK; Wilenska, B; Witkowska, E; Misicka, A Design, synthesis and in vitro biological evaluation of a small cyclic peptide as inhibitor of vascular endothelial growth factor binding to neuropilin-1. Bioorg Med Chem Lett 26: 2843 -2846 (2016) Devkota, L; Lin, CM; Strecker, TE; Wang, Y; Tidmore, JK; Chen, Z; Guddneppanavar, R; Jelinek, CJ; Lopez, R; Liu, L; Hamel, E; Mason, RP; Chaplin, DJ; Trawick, ML; Pinney, KG Design, synthesis, and biological evaluation of water-soluble amino acid prodrug conjugates derived from combretastatin, dihydronaphthalene, and benzosuberene-based parent vascular disrupting agents. Bioorg Med Chem 24: 938 -56 (2016) Inoue, T; Morita, M; Tojo, T; Nagashima, A; Moritomo, A; Miyake, H Novel 1H-imidazol-2-amine derivatives as potent and orally active vascular adhesion protein-1 (VAP-1) inhibitors for diabetic macular edema treatment. Bioorg Med Chem 21: 3873 -81 (2013) Goncalves, V; Gautier, B; Regazzetti, A; Coric, P; Bouaziz, S; Garbay, C; Vidal, M; Inguimbert, N On-resin cyclization of peptide ligands of the Vascular Endothelial Growth Factor Receptor 1 by copper(I)-catalyzed 1,3-dipolar azide-alkyne cycloaddition. Bioorg Med Chem Lett 17: 5590 -4 (2007) Inoue, T; Morita, M; Tojo, T; Yoshihara, K; Nagashima, A; Moritomo, A; Ohkubo, M; Miyake, H Synthesis and SAR study of new thiazole derivatives as vascular adhesion protein-1 (VAP-1) inhibitors for the treatment of diabetic macular edema. Bioorg Med Chem 21: 1219 -33 (2013) Dolle, RE; Dunn, JA; Bobko, M; Singh, B; Kuster, JE; Baizman, E; Harris, AL; Sawutz, DG; Miller, D; Wang, S 5,7-Dimethoxy-3-(4-pyridinyl)quinoline is a potent and selective inhibitor of human vascular beta-type platelet-derived growth factor receptor tyrosine kinase. J Med Chem 37: 2627 -9 (1994) Tian, C; Wang, M; Shi, X; Chen, X; Wang, X; Zhang, Z; Liu, J Discovery of (2-(pyrrolidin-1-yl)thieno[3,2-d]pyrimidin-4-yl)(3,4,5-trimethoxyphenyl)methanone as a novel potent tubulin depolymerizing and vascular disrupting agent. Eur J Med Chem 238: (2022) Li, Y; Yang, G; Zhang, J; Tang, P; Yang, C; Wang, G; Chen, J; Liu, J; Zhang, L; Ouyang, L Discovery, Synthesis, and Evaluation of Highly Selective Vascular Endothelial Growth Factor Receptor 3 (VEGFR3) Inhibitor for the Potential Treatment of Metastatic Triple-Negative Breast Cancer. J Med Chem 64: 12022 -12048 (2021) Inoue, T; Morita, M; Tojo, T; Nagashima, A; Moritomo, A; Imai, K; Miyake, H Synthesis and SAR study of new thiazole derivatives as vascular adhesion protein-1 (VAP-1) inhibitors for the treatment of diabetic macular edema: part 2. Bioorg Med Chem 21: 2478 -94 (2013) Liu, X; Wilson, MW; Liu, K; Lee, P; Yeomans, L; Hagen, SE; Lin, CM; Wen, B; Sun, D; White, AD; Showalter, HD; Antonetti, DA Synthesis and structure-activity relationships of thieno[2,3-d]pyrimidines as atypical protein kinase C inhibitors to control retinal vascular permeability and cytokine-induced edema. Bioorg Med Chem 28: (2020) Pöstges, T; Galster, F; Kampschulze, J; Hanekamp, W; Lehr, M ω-(5-Phenyl-2H-tetrazol-2-yl)alkyl-substituted glycine amides and related compounds as inhibitors of the amine oxidase vascular adhesion protein-1 (VAP-1). Bioorg Med Chem 98: Kettle, JG; Anjum, R; Barry, E; Bhavsar, D; Brown, C; Boyd, S; Campbell, A; Goldberg, K; Grondine, M; Guichard, S; Hardy, CJ; Hunt, T; Jones, RDO; Li, X; Moleva, O; Ogg, D; Overman, RC; Packer, MJ; Pearson, S; Schimpl, M; Shao, W; Smith, A; Smith, JM; Stead, D; Stokes, S; Tucker, M; Ye, Y Discovery of N-(4-{[5-Fluoro-7-(2-methoxyethoxy)quinazolin-4-yl]amino}phenyl)-2-[4-(propan-2-yl)-1 H-1,2,3-triazol-1-yl]acetamide (AZD3229), a Potent Pan-KIT Mutant Inhibitor for the Treatment of Gastrointestinal Stromal Tumors. J Med Chem 61: 8797 -8810 (2018) Li, Y; Liu, Y; Zhang, D; Chen, J; Yang, G; Tang, P; Yang, C; Liu, J; Zhang, J; Ouyang, L Discovery, Synthesis, and Evaluation of Novel Dual Inhibitors of a Vascular Endothelial Growth Factor Receptor and Poly(ADP-Ribose) Polymerase for BRCA Wild-Type Breast Cancer Therapy. J Med Chem 66: 12069 -12100 (2023) Perspicace, E; Jouan-Hureaux, V; Ragno, R; Ballante, F; Sartini, S; La Motta, C; Da Settimo, F; Chen, B; Kirsch, G; Schneider, S; Faivre, B; Hesse, S Design, synthesis and biological evaluation of new classes of thieno[3,2-d]pyrimidinone and thieno[1,2,3]triazine as inhibitor of vascular endothelial growth factor receptor-2 (VEGFR-2). Eur J Med Chem 63: 765 -81 (2013) Peifer, C; Selig, R; Kinkel, K; Ott, D; Totzke, F; Schächtele, C; Heidenreich, R; Röcken, M; Schollmeyer, D; Laufer, S Design, synthesis, and biological evaluation of novel 3-aryl-4-(1H-indole-3yl)-1,5-dihydro-2H-pyrrole-2-ones as vascular endothelial growth factor receptor (VEGF-R) inhibitors. J Med Chem 51: 3814 -24 (2008) Wang, DP; Liu, KL; Li, XY; Lu, GQ; Xue, WH; Qian, XH; Mohamed O, K; Meng, FH Design, synthesis, and in vitro and in vivo anti-angiogenesis study of a novel vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitor based on 1,2,3-triazole scaffold. Eur J Med Chem 211: (2021) Harris, PA; Boloor, A; Cheung, M; Kumar, R; Crosby, RM; Davis-Ward, RG; Epperly, AH; Hinkle, KW; Hunter, RN; Johnson, JH; Knick, VB; Laudeman, CP; Luttrell, DK; Mook, RA; Nolte, RT; Rudolph, SK; Szewczyk, JR; Truesdale, AT; Veal, JM; Wang, L; Stafford, JA Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem 51: 4632 -40 (2008) Bilodeau, MT; Balitza, AE; Koester, TJ; Manley, PJ; Rodman, LD; Buser-Doepner, C; Coll, KE; Fernandes, C; Gibbs, JB; Heimbrook, DC; Huckle, WR; Kohl, N; Lynch, JJ; Mao, X; McFall, RC; McLoughlin, D; Miller-Stein, CM; Rickert, KW; Sepp-Lorenzino, L; Shipman, JM; Subramanian, R; Thomas, KA; Wong, BK; Yu, S; Hartman, GD Potent N-(1,3-thiazol-2-yl)pyridin-2-amine vascular endothelial growth factor receptor tyrosine kinase inhibitors with excellent pharmacokinetics and low affinity for the hERG ion channel. J Med Chem 47: 6363 -72 (2004) Grabowska, K; Puszko, AK; Lipinski, PF; Laskowska, AK; Wilenska, B; Witkowska, E; Perret, GY; Misicka, A Structure-activity relationship study of a small cyclic peptide H-c[Lys-Pro-Glu]-Arg-OH: a potent inhibitor of Vascular Endothelial Growth Factor interaction with Neuropilin-1. Bioorg Med Chem 25: 597 -602 (2017) Gangjee, A; Zaware, N; Raghavan, S; Disch, BC; Thorpe, JE; Bastian, A; Ihnat, MA Synthesis and biological activity of 5-chloro-N4-substituted phenyl-9H-pyrimido[4,5-b]indole-2,4-diamines as vascular endothelial growth factor receptor-2 inhibitors and antiangiogenic agents. Bioorg Med Chem 21: 1857 -64 (2013) Moloney, GP; Martin, GR; Mathews, N; Milne, A; Hobbs, H; Dodsworth, S; Sang, PY; Knight, C; Williams, M; Maxwell, M; Glen, RC Synthesis and serotonergic activity of substituted 2, N-benzylcarboxamido-5-(2-ethyl-1-dioxoimidazolidinyl)-N, N-dimethyltryptamine derivatives: novel antagonists for the vascular 5-HT(1B)-like receptor. J Med Chem 42: 2504 -26 (1999) Foot, JS; Deodhar, M; Turner, CI; Yin, P; van Dam, EM; Silva, DG; Olivieri, A; Holt, A; McDonald, IA The discovery and development of selective 3-fluoro-4-aryloxyallylamine inhibitors of the amine oxidase activity of semicarbazide-sensitive amine oxidase/vascular adhesion protein-1 (SSAO/VAP-1). Bioorg Med Chem Lett 22: 3935 -40 (2012) Polverino, A; Coxon, A; Starnes, C; Diaz, Z; DeMelfi, T; Wang, L; Bready, J; Estrada, J; Cattley, R; Kaufman, S; Chen, D; Gan, Y; Kumar, G; Meyer, J; Neervannan, S; Alva, G; Talvenheimo, J; Montestruque, S; Tasker, A; Patel, V; Radinsky, R; Kendall, R AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res 66: 8715 -21 (2006) Okaniwa, M; Imada, T; Ohashi, T; Miyazaki, T; Arita, T; Yabuki, M; Sumita, A; Tsutsumi, S; Higashikawa, K; Takagi, T; Kawamoto, T; Inui, Y; Yoshida, S; Ishikawa, T Design and synthesis of novel DFG-out RAF/vascular endothelial growth factor receptor 2 (VEGFR2) inhibitors: 2. Synthesis and characterization of a novel imide-type prodrug for improving oral absorption. Bioorg Med Chem 20: 4680 -92 (2012) Samadi, A; Valderas, C; de los Ríos, C; Bastida, A; Chioua, M; González-Lafuente, L; Colmena, I; Gandía, L; Romero, A; Del Barrio, L; Martín-de-Saavedra, MD; López, MG; Villarroya, M; Marco-Contelles, J Cholinergic and neuroprotective drugs for the treatment of Alzheimer and neuronal vascular diseases. II. Synthesis, biological assessment, and molecular modelling of new tacrine analogues from highly substituted 2-aminopyridine-3-carbonitriles. Bioorg Med Chem 19: 122 -33 (2011) Kawakami, JK; Martinez, Y; Sasaki, B; Harris, M; Kurata, WE; Lau, AF Investigation of a novel molecular descriptor for the lead optimization of 4-aminoquinazolines as vascular endothelial growth factor receptor-2 inhibitors: application for quantitative structure-activity relationship analysis in lead optimization. Bioorg Med Chem Lett 21: 1371 -5 (2011) Hirose, M; Okaniwa, M; Miyazaki, T; Imada, T; Ohashi, T; Tanaka, Y; Arita, T; Yabuki, M; Kawamoto, T; Tsutsumi, S; Sumita, A; Takagi, T; Sang, BC; Yano, J; Aertgeerts, K; Yoshida, S; Ishikawa, T Design and synthesis of novel DFG-out RAF/vascular endothelial growth factor receptor 2 (VEGFR2) inhibitors: 3. Evaluation of 5-amino-linked thiazolo[5,4-d]pyrimidine and thiazolo[5,4-b]pyridine derivatives. Bioorg Med Chem 20: 5600 -15 (2012) Gangjee, A; Kurup, S; Ihnat, MA; Thorpe, JE; Shenoy, SS Synthesis and biological activity of N(4)-phenylsubstituted-6-(2,4-dichloro phenylmethyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines as vascular endothelial growth factor receptor-2 inhibitors and antiangiogenic and antitumor agents. Bioorg Med Chem 18: 3575 -87 (2010) Borzilleri, RM; Zheng, X; Qian, L; Ellis, C; Cai, ZW; Wautlet, BS; Mortillo, S; Jeyaseelan, R; Kukral, DW; Fura, A; Kamath, A; Vyas, V; Tokarski, JS; Barrish, JC; Hunt, JT; Lombardo, LJ; Fargnoli, J; Bhide, RS Design, synthesis, and evaluation of orally active 4-(2,4-difluoro-5-(methoxycarbamoyl)phenylamino)pyrrolo[2,1-f][1,2,4]triazines as dual vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 inhibitors. J Med Chem 48: 3991 -4008 (2005) Borzilleri, RM; Bhide, RS; Barrish, JC; D'Arienzo, CJ; Derbin, GM; Fargnoli, J; Hunt, JT; Jeyaseelan, R; Kamath, A; Kukral, DW; Marathe, P; Mortillo, S; Qian, L; Tokarski, JS; Wautlet, BS; Zheng, X; Lombardo, LJ Discovery and evaluation of N-cyclopropyl- 2,4-difluoro-5-((2-(pyridin-2-ylamino)thiazol-5- ylmethyl)amino)benzamide (BMS-605541), a selective and orally efficacious inhibitor of vascular endothelial growth factor receptor-2. J Med Chem 49: 3766 -9 (2006) Huang, A; Moretto, A; Janz, K; Lowe, M; Bedard, PW; Tam, S; Di, L; Clerin, V; Sushkova, N; Tchernychev, B; Tsao, DH; Keith, JC; Shaw, GD; Schaub, RG; Wang, Q; Kaila, N Discovery of 2-[1-(4-chlorophenyl)cyclopropyl]-3-hydroxy-8-(trifluoromethyl)quinoline-4-carboxylic acid (PSI-421), a P-selectin inhibitor with improved pharmacokinetic properties and oral efficacy in models of vascular injury. J Med Chem 53: 6003 -17 (2010) Samadi, A; Marco-Contelles, J; Soriano, E; Alvarez-Pérez, M; Chioua, M; Romero, A; González-Lafuente, L; Gandía, L; Roda, JM; López, MG; Villarroya, M; García, AG; Ríos, Cde L Multipotent drugs with cholinergic and neuroprotective properties for the treatment of Alzheimer and neuronal vascular diseases. I. Synthesis, biological assessment, and molecular modeling of simple and readily available 2-aminopyridine-, and 2-chloropyridine-3,5-dicarbonitriles. Bioorg Med Chem 18: 5861 -72 (2010) Sun, L; Liang, C; Shirazian, S; Zhou, Y; Miller, T; Cui, J; Fukuda, JY; Chu, JY; Nematalla, A; Wang, X; Chen, H; Sistla, A; Luu, TC; Tang, F; Wei, J; Tang, C Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem 46: 1116 -9 (2003) Thompson, AM; Delaney, AM; Hamby, JM; Schroeder, MC; Spoon, TA; Crean, SM; Showalter, HD; Denny, WA Synthesis and structure-activity relationships of soluble 7-substituted 3-(3,5-dimethoxyphenyl)-1,6-naphthyridin-2-amines and related ureas as dual inhibitors of the fibroblast growth factor receptor-1 and vascular endothelial growth factor receptor-2 tyrosine kinases. J Med Chem 48: 4628 -53 (2005) Gingrich, DE; Reddy, DR; Iqbal, MA; Singh, J; Aimone, LD; Angeles, TS; Albom, M; Yang, S; Ator, MA; Meyer, SL; Robinson, C; Ruggeri, BA; Dionne, CA; Vaught, JL; Mallamo, JP; Hudkins, RL A new class of potent vascular endothelial growth factor receptor tyrosine kinase inhibitors: structure-activity relationships for a series of 9-alkoxymethyl-12-(3-hydroxypropyl)indeno[2,1-a]pyrrolo[3,4-c]carbazole-5-ones and the identification of CEP-5214 and its dimethylglycine ester prodrug clin J Med Chem 46: 5375 -88 (2003) Hudkins, RL; Becknell, NC; Zulli, AL; Underiner, TL; Angeles, TS; Aimone, LD; Albom, MS; Chang, H; Miknyoczki, SJ; Hunter, K; Jones-Bolin, S; Zhao, H; Bacon, ER; Mallamo, JP; Ator, MA; Ruggeri, BA Synthesis and biological profile of the pan-vascular endothelial growth factor receptor/tyrosine kinase with immunoglobulin and epidermal growth factor-like homology domains 2 (VEGF-R/TIE-2) inhibitor 11-(2-methylpropyl)-12,13-dihydro-2-methyl-8-(pyrimidin-2-ylamino)-4H-indazolo[5,4-a]pyrrolo[3,4-c J Med Chem 55: 903 -13 (2012) Cai, ZW; Zhang, Y; Borzilleri, RM; Qian, L; Barbosa, S; Wei, D; Zheng, X; Wu, L; Fan, J; Shi, Z; Wautlet, BS; Mortillo, S; Jeyaseelan, R; Kukral, DW; Kamath, A; Marathe, P; D'Arienzo, C; Derbin, G; Barrish, JC; Robl, JA; Hunt, JT; Lombardo, LJ; Fargnoli, J; Bhide, RS Discovery of brivanib alaninate ((S)-((R)-1-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yloxy)propan-2-yl)2-aminopropanoate), a novel prodrug of dual vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 kinase inhibitor (BMS-540215 J Med Chem 51: 1976 -80 (2008)
ChEBML_156450 Inhibition of bovine cardiac phosphodiesterase fraction III ChEBML_213951 Inhibitory activity against Vascular endothelial growth factor receptor ChEBML_213959 Inhibition of vascular endothelial growth factor receptor 1 ChEBML_213960 Inhibition of Vascular endothelial growth factor receptor 1 ChEBML_213967 Inhibition of Vascular endothelial growth factor receptor 2 ChEBML_213970 Inhibition of Vascular endothelial growth factor receptor 2 ChEBML_214231 Inhibition of Vascular endothelial growth factor receptor 2 ChEBML_214243 Inhibition of Vascular endothelial growth factor receptor 2 ChEBML_214253 Inhibition of Vascular endothelial growth factor receptor 3 ChEMBL_213950 (CHEMBL811928) Inhibition of Vascular endothelial growth factor receptor ChEMBL_306570 (CHEMBL832087) Inhibition of Vascular endothelial growth factor receptor ChEBML_155549 Inhibition of fraction II of guinea pig phosphodiesterase ChEBML_176345 Inhibition of dopamine uptake in rat synaptosomal fraction ChEBML_176350 Inhibition of noradrenaline uptake in rat synaptosomal fraction ChEBML_214094 Inhibition of human vascular endothelial growth factor receptor 2 ChEBML_214117 Inhibitory activity against Vascular endothelial growth factor receptor 2 ChEMBL_213947 (CHEMBL811925) Inhibition of Vascular endothelial growth factor receptor 3 ChEMBL_213951 (CHEMBL811929) Inhibitory activity against Vascular endothelial growth factor receptor ChEMBL_213964 (CHEMBL811941) Inhibition of Vascular endothelial growth factor receptor 1 ChEMBL_214113 (CHEMBL820699) Inhibition of Vascular endothelial growth factor receptor 2 ChEMBL_214115 (CHEMBL820701) Inhibition of Vascular endothelial growth factor receptor 2 ChEMBL_214240 (CHEMBL819558) Inhibition of Vascular endothelial growth factor receptor 2 ChEMBL_214243 (CHEMBL820211) Inhibition of Vascular endothelial growth factor receptor 2 ChEMBL_214250 (CHEMBL820849) Inhibition of Vascular endothelial growth factor receptor 3 ChEMBL_305183 (CHEMBL876146) Inhibition of vascular endothelial growth factor receptor 2 ChEMBL_305535 (CHEMBL828555) Inhibition of vascular endothelial growth factor receptor 2 ChEMBL_877987 (CHEMBL2189053) Inhibition of vascular endothelial growth factor receptor 2 ChEBML_176338 Inhibition of 5-HT uptake in rat synaptosomal fraction ChEMBL_155544 (CHEMBL759912) Inhibition of fraction I of guinea pig phosphodiesterase ChEMBL_155561 (CHEMBL762673) Inhibition of fraction III of guinea pig phosphodiesterase ChEMBL_176345 (CHEMBL783267) Inhibition of dopamine uptake in rat synaptosomal fraction ChEMBL_176350 (CHEMBL781943) Inhibition of noradrenaline uptake in rat synaptosomal fraction ChEBML_213807 Ability to inhibit expression of Vascular cell adhesion molecule 1 ChEMBL_214107 (CHEMBL820693) Inhibition of human Vascular endothelial growth factor receptor 2 ChEMBL_305511 (CHEMBL831112) inhibitory activity against Vascular endothelial growth factor receptor 2 ChEMBL_305679 (CHEMBL828051) Inhibition of human Vascular endothelial growth factor receptor 2 ChEMBL_306103 (CHEMBL830878) Inhibitory concentration against Vascular endothelial growth factor receptor 2 ChEMBL_65317 (CHEMBL678090) Inhibitory activity against human vascular endothelial nitric oxide synthase. ChEBML_213820 Inhibition of TNF-alpha inducible Vascular cell adhesion molecule-1 expression. ChEMBL_213948 (CHEMBL811926) Inhibitory activity against vascular endothelial growth factor receptor 2 (FLK1) ChEMBL_213949 (CHEMBL811927) Inhibitory activity against vascular endothelial growth factor receptor 2 (FLK1) ChEMBL_214241 (CHEMBL819559) Inhibition of Vascular endothelial growth factor receptor 2 (VEGFR-2) ChEMBL_305774 (CHEMBL827963) In vitro inhibition of Vascular endothelial growth factor receptor 2 ChEMBL_321426 (CHEMBL881957) Inhibitory concentration against human Vascular endothelial growth factor receptor 2 ChEMBL_1461693 (CHEMBL3395231) Inhibition of 11beta-HSD1 in HEK293 cell microsomal fraction ChEMBL_176338 (CHEMBL784031) Inhibition of 5-HT uptake in rat synaptosomal fraction ChEMBL_511117 (CHEMBL1001316) Inhibition of human PDE4B1 in cytosolic fraction of lung ChEMBL_511118 (CHEMBL1001317) Inhibition of human PDE4B1 in particular fraction of lung ChEMBL_511119 (CHEMBL1001318) Inhibition of human PDE4B2 in cytosolic fraction of lung ChEMBL_511120 (CHEMBL1001319) Inhibition of human PDE4B2 in particular fraction of lung ChEMBL_511121 (CHEMBL1001321) Inhibition of human PDE4B3 in cytosolic fraction of lung ChEMBL_511122 (CHEMBL1001322) Inhibition of human PDE4B3 in particular fraction of lung ChEMBL_511124 (CHEMBL999595) Inhibition of human PDE4D1 in cytosolic fraction of lung ChEMBL_511125 (CHEMBL999596) Inhibition of human PDE4D2 in cytosolic fraction of lung ChEMBL_511126 (CHEMBL999597) Inhibition of human PDE4D3 in cytosolic fraction of lung ChEMBL_511127 (CHEMBL999598) Inhibition of human PDE4D3 in particular fraction of lung ChEMBL_511128 (CHEMBL999599) Inhibition of human PDE4D4 in cytosolic fraction of lung ChEMBL_511129 (CHEMBL999600) Inhibition of human PDE4D4 in particular fraction of lung ChEMBL_511130 (CHEMBL999601) Inhibition of human PDE4D5 in cytosolic fraction of lung ChEMBL_511131 (CHEMBL999602) Inhibition of human PDE4D5 in particular fraction of lung ChEBML_214091 Inhibition of Vascular endothelial growth factor receptor 2 at 5 uM ATP ChEBML_214114 Inhibition of vascular endothelial growth factor receptor 2 autophosphorylation in intact cells ChEMBL_213973 (CHEMBL812622) Inhibition of Vascular endothelial growth factor receptor 2 (VEGFR-2) autophosphorylation ChEMBL_305591 (CHEMBL828038) Inhibition of human vascular endothelial growth factor receptor 2 (Flk-1) ChEMBL_305649 (CHEMBL829507) Inhibition of human vascular endothelial growth factor receptor 2 (VEGFR-2) ChEMBL_305849 (CHEMBL829589) Inhibition of human vascular endothelial growth factor receptor 2 kinase activity ChEMBL_155552 (CHEMBL759850) Inhibition of phosphodiesterase fraction III in anesthetized open chest dogs ChEMBL_155556 (CHEMBL760020) Inhibition of phosphodiesterase fraction III in anesthetized open chest dogs ChEMBL_213976 (CHEMBL820672) In vitro inhibition of Vascular endothelial growth factor receptor 2 (VEGFR-2) ChEMBL_213978 (CHEMBL817492) In vitro inhibitory concentration against human vascular endothelial growth factor receptor 2 ChEMBL_305757 (CHEMBL829528) Inhibition of vascular endothelial growth factor receptor 2 in cell-free assay ChEMBL_305820 (CHEMBL829563) Inhibition of vascular endothelial growth factor receptor 2 in cell-intact assay ChEMBL_306571 (CHEMBL832088) Inhibition of Vascular endothelial growth factor receptor 2(KDR) without DTT (dithiothreitol) ChEMBL_306585 (CHEMBL832932) Inhibition of Vascular endothelial growth factor receptor 2(KDR) with DTT (dithiothreitol) ChEMBL_63185 (CHEMBL677396) Binding affinity against endothelin A receptor from rabbit renal vascular smooth muscles ChEMBL_64033 (CHEMBL671552) Binding affinity against endothelin B receptor from rabbit renal vascular smooth muscles ChEMBL_763592 (CHEMBL1820033) Displacement of [3H]PK11195 from TSPO in rat kidney mitochondrial fraction ChEMBL_763593 (CHEMBL1820034) Displacement of [3H]PK11195 from TSPO in human HEK293 mitochondrial fraction ChEBML_65825 Effective concentration against ET A receptor from rabbit renal artery vascular smooth muscle cells ChEMBL_217453 (CHEMBL823502) Inhibitory activity against alpha4-beta1 integrin (vascular cell adhesion molecule) in ELISA assay ChEMBL_65826 (CHEMBL877809) Ability to inhibit [125I]ET1 binding to vascular smooth muscle (vsm)- A10 cells ChEBML_211949 Inhibition of Tripeptidyl-peptidase II purified from a rat liver post-lysosomal fraction. ChEBML_217441 Inhibition of VCAM (vascular cell adhesion molecule) adhesion to alpha4-beta1 integrin of leukocyte cells ChEMBL_213408 (CHEMBL878745) Inhibition of Very late antigen 4/vascular cell adhesion molecule 1 interaction in ELISA ChEMBL_306228 (CHEMBL831136) In vitro inhibition of Vascular endothelial growth factor receptor 2 at 10 uM ATP ChEMBL_313055 (CHEMBL835746) Inhibition of vascular endothelial growth factor (VEGF)-stimulated human umbilical vein endothelial cell proliferation ChEMBL_321627 (CHEMBL871636) Inhibition of Vascular endothelial growth factor receptor 2 kinase phosphorylation of pGAT-biotin peptide ChEMBL_63188 (CHEMBL679791) Antagonistic activity against endothelin A receptor in rabbit renal artery vascular smooth muscle cells. ChEMBL_63196 (CHEMBL679799) Inhibition of binding to Endothelin A receptor of rabbit renal vascular smooth muscle cells ChEMBL_65825 (CHEMBL682959) Effective concentration against ET A receptor from rabbit renal artery vascular smooth muscle cells ChEMBL_65829 (CHEMBL682962) Binding affinity against ET A receptor from rabbit renal artery vascular smooth muscle cells ChEMBL_92772 (CHEMBL700072) Inhibition of L-type [Ca2+] channel (VSCC) in rat A7r5 vascular smooth muscle cells ChEBML_161419 Kinetic constant was measured for PMII of Leishmania donovani promastigotes using supernatant (S12) fraction ChEMBL_211945 (CHEMBL816535) Inhibition of Tripeptidyl-peptidase II purified from a rat liver post-lysosomal fraction. ChEMBL_424403 (CHEMBL911334) Displacement of [3H]DAMGO from mu opioid receptor in rat synaptosomes P2 fraction ChEMBL_539041 (CHEMBL1035715) Inhibition of HO1 in rat spleen microsomal fraction assessed as carbon monoxide production ChEMBL_1622 (CHEMBL616647) Tested for 5-hydroxytryptamine 1D like receptor-mediated vascular effect in rabbit saphenous vein (RSV) ChEMBL_213957 (CHEMBL811935) In vitro inhibition of Vascular endothelial growth factor receptor 1 (VEGFR-1) expressed in baculovirus ChEMBL_213958 (CHEMBL811936) In vitro inhibition of Vascular endothelial growth factor receptor 1 (VEGFR-1) expressed in baculovirus ChEMBL_213975 (CHEMBL812788) In vitro inhibition of Vascular endothelial growth factor receptor 2 (VEGFR-2) expressed in baculovirus ChEMBL_217305 (CHEMBL823926) Inhibition of alpha4-beta1 interaction to vascular cell adhesion molecule-1 (VCAM-1) was determined ChEMBL_329316 (CHEMBL863412) Activity against 5HT2A mediated intracellular calcium mobilization by FLIPR in rat vascular smooth muscle cells ChEMBL_63193 (CHEMBL679796) Inhibition of ET-1 stimulated arachidonic acid release in rabbit renal vascular smooth muscle cells ChEMBL_1453699 (CHEMBL3364101) Inhibition of rat MAO-B in rat-liver mitochondrial-fraction using [14C]-PEA substrate ChEMBL_161419 (CHEMBL766687) Kinetic constant was measured for PMII of Leishmania donovani promastigotes using supernatant (S12) fraction ChEMBL_216037 (CHEMBL819744) Evaluated for beta-receptor affinity determined in rat brain membrane fraction with [3H]- dihydroalprenolol ChEMBL_216038 (CHEMBL819745) Evaluated for beta-receptor affinity, determined in rat brain membrane fraction with [3H]- dihydroalprenolol ChEMBL_424404 (CHEMBL911335) Displacement of [3H]deltorphin II from delta opioid receptor in rat synaptosomes P2 fraction ChEMBL_430079 (CHEMBL918101) Displacement of [3H]DPDPE from delta opioid receptor in rat brain synaptosomes P2 fraction ChEMBL_430080 (CHEMBL918102) Displacement of [3H]DAMGO from mu opioid receptor in rat brain synaptosomes P2 fraction ChEBML_214087 Inhibition of vascular endothelial growth factor receptor 2 activity with 1 mM ATP and biotinylated lck peptide ChEBML_214092 Inhibitory activity against vascular endothelial growth factor receptor 2 (VEGFR2) at a concentration of 5 uM ATP. ChEBML_63191 Displacement of [125I]-ET-1 from Endothelin A receptor of rabbit renal artery vascular smooth muscle cells ChEMBL_213547 (CHEMBL816251) Inhibition of Very late antigen 4 of Ramos cell adhesion to vascular cell adhesion molecule 1 ChEMBL_217312 (CHEMBL824471) Inhibition of very late antigen-4 (VLA-4)/vascular cell adhesion molecule-1(VCAM -1) interaction ChEMBL_65830 (CHEMBL682963) Binding affinity against Endothelin A receptor in rabbit renal artery vascular smooth muscle membrane (ET-A) ChEBML_1540144 Inhibition of human MAO-B expressed in baculovirus infected BT1 cells microsome fraction by fluorescence spectrometry ChEBML_1540145 Inhibition of human MAO-A expressed in baculovirus infected BT1 cells microsome fraction by fluorescence spectrometry ChEMBL_1453698 (CHEMBL3364100) Inhibition of rat MAO-A in rat-liver mitochondrial-fraction using [14C]-5-HT substrate ChEMBL_757264 (CHEMBL1803640) Inhibition of ICMT assessed as accumulation of ras in cytosolic fraction by cell based assay ChEBML_63192 Inhibition of ET-1 binding to Endothelin A receptor in cultured rabbit renal artery vascular smooth muscle cells ChEMBL_1621 (CHEMBL616646) Compound was tested for 5-hydroxytryptamine 1D like receptor-mediated vascular effect in rabbit saphenous vein (RSV) ChEMBL_1880 (CHEMBL616477) Compound was tested for 5-hydroxytryptamine 1D like receptor-mediated vascular effect in rabbit saphenous vein (RSV) ChEMBL_214249 (CHEMBL820848) In vitro inhibition of Vascular endothelial growth factor receptor 3 [VEGFR-3(Flt-4)] expressed in baculovirus ChEMBL_63191 (CHEMBL679794) Displacement of [125I]-ET-1 from Endothelin A receptor of rabbit renal artery vascular smooth muscle cells ChEMBL_63197 (CHEMBL873588) Tested for antagonistic activity against Endothelin A receptor in the rabbit renal artery vascular smooth muscle cells. ChEMBL_1499884 (CHEMBL3583216) Displacement of [3H]-dexamethasone from cytosolic fraction of human recombinant glucocorticoid receptor by scintillation counting analysis ChEMBL_1499890 (CHEMBL3583484) Displacement of [3H]-aldosterone from cytosolic fraction of human recombinant mineralocorticoid receptor by scintillation counting analysis ChEMBL_1540144 (CHEMBL3738353) Inhibition of human MAO-B expressed in baculovirus infected BT1 cells microsome fraction by fluorescence spectrometry ChEMBL_154395 (CHEMBL759157) Inhibition of canine cardiac fraction III cAMP phosphodiesterase (PDE 3) at 0.875 mg/kg intravenous dose ChEMBL_154396 (CHEMBL759158) Inhibition of canine cardiac fraction III cAMP phosphodiesterase (PDE 3) at 1.875 mg/kg intravenous dose ChEMBL_161421 (CHEMBL766689) Kinetic constant was measured for Protein Methylase II of Leishmania donovani promastigotes using supernatant (S12) fraction ChEMBL_1921322 (CHEMBL4424167) Inhibition of MAO-A in mouse brain mitochondrial fraction using kynuramine as substrate by fluorometric analysis ChEBML_1682364 Inhibition of MAGL in human brain vascular pericytes after 1 hr by TAMRA azide-based In-gel fluorescence method ChEMBL_213979 (CHEMBL817493) Inhibition of VEGFR induced autophosphorylation of human Vascular endothelial growth factor receptor 2 (VEGFR2) transfected in CHO cells ChEMBL_2565 (CHEMBL617112) In vitro relative agonist activity against 5-hydroxytryptamine 2A using PI assay in rat vascular smooth muscle cells ChEBML_159280 In vitro inhibitory activity against prostaglandin G/H synthase 1 from the microsomal fraction of ram seminal vesicles ChEMBL_1499885 (CHEMBL3583217) Displacement of [3H]-progesterone from cytosolic fraction of human recombinant progesterone-B receptor by scintillation counting analysis ChEMBL_1682364 (CHEMBL4032641) Inhibition of MAGL in human brain vascular pericytes after 1 hr by TAMRA azide-based In-gel fluorescence method ChEMBL_63187 (CHEMBL679790) Compound was evaluated for the receptor binding activity in rabbit artery vascular smooth muscle cells expressing Endothelin A receptor ChEMBL_63217 (CHEMBL674069) Antagonism of [125 I]ET-1 binding to the rat endothelin receptor in vascular smooth muscle VSM-A10 cells. ChEMBL_2372413 Antagonist activity at alpha4beta1 integrin receptor in human Jurkat E6.1 cells assessed as inhibition of vascular cell adhesion molecule 1-induced cell adhesion pre-incubated for 30 mins followed by 30 mins incubation in vascular cell adhesion molecule 1 coated wells by fluorescence plate reader assay ChEMBL_2372414 Agonist activity at alpha4beta1 integrin receptor in human Jurkat E6.1 cells assessed as inhibition of vascular cell adhesion molecule 1-induced cell adhesion pre-incubated for 30 mins followed by 30 mins incubation in vascular cell adhesion molecule 1 coated wells by fluorescence plate reader assay ChEMBL_154397 (CHEMBL759159) Inhibitory activity against canine cardiac fraction III cyclic AMP phosphodiesterase (PDE III) at 8.75 mg/kg intravenous dose ChEMBL_2308592 Inhibition of human PDE10A2 transfected in human AD293 cells cytosolic fraction using cAMP as substrate by fluorescence polarization assay ChEMBL_940036 (CHEMBL2327845) Displacement of DAGO from MOR in guinea pig brain membrane fraction after 30 mins by scintillation counting analysis ChEBML_63195 Inhibition of [125I]endothelin-1 [ET-1] binding to endothelin A receptor (ETA) of rabbit renal artery vascular smooth muscle cells ChEMBL_199704 (CHEMBL803587) In vitro potency against TNF alpha induced expression of Selectin E on human vascular endothelial cells using CAM ELISA assay ChEMBL_213542 (CHEMBL816989) Inhibition of Very late antigen 4 (VLA-4) of Ramos cell adhesion to vascular cell adhesion molecule 1 (VCAM-1) ChEMBL_213544 (CHEMBL816991) Inhibition of binding of Very late antigen 4/vascular cell adhesion molecule 1 in a protein-based ligand binding assay ChEMBL_213817 (CHEMBL815900) In vitro potency against TNF alpha induced expression of VCAM-1 on human vascular endothelial cells using CAM ELISA assay ChEMBL_63194 (CHEMBL679797) Inhibition of Endothelin A receptor mediated (ET-1) release of arachidonic acid from rabbit renal artery vascular smooth muscle cells ChEBML_58162 Displacement of [3H]-SCH- 23390 (0.3 nM) from dopamine receptor D1 in crude membrane fraction of rat brain corpus striatum ChEMBL_1921320 (CHEMBL4424165) Inhibition of MAO-A in mouse brain mitochondrial fraction using kynuramine as substrate after 30 mins by fluorometric analysis ChEBML_225759 Inhibition of ET- 1 stimulated arachidonic acid release (AAR) in cultured rabbit renal vascular smooth muscle cells expressing the endothelin A receptor. ChEBML_58396 Displacement of [3H]YM-09151-2 (60 pm) from dopamine receptor D2 in crude membrane fraction of rat brain corpus striatum ChEMBL_1658426 (CHEMBL4008038) Displacement of [3H]-Aldosterone from MR in rat kidney cytosolic fraction measured after 18 hrs by liquid scintillation counting method ChEMBL_1752866 (CHEMBL4187626) Inhibition of human FAAH expressed in baculovirus-infected High Five cells S9 fraction using OMP substrate by fluorescence based assay ChEMBL_726920 (CHEMBL1686709) Inhibition of mPGES-1 expressed in LPS-stimulated human A549 cells mitochondrial fraction assessed as conversion of PGH2 to PGE2 ChEMBL_940038 (CHEMBL2327847) Displacement of [3H]Cl-977 from KOR in guinea pig brain membrane fraction after 30 mins by scintillation counting analysis ChEBML_63190 Compound was tested for its ability to inhibit Endothelin A receptor induced arachidonic acid release(AARA) in rabbit renal artery vascular smooth muscles. ChEMBL_213543 (CHEMBL816990) Inhibition of binding of Very late antigen 4/vascular cell adhesion molecule 1 in a cell based ligand binding assay (Jurkat cells) ChEMBL_217611 (CHEMBL819426) Inhibition of alpha4-beta7 integrin binding to radiolabeled vascular cell adhesion molecule-1 (VCAM-1) Ig fusion protein in human Jurkat cells ChEMBL_225759 (CHEMBL848083) Inhibition of ET- 1 stimulated arachidonic acid release (AAR) in cultured rabbit renal vascular smooth muscle cells expressing the endothelin A receptor. ChEMBL_225760 (CHEMBL848084) Inhibition of ET- 1 stimulated arachidonic acid release (AAR) in cultured rabbit renal vascular smooth muscle cells expressing the endothelin B receptor. ChEMBL_2372423 Antagonist activity at alpha4beta1 integrin receptor in human Jurkat E6.1 cells assessed as inhibition of vascular cell adhesion molecule 1-induced cell adhesion ChEMBL_89064 (CHEMBL697203) In vitro potency against TNF alpha induced expression of intercellular adhesion molecule-1 on human vascular endothelial cells using CAM ELISA assay ChEMBL_2372415 Antagonist activity at alpha4beta7 integrin receptor in human RPMI-8866 cells assessed as inhibition of mucosal vascular add-ressin cell adhesion molecule 1-induced cell adhesion pre-incubated for 30 mins followed by 30 mins incubation in mucosal vascular add-ressin cell adhesion molecule 1 coated wells by fluorescence plate reader assay ChEMBL_2372416 Agonist activity at alpha4beta7 integrin receptor in human RPMI-8866 cells assessed as inhibition of mucosal vascular add-ressin cell adhesion molecule 1-induced cell adhesion pre-incubated for 30 mins followed by 30 mins incubation in mucosal vascular add-ressin cell adhesion molecule 1 coated wells by fluorescence plate reader assay ChEMBL_1363487 (CHEMBL3291709) Inhibition of human placental 17beta-HSD2 microsomal fraction using tritiated estradiol as substrate assessed as formation of estrone by HPLC analysis ChEMBL_1363489 (CHEMBL3291711) Inhibition of human placental 17beta-HSD1 cytosolic fraction using tritiated estrone as substrate assessed as formation of estradiol by HPLC analysis ChEMBL_1363494 (CHEMBL3291985) Inhibition of mouse liver 17beta-HSD2 microsomal fraction using tritiated estradiol as substrate assessed as formation of estrone by HPLC analysis ChEMBL_1466123 (CHEMBL3404741) Inhibition of human 11beta-HSD1 using microsomal fraction and NADPH assessed as conversion of cortisone to cortisol by biochemical enzyme assay ChEMBL_1466125 (CHEMBL3404743) Inhibition of mouse 11beta-HSD1 using microsomal fraction and NADPH assessed as conversion of cortisone to cortisol by biochemical enzyme assay ChEMBL_1466126 (CHEMBL3404744) Inhibition of human 11beta-HSD2 using microsomal fraction and NADPH assessed as conversion of cortisone to cortisol by biochemical enzyme assay ChEMBL_1499893 (CHEMBL3583487) Displacement of [3H]methyltrienolone from cytosolic fraction of androgen receptor in human LANCAP cells after 24 hrs by scintillation counting analysis ChEMBL_1541061 (CHEMBL3744292) Inhibition of 17beta-HSD2 in human placental microsomal fraction using E2/NAD+ as substrate/cofactor after 20 mins by HPLC analysis ChEMBL_1541062 (CHEMBL3744293) Inhibition of 17beta-HSD1 in human placental cytosolic fraction using E1/NADH as substrate/cofactor after 10 mins by HPLC analysis ChEMBL_1861097 (CHEMBL4361953) Inhibition of human placental microsomal fraction 17beta-HSD2 using [3H]-E2 as substrate in presence of NAD+ by radio-HPLC analysis ChEMBL_1861098 (CHEMBL4361954) Inhibition of human placental cytosolic fraction 17beta-HSD1 using [3H]-E1 as substrate in presence of NAD+ by radio-HPLC analysis ChEMBL_1863749 (CHEMBL4364724) Inhibition of human placental microsomal fraction 17beta-HSD2 using [3H]-E2 as substrate in presence of NAD+ by radio-HPLC analysis ChEMBL_1863752 (CHEMBL4364727) Inhibition of human placental cytosolic fraction 17beta-HSD1 using [3H]-E1 as substrate in presence of NAD+ by radio-HPLC analysis ChEMBL_1898526 (CHEMBL4400641) Inhibition of human placental microsomal fraction 17beta-HSD2 using [3H]-E2 as substrate in presence of NAD+ by radio-HPLC analysis ChEMBL_1898527 (CHEMBL4400642) Inhibition of human placental cytosolic fraction 17beta-HSD1 using [3H]-E1 as substrate in presence of NAD+ by radio-HPLC analysis ChEMBL_63190 (CHEMBL679793) Compound was tested for its ability to inhibit Endothelin A receptor induced arachidonic acid release(AARA) in rabbit renal artery vascular smooth muscles. ChEMBL_1446069 (CHEMBL3380296) Inhibition of human recombinant MDR1 in cell membrane fraction preincubated for 5 mins measured after 40 mins by Pgp-Glo luciferase assay ChEMBL_1463279 (CHEMBL3399468) Displacement of [3H]MPPF from 5HT1A receptor in Sprague-Dawley rat hippocampal membrane fraction incubated for 60 mins by scintillation counting method ChEMBL_1572346 (CHEMBL3795929) Displacement of [125I]-CRF from human CRF1 receptor expressed in CHO cellular membrane fraction after 1.5 hrs by liquid scintillation counting method ChEMBL_1658427 (CHEMBL4008039) Displacement of [3H]-Aldosterone from MR in Sprague-Dawley rat kidney cytosolic fraction measured after overnight incubation by liquid scintillation counting method ChEMBL_606059 (CHEMBL1071958) Inhibition of SCD1 in mouse microsomal liver S9 microsomal fraction assessed as conversion of [14C]stearate to [14C]oleate after 60 mins ChEMBL_798594 (CHEMBL1942485) Inhibition of alpha-glucosidase activity of maltase in rat small intestinal brush border membrane fraction using maltose as substrate after 30 mins ChEMBL_798596 (CHEMBL1942487) Inhibition of alpha-glucosidase activity of sucrase in rat small intestinal brush border membrane fraction using maltose as substrate after 30 mins ChEMBL_938947 (CHEMBL2328436) Inhibition of 17beta-HSD1 in human placental cytosolic fraction using [3H]E1 as substrate assessed as formation of E2 by HPLC analysis ChEMBL_938948 (CHEMBL2328437) Inhibition of 17beta-HSD2 in human placental microsomal fraction using [3H]E2 as substrate assessed as formation of E1 by HPLC analysis ChEMBL_1566203 (CHEMBL3788344) Displacement of [125I]-Ang II from Angiotensin 2 type-1A receptor in rat vascular smooth muscle cells after 150 mins by gamma counting method ChEMBL_1367282 (CHEMBL3300203) Inhibition of human placental 17beta-HSD1 cytosolic fraction using [2, 4, 6, 7-3H]-estrone as substrate after 10 mins by HPLC analysis ChEMBL_1367283 (CHEMBL3300204) Inhibition of human placental 17beta-HSD2 microsomal fraction using [2, 4, 6, 7-3H]-estradiol as substrate after 20 mins by HPLC analysis ChEMBL_1752865 (CHEMBL4187625) Inhibition of human recombinant soluble epoxide hydrolase expressed in baculovirus-infected High Five cells S9 fraction using CMNPC substrate by fluorescence based assay ChEMBL_1795802 (CHEMBL4267919) Displacement of [3H]DTG from S1R in P2 fraction of Hartley guinea pig whole brain after 90 mins by liquid scintillation counting method ChEMBL_1849337 (CHEMBL4349878) Inhibition of SOAT1 in CHO cell microsomal fraction using [1-14C]oleoyl-CoA as substrate measured after 5 mins by TLC based method ChEMBL_1866205 (CHEMBL4367180) Inhibition of isopentenyl diphosphate isomerase in Sprague-Dawley rat liver S100 fraction preincubated for 10 min followed by MVA addition by HPLC analysis ChEMBL_2324327 Inhibition of human placental cytosolic fraction 17beta-HSD1 using [3H]E1 as substrate measured after 10 mins in presence of NADH by HPLC analysis ChEMBL_2324328 Inhibition of human placental microsomal fraction 17beta-HSD2 using [3H]E2 as substrate measured after 20 mins in presence of NAD+ by HPLC analysis ChEMBL_736715 (CHEMBL1695096) Inhibition of Prostaglandin E2 synthase-1 in IL1-beta stimulated microsomal fraction of human A549 cell assessed as PGE2 level by RP-HPLC ChEMBL_141416 (CHEMBL752320) Displacement of [3H]ifendropil from binding site of N-methyl-D-aspartate glutamate receptor was evaluated employing synaptosomal fraction of porcine hippocampal brain membranes ChEMBL_1470660 (CHEMBL3420247) Inhibition of HO1 in Sprague-Dawley rat spleen microsomal fraction assessed as reduction in bilirubin formation incubated for 60 mins by double beam spectrophotometry ChEMBL_1470661 (CHEMBL3420440) Inhibition of HO2 in Sprague-Dawley rat brain microsomal fraction assessed as reduction in bilirubin formation incubated for 60 mins by double beam spectrophotometry ChEMBL_1702993 (CHEMBL4054226) Inhibition of full-length mouse 11beta-HSD1 expressed in HEK293 microsomal fraction using [3H]cortisone as substrate after 1 hr by scintillation proximity assay ChEMBL_1702996 (CHEMBL4054229) Inhibition of full-length human 11beta-HSD1 expressed in HEK293 microsomal fraction using [3H]cortisone as substrate after 2 hr by scintillation proximity assay ChEMBL_1702997 (CHEMBL4054230) Inhibition of full-length mouse 11beta-HSD2 expressed in HEK293 microsomal fraction using [3H]cortisone as substrate after 1 hr by scintillation proximity assay ChEMBL_1702998 (CHEMBL4054231) Inhibition of full-length human 11beta-HSD2 expressed in HEK293 microsomal fraction using [3H]cortisone as substrate after 2 hr by scintillation proximity assay ChEMBL_1921326 (CHEMBL4424171) Inhibition of MAO-A in Sprague-Dawley rat brain mitochondrial fraction after 10 mins in presence of [14C]5-hydroxytryptamine by liquid scintillation analysis ChEMBL_2173334 (CHEMBL5058468) Inhibition of human placental cytosolic fraction 17beta-HSD1 using [3H]-E1 as substrate in presence of NADH measured after 10 mins by HPLC method ChEMBL_2173335 (CHEMBL5058469) Inhibition of human placental microsomal fraction 17beta-HSD2 using [3H]-E2 as substrate in presence of NAD+ measured after 20 mins by HPLC method ChEMBL_2217338 (CHEMBL5130470) Inhibition of human placental cytosolic fraction 17beta-HSD1 using [3H]-E1 as substrate in presence of NADH measured after 10 mins by HPLC method ChEMBL_2217339 (CHEMBL5130471) Inhibition of human placental microsomal fraction 17beta-HS2 using [3H]-E2 as substrate in presence of NAD+ measured after 10 mins by HPLC method ChEMBL_2261405 (CHEMBL5216416) Displacement of [14C]-ethanolamine from FAAH derived from rat brain membrane fraction preincubated for 20 mins followed by susbstrate addition by scintillation counting method ChEMBL_606061 (CHEMBL1071960) Inhibition of SCD1 in human liver S9 microsomal fraction assessed as conversion of [14C]stearate to [14C]oleate after 60 mins at 0.4 uM ChEMBL_64015 (CHEMBL671536) Ability to block endothelin-1 (ET-1) stimulated arachidonic acid release in rabbit renal vascular smooth muscle cells (AARA) expressing recombinant rat endothelin B (ETB) receptor ChEMBL_1358464 (CHEMBL3281632) Displacement of 1 x 10'-8 M of [1,2,3-3H]-triamcinolone acetonide from glucocorticoid receptor in soluble fraction of mouse L929 cells after 20 hrs ChEMBL_1358466 (CHEMBL3281634) Displacement of 2 x 10'-8 M of [1,2,3-3H]-triamcinolone acetonide from glucocorticoid receptor in soluble fraction of mouse L929 cells after 20 hrs ChEMBL_1474763 (CHEMBL3424220) Inhibition of mouse 11beta-HSD2 overexpressed in microsomal fraction of HEK293 cells using [3H]-cortisol as substrate by scintillation proximity assay in presence of NAD+ ChEMBL_1474766 (CHEMBL3424223) Inhibition of human 11beta-HSD2 overexpressed in microsomal fraction of HEK293 cells using [3H]-cortisol as substrate by scintillation proximity assay in presence of NAD+ ChEMBL_1789617 (CHEMBL4261351) Inhibition of mouse microsomal fraction 17beta-HSD2 using [3H]-E2 as substrate after 20 mins in presence of NAD+ by radio-flow detector based analysis ChEMBL_606058 (CHEMBL1071957) Inhibition of SCD1 in human 293A cells assessed as conversion of [14C]stearate to [14C]oleate after 60 mins in presence of S9 microsomal fraction ChEMBL_988473 (CHEMBL2438122) Inhibition of human placenta 17beta-HSD1 cytosolic fraction using unlabeled, [2,4,6,7-3H]-estrone as substrate after 20 mins by HPLC analysis in presence of NADH ChEMBL_988475 (CHEMBL2438124) Inhibition of human placenta 17beta-HSD2 microsomal fraction using unlabeled, [2,4,6,7-3H]-estradiol as substrate after 20 mins by HPLC analysis in presence of NAD+ ChEMBL_1752869 (CHEMBL4187629) Competitive inhibition of human recombinant soluble epoxide hydrolase expressed in baculovirus-infected High Five cells S9 fraction using varying CMNPC substrate levels by fluorescence based assay ChEMBL_1789612 (CHEMBL4261346) Inhibition of human placental microsomal fraction 17beta-HSD2 using [3H]-E2 as substrate after 20 mins in presence of NAD+ by radio-flow detector based analysis ChEMBL_1882121 (CHEMBL4383620) Inhibition of FAAH in rat liver S9 fraction using N-(2-hydroxyethyl)-4-pyren-1-ylbutanamide as substrate after 60 mins by fluorescence-based HPLC analysis ChEMBL_1994033 (CHEMBL4627928) Inhibition of human SOAT1-mediated [14C]Cholesterol ester biosynthesis in CHO microsomal fraction after 5 mins in presence of [1-14C]oleoyl-CoA by TLC analysis ChEMBL_1994034 (CHEMBL4627929) Inhibition of human SOAT2-mediated [14C]Cholesterol ester biosynthesis in CHO microsomal fraction after 5 mins in presence of [1-14C]oleoyl-CoA by TLC analysis ChEMBL_861317 (CHEMBL2173705) Inhibition of human placenta 17beta-HSD1 cytosolic fraction using unlabelled and [2,4,6,7-3H]estrone as substrate after 10 mins by HPLC analysis in presence of NADH ChEMBL_861318 (CHEMBL2173706) Inhibition of human placenta 17beta-HSD2 microsomal fraction using unlabelled and [2,4,6,7-3H]estradiol as substrate after 20 mins by HPLC analysis in presence of NAD+ ChEMBL_1866206 (CHEMBL4367181) Inhibition of farnesyl diphosphate synthase in Sprague-Dawley rat liver S100 fraction using MVA as substrate preincubated for 10 min followed by MVA addition by HPLC analysis ChEMBL_1274566 (CHEMBL3091166) Inhibition of 17beta-HSD3 in rat testes microsomal fraction using [14C]-4-androstene-3,17-dione as substrate assessed as testosterone formation after 2 hrs by thin layer chromatography ChEMBL_141414 (CHEMBL751708) Compound was tested for inhibition of [3H]MK-801 binding to N-methyl-D-aspartate glutamate receptor at NR2B subunit in high affinity fraction of porcine brain membranes ChEMBL_141415 (CHEMBL751709) Compound was tested for inhibition of [3H]MK-801 binding to N-methyl-D-aspartate glutamate receptor lacking NR2B subunit in low affinity fraction of porcine brain membranes ChEMBL_1658402 (CHEMBL4008014) Displacement of [3H]-Aldosterone from MR in rat renal soluble fraction measured after 18 hrs in presence of glucocorticoid receptor antagonist RU-486 by liquid scintillation counting method ChEMBL_1904878 (CHEMBL4407236) Inhibition of 17beta-HSD3 in Sprague-Dawley rat testes microsomal fraction assessed as reduction in [14C]-testosterone formation from [14C]-4-androstene-3,17-dione measured after 2 hrs ChEMBL_306909 (CHEMBL828352) Inhibition of Very late antigen-4 (VLA-4) expressing human leukemia cells (HL-60) aggregation with human Vascular cell adhesion molecule-1 (VCAM-1) expressing chinese hamster ovary (CHO) cells ChEMBL_1474762 (CHEMBL3424219) Inhibition of human 11beta-HSD1 overexpressed in microsomal fraction of HEK293 cells assessed as formation of [3H]-cortisol from [3H]-cortisone by scintillation proximity assay in presence of NADPH ChEMBL_1474764 (CHEMBL3424221) Inhibition of mouse 11beta-HSD1 overexpressed in microsomal fraction of HEK293 cells assessed as formation of [3H]-cortisol from [3H]-cortisone by scintillation proximity assay in presence of NADPH ChEMBL_1796598 (CHEMBL4268715) Inhibition of heme oxygenase 1 in Sprague-Dawley rat spleen microsomal fraction assessed as decrease in bilirubin formation using biliverdin reductase as substrate after 60 mins by spectrophotometric method ChEMBL_2372426 Binding affinity to alpha4beta1 integrin receptor (unknown origin) preincubated for 30 mins followed by 30 mins incubation in vascular cell adhesion molecule 1-induced cell adhesion coated wells by fluorescence based assay ChEMBL_2372427 Binding affinity to alpha4beta7 integrin receptor (unknown origin) preincubated for 30 mins followed by 30 mins incubation in mucosal vascular add-ressin cell adhesion molecule 1 coated wells by fluorescence based assay ChEMBL_1856724 (CHEMBL4357453) Inhibition of human BK alpha/beta4 channel expressed in HEK293T cells assessed as inhibition of K+ outward current fraction at -80 mV holding potential by whole cell patch clamp technique ChEMBL_2018315 (CHEMBL4671893) Inhibition of FP-Rh binding to ABHD6 in mouse brain cytosolic fraction preincubated for 1 hr followed by FP-Rh addition and measured after 1 hr by SDS-PAGE analysis ChEMBL_1337889 (CHEMBL3240916) Inhibition of CYP3A4 in human liver microsomal fraction assessed as midazolam-1'-hydroxylation preincubated for 10 mins followed by NADPH addition measured after 5 to 20 mins by RF-MS analysis ChEMBL_1337890 (CHEMBL3240917) Inhibition of CYP2C9 in human liver microsomal fraction assessed as diclofenac-4'-hydroxylation preincubated for 10 mins followed by NADPH addition measured after 5 to 20 mins by RF-MS analysis ChEMBL_1337891 (CHEMBL3240918) Inhibition of CYP2D6 in human liver microsomal fraction assessed as dextromethorphan O-demethylase preincubated for 10 mins followed by NADPH addition measured after 5 to 20 mins by RF-MS analysis ChEMBL_1798975 (CHEMBL4271267) Inhibition of partially purified HDAC isolated from human M8 cells enzyme fraction pre-incubated for 5 mins before [3H]acetylated histone addition and measured after 30 mins by scintillation counting method ChEMBL_2244526 (CHEMBL5158736) Inhibition of human recombinant LTC4S expressed in HEK293 cell microsomal fraction preincubated for 10 mins followed by LTA4-methyl ester addition and measured after 10 mins by UPLC-MS/MS analysis ChEMBL_956682 (CHEMBL2379079) Inhibition of MAGL (unknown origin) expressed in African green monkey COS7 cell cytosolic fraction assessed as [3H]2-AG hydrolysis to [3H]arachidonic acid after 20 mins by beta counting analysis ChEMBL_1343944 (CHEMBL3259253) Inhibition of HMG-coA reductase in CD rat liver microsomal-cytosol fraction assessed as inhibition of total lipid synthesis using [2-14C]acetate as substrate after 90 mins by scintillation counting analysis ChEMBL_1343945 (CHEMBL3259254) Inhibition of HMG-coA reductase in CD rat liver microsomal-cytosol fraction assessed as inhibition of nonsaponifiable lipid synthesis using [2-14C]acetate as substrate after 90 mins by scintillation counting analysis ChEMBL_1343946 (CHEMBL3259255) Inhibition of HMG-coA reductase in CD rat liver microsomal-cytosol fraction assessed as inhibition of fatty acid synthesis using [2-14C]acetate as substrate after 90 mins by scintillation counting analysis ChEMBL_2018313 (CHEMBL4671891) Inhibition of FP-Rh binding to APT-1/2 in mouse brain cytosolic fraction preincubated for 1 hr followed by FP-Rh addition and measured after 1 hr by SDS-PAGE analysis ChEMBL_808211 (CHEMBL1961123) Inhibition of 5-lipoxygenase in S100 cell free fraction of human polymorphonuclear leukocytes assessed as reduction in 5-LO product formation preincubated for 15 mins measured after 10 mins by HPLC analysis ChEBML_1716382 Inhibition of human full length NPP1 expressed in African green monkey COS7 cell membrane fraction using p-nitrophenyl-5'-thymidine monophosphate as substrate preincubated for 10 mins followed by substrate addition measured after 30 mins ChEBML_1716383 Inhibition of human full length NPP3 expressed in African green monkey COS7 cell membrane fraction using p-nitrophenyl-5'-thymidine monophosphate as substrate preincubated for 10 mins followed by substrate addition measured after 30 mins ChEBML_1716384 Inhibition of human NTPDase1 expressed in African green monkey COS7 cell membrane fraction using ATP as substrate preincubated for 10 mins followed by substrate addition measured after 10 mins by malachite green reagent-based assay ChEBML_1716385 Inhibition of human NTPDase2 expressed in African green monkey COS7 cell membrane fraction using ATP as substrate preincubated for 10 mins followed by substrate addition measured after 10 mins by malachite green reagent-based assay ChEBML_1716386 Inhibition of human NTPDase3 expressed in African green monkey COS7 cell membrane fraction using ATP as substrate preincubated for 10 mins followed by substrate addition measured after 10 mins by malachite green reagent-based assay ChEBML_1716387 Inhibition of human NTPDase8 expressed in African green monkey COS7 cell membrane fraction using ATP as substrate preincubated for 10 mins followed by substrate addition measured after 10 mins by malachite green reagent-based assay ChEMBL_1699174 (CHEMBL4050156) Inhibition of human DGAT1 expressed in yeast membrane fraction assessed as inhibition of triglyceride formation using diolein/oleoyl-CoA as substrate measured after 1 hr by 7-diethylamino-3-(4'-maleimidylphenyl)-4-methylcoumarin based fluorescence analysis ChEMBL_826632 (CHEMBL2050886) Inhibition of mPGES-1 derived from IL1beta and TNFalpha stimulated human HeLa cells microsomal fraction using PGH2 as substrate preincubated for 30 mins prior to substrate addition measured after 1 min by LC-MS/MS analysis ChEMBL_1793492 (CHEMBL4265411) Inhibition of RALDH2 in choroidal cytosol fraction from 4 day recovering chick assessed as inhibition of ATRA synthesis pre-incubated for 20 mins followed by ATRA and NAD+ addition and measured after 30 mins by HPLC analysis ChEMBL_1567868 (CHEMBL3789263) Inhibition of microsomal fraction of human CYP26B1 expressed in Sf9 cells using 9-cis-RA as substrate preincubated for 5 mins followed by NADPH addition measured after 5 mins by HPLC analysis in presence of rat P450 reductase ChEMBL_1567869 (CHEMBL3789264) Inhibition of microsomal fraction of human CYP26A1 expressed in Sf9 cells using 9-cis-RA as substrate preincubated for 5 mins followed by NADPH addition measured after 1 min by HPLC analysis in presence of rat P450 reductase ChEMBL_1882122 (CHEMBL4383621) Inhibition of FAAH in rat liver S9 fraction using N-(2-hydroxyethyl)-4-pyren-1-ylbutanamide as substrate preincubated for 45 mins in absence of NADPH followed by substrate addition and measured after 60 mins by fluorescence-based HPLC analysis Inhibition Assay An MGAT2 inhibition test for test compounds was conducted in accordance with a partially modified version of the method described in John F. Lockwood et al., Am. J. Physiol. Endocrinol. Metab., 2003, 285, E927.A 10 mg/mL microsomal fraction of human small intestine (duodenum- and jejunum-derived fraction purchased from Becton Dickinson Japan) was used as an enzyme source.The test was conducted using a flat-bottom 96-well plate (Corning).A buffer solution with final concentrations of 5 mM magnesium chloride, 1.25 mg/ml bovine serum albumin and 100 mM Tris-HCl (pH=7.5) was prepared and the microsomal fraction of human small intestine was added thereto to give a final concentration of 5 .mu.g/ml. Test compounds prepared in various concentrations using dimethyl sulfoxide were added thereto to give a final dimethyl sulfoxide concentration of 1% and a reaction was performed at room temperature for 5 min. In Vitro Activity Assay In Vitro Activity: Evaluation of NMDAR Blocking Activity Using Patch-ClampPrevious studies have shown that the NMDAR exists in the peripheral vasculature.All NMDAR subunits were examined by RT-PCR and sequencing in the peripheral endothelium and peripheral vascular smooth muscle cells. The sequences of these NMDAR subunits in both vascular cells showed a high similarity if not identity to the sequences of brain NMDAR (Chen H et al, J Vasc Surg 2005, Qureshi I et al Vasc Med 2005).The molecules described herein were tested in serial concentrations ranging from 1 nM to 100 μM for their NMDAR blocking activity using patch-clamp. ChEMBL_1882123 (CHEMBL4383622) Inhibition of FAAH in rat liver S9 fraction using N-(2-hydroxyethyl)-4-pyren-1-ylbutanamide as substrate preincubated for 15 mins followed by NADPH addition and subsequent substrate addition at 30 mins post NADPH addition and measured after 60 mins by fluorescence-based HPLC analysis Inhibition Assay Receptor-expressing HEK 293 cell membrane fraction was analyzed by modifying the methods of C. J. Langmead et al. (see Langmead C J, Szekeres P G; Chambers J K, Ratcliffe S J, Jones D N C, Hirst W D, et al. Characterization of the binding of [125I]-human prolactin releasing peptide (PrRP) to GPR10, a novel G protein coupled receptor. Br J Pharmacol 2000; 131: 683-688.). To each well of a 96-well plate, 150 μl of assay buffer (20 mM HEPES, 10 mM EDTA, 1 μl/ml protease inhibiting agent cocktail, pH 7.4), 20 μl of cell membrane fraction, 10 μl of test compound and [3H]-PrRP (final concentration 1 nM) were added and incubated at room temperature for 90 minutes. After completion of the reaction, using a cell harvester, the cell-membrane fraction sample product was filtered under aspiration by a glass fiber filter plate (Unifilter; GF/B) previously treated with 0.5% polyethylene imine. The filter was washed with a 50 mM Tris hydrochloric acid buffer (pH 7.4) three times. hERG Inhibition Assay The determination of plasma protein binding (PPB) of a compound is enabled by equilibrium dialysis, an accepted and standard method for reliable estimation of the non-bound drug fraction in plasma. RED (Rapid Equilibrium Dialysis) device insert is made of two side-by-side chambers separated by an O-ring-sealed vertical cylinder of dialysis membrane (MWCO 8,000). Plasma containing drug (at 5 μM or blood concentrations otherwise corresponding to efficacious doses, if known) is added to one chamber while buffer is added to the second. After 4 hours incubation at 37° C. under shaking, an aliquot is removed from each chamber and analyzed by a LC-MS/MS procedure enables the determination of both free and bound drug.The percentages provided in Table 4 represent, for the compounds of the invention, the bound drug fraction to the plasma protein. The free fraction may be calculated as 100%−% rPPB (i.e. the complementary percentage of that disclosed in Table 4, corresponding to the drug concentration that is unbound and therefore available to engage biological target and elicit pharmacological activity). Enzyme Inhibition Assay Time-dependent optical density change was followed at 405 nm using a kinetic microplate reader at room temperature. Enzyme activity in the presence of inhibitor was expressed as fraction of a DMSO control and curve fit to the equation: activity= control activity / (1 + [I]/ IC50) using Excel Fit. The IC50 value is that concentration causing half maximal inhibition. HDAC Enzyme Activity Assay For the enzyme assay, the enzyme fraction was added to fluorescent substrate in the presence of test compounds, and the mixture was incubated for additional 30 min. The released aminomethyl coumarin (AMC) was measured using a fluorescence plate reader. The 50% inhibitory concentrations (IC50) were determined as means with SD calculated from at least three independent dose response curves. Binding Assay A competition binding assay (DiscoveRx KINOMEscan) was used to measure the ability of the compound to compete for binding of an immobilized adenosine triphosphosphate (ATP) site directed ligand using a DNA-tagged vascular endothelial growth receptor 2 (VEGFR2) as the target. The ability of the test compound to compete with the immobilized ligand was measured using quantitative polymerase chain reaction (qPCR) of the DNA tag. sEH_DR_Inh_Infinite200_Fluorescence_01072008 Data Source: Columbia University Molecular Screening Center Source (MLSCN Center Name): Columbia University Molecular Screening Center Center Affiliation: Columbia University Molecular Screening Center Assay Provider: Dr. Bruce D. Hammock, UC, Davis, CA. Network: Molecular Library Screening Center Network (MLSCN) Grant Proposal Number: X01 MH078954-01 Hypertension and vascular inflammation are associated with cardiovascular diseases, the primary cause of death in our society. Because a large proportion of patients are not responding to current therapies, the next generation of drugs will not only need to reduce blood pressure but also treat vascular and renal inflammation as well as reduce smooth muscle cell proliferation, which in turn should also reduce hypertension related organ damage. Using inhibitors developed in the Hammock laboratory, it was shown that the inhibition of soluble epoxide hydrolase (sEH) has therapeutic application in the treatment of hypertension and several in Inhibition Test for Each Compound of the Present Invention Against 20-HETE Producing Enzymes (CYP4F2 and CYP4A11) In the CYP4F2 inhibition test, the reaction solution containing each compound [final concentration of 50 mM, KPO4 (pH 7.4), 2.5 μM luciferine derivative, and 1 mM NADPH] was added to an Escherichia coli membrane fraction (100 μg/mL protein) in which human CYP4F2 had been expressed. In the CYP4A11 inhibition test, the reaction solution containing each compound [final concentration of 100 mM, Tris-HCl (pH 7.5), 60 μM luciferine derivative, 1.3 mM NADP+, 3.3 mM Glucose 6-Phosphate, 3.3 mM MgCl2, and 0.4 U/mL Glucose 6-Phosphate dehydrogenase] was added to an Escherichia coli membrane fraction (100 μg/mL protein) in which human CYP4A11 had been expressed. Following this, the membrane fraction was left to stand at room temperature for 60 minutes to perform an enzymatic reaction. After the reaction, a luciferine detection reagent was added, and the luminescence value was measured using a plate reader. By using that value, the percent inhibition of 20-HETE producing enzyme (%) was calculated according to the equation described below, and the 50% inhibitory concentration (IC50 value) for each compound was calculated. Biochemical Activity Assay Protein was expressed and purified from E. coli BL21 DE3 Rosetta 2 (EMD Millipore) cells using standard techniques. Cells were grown in 2x yeast extract tryptone medium and expression was initiated via the addition of isopropyl -D-1-thiogalactopyranoside. Expression proceeded overnight at 18 ° C. Cells were harvested by centrifugation and subsequently lysed via sonication. Insoluble fraction was removed by centrifugation. Maltose binding protein (MBP) fusion proteins were purified on a dextrin sepharose column (GE Healthcare) and the MBP tag was removed using tobacco etch virus protease overnight during dialysis. Protein was further purified on a heparin column (GE Healthcare) and eluted using a NaCl gradient. Column fraction were pooled and further purified on a Superdex 75 gel filtration column (GE Healthcare). Protein was quantified using 280 nm absorbance. Protein was then flash frozen in liquid nitrogen and stored at 80 ° C. until use. Dose-response biochemical assay for antagonists of the interaction between the Eph receptor B4 (EphB4) and its ligand ephrin-B2 via TNYL-RAW peptide Source (MLSCN Center Name): The Scripps Research Institute Molecular Screening Center Center Affiliation: The Scripps Research Institute, TSRI Assay Provider: Peter Kuhn, TSRI Network: Molecular Library Screening Center Network (MLSCN) Grant Proposal Number: 1 X01 MH079857-01 Grant Proposal PI: Peter Kuhn External Assay ID: EphB4TNYLRAW_INH_FP_1536_IC50 Name: Dose-response biochemical assay for antagonists of the interaction between the Eph receptor B4 (EphB4) and its ligand ephrin-B2 via TNYL-RAW peptide Description: The erythropoietin-producing hepatocellular (Eph) receptor family is the largest family of receptor tyrosine kinases identified to date, with 16 structurally similar family members(1). The Eph family plays important roles in both the developing and adult tissues, and is involved in biological processes such as tissue patterning, vascular system development, axonal guidance, and neuronal development (2-4). During vascular development, the Eph receptor B4 (EphB4) is p Binding Assay Measurement of EP2 receptor binding action was performed in accordance with the method of Abramovitz et al. (Biochimica et Biophysica Acta, 1483, 285 (2000)). A test compound dissolved in dimethyl sulfoxide (final concentration: 1.0 (V/V) %) and [3H]prostaglandin E2 (NET-428, manufactured by PerkinElmer) (final concentration: 10 nM) were added to a buffer solution (10 mM MES-KOH (pH 6.0), 10 mM MgCl2, 1 mM EDTA) in which 10 μg of a membrane fraction of HEK293 cells expressing human EP2 receptor (ES-562-M, manufactured by Euroscreen) was suspended, and then incubated at 30° C. for 60 minutes. The membrane fraction was recovered on glass fiber filter paper (GF/B, manufactured by Whatman) using a cell harvester (M30R, manufactured by Brandel), and after washing with a buffer solution (10 mM MES-KOH (pH 6.0), 10 mM MgCl2), radioactivity was measured with a liquid scintillation analyzer (2000CA, manufactured by Packard). Binding Assay Measurement of EP2 receptor binding affinity was carried out according to the method of Abramovitz et al. (Biochimica et Biophysica Acta, 1483, 285 (2000)). A test compound dissolved in dimethylsulfoxide and [3H]PGE2 (NET-428, available from PerkinElmer Inc.) (final concentration: 10 nM) were added to a buffer solution (10 mM MES-KOH (pH 6.0), 10 mM MgCl2, 1 mM EDTA) in which 10 μg of a membrane fraction (ES-562-M, available from Euroscreen S.A.) of HEK293 cells expressing human EP2 receptor had been suspended, followed by incubation at 30° C. for 60 minutes. The membrane fraction was recovered on glass fiber filter paper (GF/B, available from Whatman PLC) using a cell harvester (M30R, available from Brandel Inc.), and after washing with a buffer solution (10 mM MES-KOH (pH 6.0), 10 mM MgCl2), radioactivity was measured with a liquid scintillation analyzer (2000CA, available from Packard). Inhibition Assay A test for MGAT2 inhibitory action of test compounds was conducted in accordance with a partially modified version of the method described in John F. Lockwood et al., Am. J. Physiol. Endocrinol. Metab., 2003, 285, E927.Bac-to-Bac Baculovirus Expression System (Life Technologies Japan) was used to express human MGAT2 in insect cells (Sf9) (Life Technologies Japan). These cells were sonicated and centrifuged at 100,000 g.times.1 hr to give a precipitate. The precipitate was used as a human MGAT2 enzyme fraction in this assay.This test was conducted using a black flat-bottom 96-well plate (Corning). A buffer solution with final concentrations of 5 mM magnesium chloride, 100 mM sucrose and 100 mM Tris-HCl (pH=7.5) was prepared and test compounds prepared in various concentrations using dimethyl sulfoxide were added thereto to give a final dimethyl sulfoxide concentration of 1%. The MGAT2 enzyme fraction was added thereto to give a final concentration of 0.5 .mu.g/ml. Inhibition Assay The PDE 4 (phosphodiesterase IV) inhibiting effect of the compounds of the present invention was performed by the following method, which was modified assay method described in Biochemical. Pharmacol. 48(6), 1219-1223 (1994). (1) The active fraction of PDE 4 (phosphodiesterase IV) was obtained. That is, the livers obtained from three Balb/c mice (male, 12 weeks: obtainable from CLEA Japan, Inc.) were suspended with 30 mL of buffer solution B [20 mM of bis-tris, 5 mM of 2-mercaptoethnol, 2 mM of benzamidine, 2 mM of EDTA, 0.1 mM of 4-(2-aminoethyl)benzensulfonyl hydrochloride, 50 mM of sodium acetate; pH 6.5], then homogenized by Polytron homogenizer. The homogenate were centrifuged under 25,000xG for 10 minutes at 4 C. The supernatant was separated and thus obtained supernatant was further centrifuged under 100,000xG for 60 minutes at 4 ° C., and then filtrated with 0.2 um filter to obtain the soluble fraction. TSP1 inhibitory activity As referred to herein, the TSP1 inhibitory activity refers to the activity to inhibit one or more effects of TSP1, including angiostatic effect. The TSP1 inhibitory activity is measured by a cell adhesion inhibition assay using human TSP1 and vascular endothelial cells as described hereinbelow in the Examples section. In this assay, when the 50% inhibitory concentration (IC5O) of a test substance is 200 nM or less, the test substance is determined to have TSP1 inhibitory activity. Stimulation of sGC Enzyme Activity To carry out the assay, 29 μl of enzyme solution [0-10 nM soluble guanylate cyclase (prepared according to H nicka et al, J. Mol. Med. 77, 14-23 (1999)) in 50 mM TEA, 2 mM MgC2, 0.1% BSA (fraction V), 0.005% Brij , pH 7.5] are initially introduced into a microplate, and 1 μl of the substance to be tested (as a serially diluted solution in DMSO) is added. The mixture is incubated at room temperature for 10 min. Then 20 μl of detection mix [1.2 nM Firefly Luciferase (Photinus pyralis luciferase, Promega), 29 μM dehydroluciferin (prepared according to Bitler & McElroy, Arch. Biochem. Biophys. 72, 358 (1957)), 122 μM luciferin (Promega), 153 μM ATP (Sigma) and 0.4 mM DTT (Sigma) in 50 mM TEA, 2 mM MgCl2, 0.1% BSA (fraction V), 0.005% Brij , pH 7.5] are added. The enzyme reaction is started by adding 20 μl of substrate solution [1.25 mM guanosine 5′-triphosphate (Sigma) in 50 mM TEA, 2 mM MgC2, 0.1% BSA (fraction V), 0.005% Brij , pH 7.5] and measured continuously in a luminometer. The extent of the stimulation by the substance to be tested can be determined relative to the signal of the unstimulated reaction.The activation of haem-free guanylate cyclase is examined by addition of 25 μM of 1H-1,2,4-oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) to the enzyme solution and subsequent incubation for 30 minutes and compared to the stimulation of the native enzyme. 125I-Leuprorelin Binding Assay The monkey and human membrane fractions prepared were diluted with the assay buffer to yield a 200 g/ml dilution, each of which was then dispensed at 188 μl per tube. To a tube containing the cell membrane fraction of the CHO with monkey GnRH receptors expressed were added 2 μl of a solution of 20 mM compound in 60% DMSO and 10 μl of 38 nM 125I-leuprorelin. To a tube containing the cell membrane fraction of the CHO with human GnRH receptors expressed were added 2 μl of a solution of 2 mM compound in 60% DMSO and 10 μl of 38 nM 125I-leuprorelin. To determine maximum binding quantity, a reaction mixture containing 2 μl of 60% DMSO and 10 μl of 38 nM 125I-leuprorelin was prepared. To determine non-specific binding amount, a reaction mixture containing 2 μl of a solution of 100 μM leuprorelin in 60% DMSO and 10 μl of 38 nM 125I-leuprorelin was prepared. Inhibitory Activity Assay Factor IX is a key component of the plasma system that forms a fibrin clot at a site of vascular injury. The activity of Factor IXa is measured by monitoring the cleavage of the fluorescent peptide, CH3SO2-D-CHG-Gly-Arg-AFC.AcOH ( CHG is cyclohexyl-glycine and AFC is trifluoro aminomethyl coumarin). Factor IXa cleaves the amide bond between Arg and AFC, thereby releasing the AFC fluorophore. The free AFC can be detected with a fluorescence detector at an excitation wavelength of 405 nM and emission wavelength of 510 nM. HDAC Enzyme Activity Assay For the enzyme assay, 10 µL of the enzyme fraction was added to 1 µL of fluorescent substrate (2 mM Ac-KGLGK(Ac)-MCA) and 9 µL of histone deacetylase buffer, and the mixture was incubated at 37 °C for 30 min. The reaction was stopped by the addition of 30 µL of trypsin (20 mg/mL) and incubated at 37 °C for 15 min. The released amino methyl coumarin (AMC) was measured using a fluorescence plate reader. DTT was added simultaneously with drugs to the enzyme solutions Inhibition Assay The kinase activity of purified human MAP4K4 kinase domain was measured by monitoring the phosphorylation of a peptide substrate derived from moesin protein (Leu-Gly-Arg-Asp-Lys-Tyr-Lys-Thr-Leu-Arg-Gln-Ile-Arg-Gln) fluorescently labeled on the N-terminus with 5-carboxyfluorescein using the Caliper LabChip technology (Caliper Life Sciences, Hopkinton, Mass.). To determine inhibition constants (IC50), compounds were serially diluted in DMSO and added to 10 uL kinase reactions containing 1 nM purified MAP4K4 enzyme, 1 uM peptide substrate, 10 uM ATP, 10 mM MgCl2, 1 mM EGTA, 50 mM Hepes pH 7.2, 1 mM DTT, 0.01% Triton X-100, and 2% DMSO. Reactions were incubated at room temperature in Perkin Elmer Proxiplates for 45 minutes and stopped by the addition of 10 uL of an EDTA-containing solution (50 mM Hepes pH 7.2, 40 mM EDTA, 0.02% Triton X-100). The fraction of phosphorylated peptide was determined as a fraction of total peptide substrate using the Caliper Lab Chip 3000 according to the manufacturer's instructions. IC50 values were determined using the four-parameter non-linear fit model. MAP4K4 Inhibition Assay The kinase activity of purified human MAP4K4 kinase domain was measured by monitoring the phosphorylation of a peptide substrate derived from moesin protein (Leu-Gly-Arg-Asp-Lys-Tyr-Lys-Thr-Leu-Arg-Gln-Ile-Arg-Gln) fluorescently labeled on the N-terminus with 5-carboxyfluorescein using the Caliper LabChip technology (Caliper Life Sciences, Hopkinton, Mass.). To determine inhibition constants (IC50), compounds were serially diluted in DMSO and added to 10 uL kinase reactions containing 1 nM purified MAP4K4 enzyme, 1 uM peptide substrate, 10 uM ATP, 10 mM MgCl2, 1 mM EGTA, 50 mM Hepes pH 7.2, 1 mM DTT, 0.01% Triton X-100, and 2% DMSO. Reactions were incubated at room temperature in Perkin Elmer Proxiplates for 45 minutes and stopped by the addition of 10 uL of an EDTA-containing solution (50 mM Hepes pH 7.2, 40 mM EDTA, 0.02% Triton X-100). The fraction of phosphorylated peptide was determined as a fraction of total peptide substrate using the Caliper Lab Chip 3000 according to the manufacturer's instructions. IC50 values were determined using the four-parameter non-linear fit model. STING-Binding Assay The above-mentioned biotinylated STING protein (wild-type (WT)) was prepared by the following method. The culture solution was centrifuged, the obtained fungus bodies were suspended in Lysis Buffer (50 mM TrisHCl, 150 mM NaCl, 20 mM Imidazole, 1 mg/mL Lysozyme, 5 U/mL SEM Nuclease, recombinant, Complete EDTA-free, pH7.6), and the protein was extracted by ultrasonic fragmentation. The reagent was added thereto so that the salt concentration of the extract was adjusted to 300 mM NaCl, and the supernatant was collected by centrifugation. The obtained supernatant was passed through NiNTA superflow Cartridge equilibrated with Wash Buffer (50 mM TrisHCl, 300 mM NaCl, 20 mM Imidazole, pH7.6), and the Cartridge was washed with Wash Buffer, and eluted with Elution Buffer (50 mM TrisHCl, 300 mM NaCl, 250 mM Imidazole, pH7.6). The eluate was passed through HiLoad 26/60 Superdex 200 pg column equilibrated with Storage Buffer (50 mM TrisHCl, 150 mM NaCl, pH7.6), and the eluted fraction was collected as biotinylated His-Avi-SUMO-FLAG-hSTING (139-379, H232R). The protein concentration was measured using BCA protein assay kit, and the fraction was cryopreserved at −80° C. until used. Stimulation of sGC Enzyme Activity Soluble guanylate cyclase (sGC) converts on stimulation GTP into cGMP and pyrophosphate (PPi). PPi is detected with the aid of the assay described below. The signal produced in the assay increases as the reaction progresses and serves as a measure of the sGC enzyme activity under the given stimulation.To carry out the assay, 29 μl of enzyme solution [0-10 nM soluble guanylate cyclase (prepared according to Honicka et al., J. Mol. Med. 77, 14-23 (1999)) in 50 mM TEA, 2 mM MgCl2, 0.1% BSA (fraction V), 0.005% Brij , pH 7.5] are initially introduced into a microplate, and 1 μl of the substance to be tested (as a serially diluted solution in DMSO) is added. The mixture is incubated at room temperature for 10 min. Then 20 μl of detection mix [1.2 nM Firefly Luciferase (Photinus pyralis luciferase, Promega), 29 μM dehydroluciferin (prepared according to Bitler & McElroy, Arch. Biochem. Biophys. 72, 358 (1957)), 122 μM luciferin (Promega), 153 μM ATP (Sigma) and 0.4 mM DTT (Sigma) in 50 mM TEA, 2 mM MgCl2, 0.1% BSA (fraction V), 0.005% Brij , pH 7.5] are added. The enzyme reaction is started by adding 20 μl of substrate solution [1.25 mM guanosine 5′-triphosphate (Sigma) in 50 mM TEA, 2 mM MgCl2, 0.1% BSA (fraction V), 0.005% Brij, pH 7.5] and measured continuously in a luminometer. The extent of the stimulation by the substance to be tested can be determined relative to the signal of the unstimulated reaction.The activation of haem-free guanylate cyclase is examined by addition of 25 μM of 1H-1,2,4-oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) to the enzyme solution and subsequent incubation for 30 minutes and compared to the stimulation of the native enzyme. Activity Assay A human acute lymphoblastic lymphoma T cell line MOLT-4 (which can be purchased under ATCC No. CRL-1582 from ATCC) was cultured in an RPMI1640 medium containing 10% fetal bovine serum to obtain 5108 MOLT-4 cells. The cells were recovered by centrifugation and suspended in 10 ml of buffer solution A (25 mM Tris-HCl, 5 mM 2-mercaptoethanol, 2 mM benzamidine, 2 mM EDTA, and 0.1 mM 4-(2-aminoethyl)benzenesulfonyl hydrochloride, pH 7.5). The cells were homogenized using a Polytron homogenizer and centrifuged (4° C., 25,000 g, 10 minutes). Then, the supernatant was further ultracentrifuged (4° C., 100,000 g, 60 minutes). The obtained supernatant was filtered through a 0.2um filter to obtain a soluble fraction.HiTrap Q HP column (5 ml2, GE Healthcare Japan Corp.) equilibrated with buffer solution A was charged with the obtained soluble fraction. Phosphodiesterase was eluted with 300 ml of buffer solution A containing a linear gradient solution of 0 to 0.8 M NaCl to recover sixty 5-ml fractions. Each fraction was tested for cAMP-metabolizing phosphodiesterase activity. In each fraction, a fraction eluted as an active peak centered around 250 mM NaCl was collected from fractions that had cAMP metabolic activity and did not lose the metabolic activity by 10uM rolipram (PDE4 selective inhibitor) and 10uM milrinone (PDE3 selective inhibitor). In order to confirm whether or not PDE10A mRNA was expressed in the MOLT-4 cells, total RNA was prepared from the MOLT-4 cells according to a standard method and analyzed by RT-PCR using PDE10A gene-specific primers (PDE10A sense primer: 5'-TGCTCCATGGTGGAAGTGGA-3' (SEQ ID NO: 1) and PDE10A antisense primer: 5'-CAACTGGAAGCATGCGGTCA-3' (SEQ ID NO: 2)). As a result, PDE10A mRNA was detected from the total RNA of the MOLT-4 cells. On the other hand, total RNA was prepared from Jurkat cells, one type of human T cell, and similarly subjected to RT-PCR. As a result, PDE10A mRNA was rarely detected. An active peak centered around 250 mM NaCl was not observed in fractions obtained by the treatment of Jurkat cells in the same way as above. Eur J Biochem. 1999, 266 (3), 1118-2 has reported that PDE10A from the rat striatum and testis is eluted as an active peak centered around 250 mM NaCl. 5α-R1 Activity Assay The activity of the 5α-R type 1 isozyme was determined by following the conversion of T to DHT at pH 7.5. 2 nM of [1,2,6,7 3H] T, and different concentrations of unlabeled T (5 × 10^-7-6.3 × 10^-6 M) or DHT (5 × 10^-7-2.7 × 10^-5M) and 2 mM NADPH11 was prepared. The reaction in duplicate was started when 5α-R type 1 isozyme fraction (60 μg protein in a volume of 7.5 μL) was added to the mixtureand incubated at 37°C for 60 min. [3H]-Gabapentin Binding Assay A test compound (20 μL) diluted to 5 times of the final concentration with Binding buffer, [3H]-gabapentin diluted to 100 nmol/L with binding buffer [20 μL (final concentration 20 nmol/L)], and the rat cerebral cortex membrane fraction (60 μL; 12 μg membrane fraction) obtained in the (1) above were added to each well of a 96-well round-bottom plate. After being sufficiently mixed, these were allowed to react at room temperature for 1 hour. After the reaction, the reaction sample was suction filtered using a filter plate with 50 μL/well of 0.3 vol % polyethyleneimine added, and a cell harvester. The filter was then washed with ice-cooled Wash buffer (100 mmol/L NaCl, 0.1 w/v % BSA). After being washed, the filter plate was dried, and a scintillation cocktail (MicroScint-20, purchased from PerkinElmer) was added (50 μL/well), and the radioactivity on the filter was measured. The radioactivity obtained in the absence of the test compound was measured as total binding amount, and the radioactivity obtained by addition of an unlabeled gabapentin (final concentration 100 mmol/L) as a test compound was measured as non-specific binding amount. CYP19 Inhibition Assay The enzyme was obtained from the microsome fraction of fresh human placenta (St. Josephs Krankenhaus, Saarbrucken-Dudweiler, Germany) according to the method of Thompson and Siiteri (Thompson, E. A. & Siiteri, P. K., J. Biol. Chem. 249: 5364-5372 (1974)). The isolated microsomes were suspended in a mini mum volume of phosphate buffer (0.05 M; pH 7.4; 20% glycerol). In addition, DTT (10 mM) and EDTA (1 mM) were added to protect the enzyme from degradation reactions. The protein concentration was determined according to Lowry et al. (Lowry, O. H. et al., J. Biol. Chem. 193: 265-275 (1951)) and should be about 35 mg/ml after the processing. In Vitro Competitive Activity-Based Protein Assay Proteomes (mouse brain membrane fraction for mouse assays; human prefrontal cortex membrane fractions for human assays) (50 mL, 1.0 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37° C. After 30 min, FP-Rh (1.0 mL, 50 mM in DMSO) was added and the mixture was incubated for another 30 min at 37° C. Reactions were quenched with SDS loading buffer (15 μL-4×) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL and FAAH using ImageJ 1.43u software. In Vitro Competitive Activity-Based Protein Profiling Assay MAGL, ABHD6 and FAAH: Proteomes (mouse brain membrane fraction or cell lysates) (50 μL, 1.0 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37° C. After 30 min, FP-Rh (1.0 μL, 50 μM in DMSO) was added and the mixture was incubated for another 30 min at 37° C. Reactions were quenched with SDS loading buffer (50 μL-4X) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL, ABHD6 and FAAH using ImageJ 1.43 u software. Receptor Binding Assay A prepared WP was homogenated and a membrane fraction was collected with high-speed centrifugation. A compound of the present invention was added to the plate and [3H]-PGD2 was also added. A platelet membrane, a protein concentration is 2 mg/mL, was added and mixed in the plate, and placed on ice for 2 hours. The reaction solution was transferred to a low protein-adsorptive filter and washed with a wash solution eight times using a cell harvester. After the final washing, water was removed sufficiently, and scintillator was added. DP inhibitory activity was investigated by measuring [3H] by using Micro Beta. [3H]-NMS Binding Assay Assays were carried out at 25° C. Membrane preparations from stably transfected chinese hamster ovary-K1 cells (CHO) expressing the genes for the human muscarinic receptors Hm3 were used. For determination of IC, membrane preparations were suspended in DPBS to a final concentration of 89 μg/ml for the Hm3 subtype. The membrane suspension was incubated with the tritiated compound for 60 min. After incubation the membrane fraction was separated by filtration and the bound radioactivity determined. Non specific binding was determined by addition of 10^−4 M atropine. At least six concentrations were assayed in duplicate to generate individual displacement curves. In vitro kinase inhibition assay An in vitro kinase assay was performed to evaluate the kinase suppression activity of the most promising cytotoxic candidates 4b, 4j against four different receptor tyrosine kinases (RTKs) - namely epidermal growth factor receptor (EGFR), human epidermal growth factor receptor (HER2), vascular endothelial growth factor receptor-2 (VEGFR-2) and platelet-derived growth factor receptor (PDGFR). The activities of the examined compounds against EGFR, HER2, PDGFR-β, and VEGFR2 were in vitro tested using Abcam's Human In cell ELISA Kit (ab 126419) for EGFR, ADP-Glo TM Kinase Assay for Her2, PDGFR-β, active, recombinant protein expressed in Sf9 cells for VEGFR2 (KDR) Kinase Assay Kit Catalog No. 40325, respectively. ERG Channel Binding Assay Fluorescence polarization assay based on the principle of fluorescence polarization where a red-shifted fluorescent tracer is displaced from the hERG channel by compounds that bind to the channel was used to establish the binding of the compounds to hERG channel. Briefly, the compounds in serial dilutions were incubated with a membrane fraction containing hERG channel protein and a high-affinity red fluorescent hERG channel ligand in black 384-well plate. The reaction was incubated for 4 h at room temperature followed by the measurement of the fluorescence polarization using Tecan Spark multimode plate reader at excitation wavelength of 535 nm and emission wavelength 590 nm. The IC50 values were calculated using GraphPad Prism. In Vitro Competitive Activity-Based Protein Assay Proteomes (mouse brain membrane fraction or cell lysates for mouse assays; human prefrontal cortex or cell membrane fractions for human assays) (50 μL, 1.0 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37° C. After 30 min, FP Rh or HT-01 (1.0 μL, 50 μM in DMSO) was added and the mixture was incubated for another 30 min at 37° C. Reactions were quenched with SDS loading buffer (15 μL-4×) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL and FAAH using ImageJ 1.43u software. In Vitro Competitive Activity-Based Protein Profiling Assay Proteomes (mouse brain membrane fraction or cell lysates for mouse assays; human prefrontal cortex or cell membrane fractions for human assays) (50 μL, 1.0 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37° C. After 30 min, FP Rh or HT-01 (1.0 μL, 50 μM in DMSO) was added and the mixture was incubated for another 30 min at 37° C. Reactions were quenched with SDS loading buffer (15 μL 4×) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL using ImageJ 1.43u software. In Vitro Competitive Activity-Based Protein Profiling Proteomes (mouse brain membrane fraction or cell lysates) (50 μL, 1.0 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37° C. After 30 min, FP Rh (1.0 μL, 50 μM in DMSO) was added and the mixture was incubated for another 30 min at 37° C. Reactions were quenched with SDS loading buffer (50 μL-4×) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL, ABHD6 and FAAH using ImageJ 1.43u software. Data from this assay is shown in Table 2 (% inhibition at 1 μM). K-RAS Inhibition Assay Table 7: Potency and efficacy of K-RAS inhibition and toxicity ofbatch 5 compounds. MDCK cells co-expressing GFP-K-RASG12V andmCherry-CAAX were grown on coverslips, treated with 0.1% vehicle (DMSO)or various concentrations of batch 5 compounds for 48 h, and fixed with 4%paraformaldehyde. The coverslips were mounted in mowiol and imaged byconfocal microscopy (Nikon A1) using a 60× objective. Using ImageJ softwarev1.42q, images were converted to 8-bit, and a threshold to a control pixel of eachimage was set. As a measure of K-Ras mislocalization, the fraction of mCherry-CAAX co-localizing with mGFP-K-RASG12V was calculated using a Manderscoefficient plugin downloaded from Wright Cell Image Facility. [35S]GTPgammaS Binding Assay The frozen cell membrane fraction prepared as described above was thawed before use, and the resultant was diluted with a buffer for a binding assay (final concentrations: 20 mM HEPES, 100 mM NaCl, 1 mM MgCl2, 3 uM GDP, 300 ug/mL saponin, 0.1% BSA). The compound of each example was added to a cell membrane fraction containing 1.8 to 3 ug/assay of membrane protein on a plate, followed by incubation at room temperature for 30 minutes. Thereafter, with glutamic acid (in a final concentration of 10 uM) added thereto, incubation was performed at room temperature for 15 minutes, and thereafter, [35S]GTPγS (in a final concentration of 0.8 kBq) and 588 ug WGA-SPA beads were added thereto, followed by incubation at room temperature for 1 hour. After the incubation, the plate was subjected to the centrifugal separation at 2,500 rpm and room temperature, and then, membrane cell binding radioactivity was measured with a top count. A [35S]GTPγS binding amount obtained in the presence of glutamic acid was defined as specific binding. On the basis of ratios of inhibiting the specific binding at various concentrations of the compounds of the respective examples, inhibition curves were obtained. Concentrations of the compounds of the respective examples at which the specific [35S]GTPγS binding amount was suppressed by 50% (IC50 values) were calculated on the basis of the inhibition curves. K-RAS Inhibition Assay Table 1: Potency and efficacy of K-RAS inhibition and toxicity of batch1 and 2 compounds. MDCK cells co-expressing GFP-K-RASG12V and mCherry-CAAX were grown on coverslips, treated with 0.1% vehicle (DMSO) or variousconcentrations of drugs (A; batch 1, B; batch 2) for 48 h, and fixed with 4%paraformaldehyde. The coverslips were mounted in mowiol and imaged byconfocal microscopy (Nikon A1) using a 60× objective. Using ImageJ softwarev1.42q, images were converted to 8-bit, and a threshold to a control pixel of eachimage was set. As a measure of K-Ras mislocalization, the fraction of mCherry-CAAX co-localizing with mGFP-K-RASG12V was calculated using a Manderscoefficient plugin downloaded from Wright Cell Image Facility. LCAT Activity Measurement (In Vitro) A fraction composed of HDL3 (1.125 Measurement of the Inhibitory Action of the Compound of the Present Invention on Binding of [33P]-S1P to S1P5 (EDG-8) Compound of the Present Invention on Binding of [33P]-S1P to S1P5 (EDG-8)A reaction was carried out in a 96-well microplate by using membrane fractions of Chinese hamster ovary (CHO) cells each of which was made to overexpress human S1P1 (EDG-1) or human S1P5 gene respectively, in an amount of the membrane fraction of 1 mg protein/mL. To each of the wells, 100 μL of a vehicle (DMSO) solution or a two-fold concentration-ligand solution each of which was diluted with Binding Buffer (50 mmol/L, Tris pH 7.5, 5 mmol/L, MgCl2, 0.5% BSA and Complete EDTA free (1 tablet/50 mL)), and 50 μL of 0.16 nmol/L [33P]-S1P (manufactured by American Radiolabeled Chemicals, Inc.) diluted with Binding Buffer were added. Thereafter, the membrane fraction solutions (50 μL) was added to the wells and the reaction was carried out at room temperature for 60 minutes. After the reaction, suction filtration was carried out by using a 96-well UNIFILTER, and the 96-well microplate was washed with Wash Buffer (50 mmol/L, Tris pH 7.5, 0.5% BSA) (150 mL), and thereafter, was dried at 60° C. for 45 minutes. MicroScint (trade name) 20 (50 μL/well) was added and the plate was covered with TopSeal-A, and thereafter, the radioactivity was measured by using TopCount (manufactured by PerkinElmer Inc.). Measurement of Human MGAT2 Inhibitory Activity A full-length human MGAT2 gene to which a Flag-tag had been added at the N-terminal was inserted into pFastBac (from Invitrogen). A recombinant baculovirus was produced in accordance with the protocol for a Bac-to-Bac baculovirus expression system (produced by Invitrogen), and Sf-9 cells were infected therewith. The cells were collected and sonicated, and then the membrane fraction was collected through centrifugation. Western blotting analysis with an anti-Flag antibody was performed for the membrane fraction to confirm expression, and the membrane fraction was used as a recombinant human MGAT2 enzyme solution. Solutions of the compounds of the present invention in DMSO were each aliquoted into 0.2-μL portions in a 384-well polystyrene microplate produced by Corning Incorporated, and 5 μL of an enzyme solution prepared with an assay buffer (100 mmol/L phosphate buffer (pH 7.4) containing 2 mmol/L DTT) and 5 μL of a substrate solution (100 mmol/L phosphate buffer (pH 7.4), 30 μmol/L 2-Oleoylglycerol, 10 μmol/L Oleoyl-CoA) were added thereto, and the resultant was stirred and centrifuged, and incubated in a moist chamber at room temperature for 1 hour. After enzymatic reaction, 50 μL of a quenching solution (containing 0.2 μmol/L Diolein-d5, 0.4% formic acid, and 50% isopropanol) containing Internal Standard (IS) was added to terminate the reaction, and the resultant was sealed in a plate produced by Shimadzu GLC Ltd., and then stirred and centrifuged, and measurement was performed by using an electrospray ionization method with a RapidFire360 and Agilent 6550 Q-TOF mass spectrometer. Diolein as a reaction product (P) of 2-Oleoylglycerol as the substrate and an ammonium adduct ion of the IS were detected, and the peak intensity ratio, P/IS, was calculated from the peak heights to evaluate the inhibitory activity. Inhibitory activities with/without addition of enzyme were defined as Control (+)/Control (−), respectively, and the respective % inhibitions were defined as 0% inhibition and 100% inhibition. The inhibitory activity was calculated from the formula below with TIBCO Spotfire (produced by TIBCO Software Inc.):Inhibitory activity (%)=[1−{Sample−Control(−)}/{Control(+)−Control(−))}]*100where Sample indicates a peak intensity ratio: P/IS, when the compound of the present invention was added. Determination of Inhibitory Activity Against Factor IXa Formation of a clot to stem bleeding at a site of blood vessel injury involves the coordinated activity of a group of plasma proteins that initiate and propagate fibrin formation and subsequently protect fibrin from premature degradation. Factor IX is a key component of the plasma system that forms a fibrin clot at a site of vascular injury. The activity of Factor IXa is measured by monitoring the cleavage of the fluorescent peptide, CH3SO2-D-CHG-Gly-Arg-AFC.AcOH (CHG is cyclohexyl-glycine and AFC is trifluoro aminomethyl coumarin). Factor IXa cleaves the amide bond between Arg and AFC, thereby releasing the AFC fluorophore. The free AFC can be detected with a fluorescence detector at an excitation wavelength of 405 nM and emission wavelength of 510 nM. Biochemical Assay 3 This assay quantifies human SOS1-mediated loading of human K-RasG12C-GDP with a fluorescent GTP-analog. Detection of successful loading is achieved by measuring homogenous time-resolved fluorescence resonance energy transfer (HTRF) from antiGST-Terbium (FRET donor) bound to GST-K-RasG12C to the loaded fluorescent GTP analog (FRET-acceptor).The fluorescent GTP-analog EDA-GTP-Dy647P1 (2′/3′-O-(2-Aminoethyl-carbamoyl)-guanosine-5′-triphosphate labelled with Dy647P1 (Dyomics GmbH, Germany)) was synthesized by Jena Biosciences GmbH (Germany) and supplied as a 1 mM aqueous solution. The assay buffer contained 10 mM HEPES pH 7.4 (Applichem), 150 mM NaCl (Sigma), 5 mM MgCl2 (Sigma), 1 mM DTT (Thermofisher), 0.05% BSA Fraction V, pH 7.0, (ICN Biomedicals), 0.0025% (v/v) Igepal (Sigma). In Vitro Competitive Activity-Based Protein Profiling Proteomes (mouse brain membrane fraction for mouse assays; human prefrontal cortex membrane fractions for human assays) (50 mL, 1.0 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37° C. After 30 min, FP-Rh (1.0 mL, 50 mM in DMSO) was added and the mixture was incubated for another 30 min at 37° C. Reactions were quenched with SDS loading buffer (15 μL-4×) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL and FAAH using ImageJ 1.43u software. IC50 data from this assay is shown in Table 1. All compounds in Table 1 were more potent inhibitors of MAGL than FAAH. In Vitro Competitive Activity-Based Protein Profiling Proteomes (mouse brain membrane fraction or cell lysates for mouse assays; human prefrontal cortex or PC3 cell membrane fractions for human assays) (50 μL, 1.0 or 2.0 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37° C. After 30 min, FP-Rh or JW912 or HT-01 (1.0 μL, 50 μM in DMSO) was added and the mixture was incubated for another 30 min at 37° C. Reactions were quenched with SDS loading buffer (15 μL-4×) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL and FAAH using ImageJ 1.43u software. IC50 data from this assay is shown in Table 1. BCA Protein Assay Inhibitory activity evaluation was performed by multi-screen (registered trademark) HTS 96-well plate (Millipore) using the cell membrane fraction stably expressing S1P1 obtained by the above operation. 25 μL of assay solution (50 mM Tris, 100 mM sodium chloride, 5 mM magnesium chloride, 1 mM EDTA, 1 mM DTT (dithiothreitol), 10 μM GDP (guanosine diphosphate) and 0.5% BSA (bovine serum albumin), pH 7.4), 25 μL of test compounds solution diluted by assay solution and 25 μL of the membrane fraction (0.2 μg protein/μL) were added to each well, and the mixture was gently shaken at room temperature: for 30 minutes. Then, 25 μL of 35S-GTP, and 25 μL of 50 nM S1P1 receptor selective agonist (1-[4-(4-phenyl-5-trifluoromethylthiophen-2-ylmethoxy)benzyl]azetidine-3-carboxylic acid, J. Med. Chem. , 2004, vol. 47, pp. 6662-6665, compound 18) were added to each well, respectively, and it was reacted at room temperature for 60 minutes. After the reaction, a reaction solution of each well was suction-filtered. After suction filtration, ice-colded wash solution (50 mM Tris, 100 mM sodium chloride, 5 mL magnesium chloride and 1 mM EDTA, pH 7.4) was added to each well, and moreover filters were washed by suction filtration. This wash procedure was performed three times. A bottom of the plate containing the filters was dried at 60° C. After drying, 30 μL of MicroScinti-40 (PerkinElmer) was added to each well, and the radioactivity adsorbed on the filter was determined by TopCount NXT (registered trademark) (PerkinElmer) after shaking for 30 minutes at room temperature. Inhibitory Effect on hCYP11B2 The pcDNA3.1-human CYP11B2 plasmid was transfected into a Chinese hamster lung fibroblast V79 cell line to produce a cell line stably expressing human CYP11B2 gene.The cells were cultured and grown in the Dulbecco's modified Eagle's/Ham's medium supplemented with 10% fetal bovine serum and 1% G418 disulfate solution under the environment of 37° C., 95% air, and 5% CO2, and the grown cells were harvested.Then, the cells were fractionated to obtain mitochondria by reference to a method described in Chabre et al. JCE 86 M 85 (11) 4060-68, 2000. In particular, the cells suspended in a 5 mmol/L Tris-HCl buffer (pH 7.4) containing 250 mmol/L sucrose were homogenized in a Teflon (Registered Trademark) Potter Elvehjem homogenizer, and then the suspension was centrifuged (800×g, 15 min.). The supernatant was separated and again centrifuged (10000×g, 15 min.) to obtain a pellet (mitochondrial fraction).The mitochondrial fraction diluted with a buffer containing 10 mmol/L KH2PO4, 10 mmol/L Tris, 20 mmol/L KCl, 25 mmol/L sucrose, 5 mmol/L MgCl2, and 0.05% bovine serum albumin was dispensed to a 96-well plate. 0.5 μmol/L Deoxycorticosterone, 150 μmol/L NADPH and a compound of each concentration were added to each well, and incubated for 1.5-2 hours at room temperature to produce aldosterone. An amount of the produced aldosterone in the incubated solution was determined by using HTRF (Homogeneous Time Resolved Fluorescence) method.IC 50 (nmol/L) was calculated by analyzing the aldosterone production inhibition rate (%) of each concentration of compounds by non-linear regression to a logistic curve. GTPγS binding test CHO cells stably expressing human metabotropic glutamate receptors mGluR2 and mGluR3 were cultured at 37° C. under 5% CO2 using a Dulbecco's modified Eagle medium [1% proline, 1 mM sodium pyruvate, 1 mM succinic acid, 1 mM disodium succinate, 100 units/mL penicillin, 100 μg/mL streptomycin, 400 (mGluR2) or 300 (mGluR3) μg/mL hygromycin B, 2 mM L-glutamine (added just before use)] containing 10% dialyzed fetal bovine serum. The cells in a confluent state were washed with PBS(−), then dissociated using a cell scraper, and centrifuged at 1000 rpm for 5 minutes at 4° C. to recover the cells. The obtained pellet was suspended in a 20 mM HEPES buffer (pH 7.4) (mGluR2) or 20 mM HEPES buffer containing 1 mM EDTA (pH 7.4) (mGluR3), and the suspension was homogenized in a Teflon homogenizer and then centrifuged at 48,000×g for 20 minutes at 4° C. to obtain a pellet again. The obtained pellet was subjected to two additional cycles of washing and centrifugation and then homogenized with the buffer described above to obtain a crude membrane fraction. The crude membrane fraction was diluted with a buffer for a binding test (final concentration: 20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, 10 μM GDP, 10 ng/mL saponin, 0.1% BSA) (mGluR2) or (final concentration: 20 mM HEPES, 1 mM EDTA, 100 mM NaCl, 10 mM MgCl2, 10 μM GDP, 10 μg/mL saponin, 0.1% BSA) (mGluR3). To the crude membrane fraction containing 10 μg of membrane proteins/assay, Compounds (II)-1 to (II)-15 were each added, and the mixture was incubated at 30° C. for 20 minutes. Then, glutamate (final concentration: 20 (mGluR2) or 1 (mGluR3) μM) and [35S]GTPγS (final concentration: 0.15 nM) were added thereto, and the mixture was incubated at 30° C. for 1 hour. The solution thus incubated was filtered by suction onto Whatman GF/C filter, and the filter was washed with 1000 μL of an ice-cooled 20 mM HEPES buffer (pH 7.4) (mGluR2) or 20 mM HEPES buffer containing 1 mM EDTA (pH 7.4) (mGluR3). A scintillation cocktail was added to the obtained filter, and the membrane binding radioactivity was measured using a liquid scintillation counter. The residual radioactivity in the absence of glutamate was defined as nonspecific binding, and the difference from the residual radioactivity in the presence of glutamate was defined as specific binding. Counterscreen for S1P2 Antagonists: Dose Response Cell-Based Screen to Identify Antagonists of CRE-BLA Source (MLSCN Center Name): The Scripps Research Institute Molecular Screening Center Center Affiliation: The Scripps Research Institute (TSRI) Assay Provider: Hugh Rosen, TSRI Network: Molecular Library Screening Center Network (MLSCN) Grant Proposal Number: 1 R03 MH076533-01 Grant Proposal PI: Germana Sanna External Assay ID: CRE _ANT_BLA_384_IC50_Counterscreen_ S1P2_Hits Name: Counterscreen for S1P2 Antagonists: Dose Response Cell-Based Screen to Identify Antagonists of CRE-BLA Description: Sphingosine 1-phosphate (S1P) influences heart rate [1,2], coronary artery caliber, endothelial integrity, lung epithelial integrity [3] and lymphocyte recirculation [2,4] through five related high affinity G-protein coupled receptors [5]. Subtype-selective modulators of S1P receptors will be of broad utility in understanding cell functions in vitro and vascular physiology in vivo, as well as de-convoluting the role of individual subtypes in cellular processes. The S1P receptor 2 (S1P2) Dose Response Cell Based Assay for Antagonists of the S1P2 Receptor Source (MLSCN Center Name): The Scripps Research Institute Molecular Screening Center Center Affiliation: The Scripps Research Institute (TSRI) Assay Provider: Hugh Rosen, TSRI Network: Molecular Library Screening Center Network (MLSCN) Grant Proposal Number: 1 R03 MH076533-01 Grant Proposal PI: Germana Sanna External Assay ID: S1P2_ANT_BLA_384_IC50 Name: Dose Response Cell Based Assay for Antagonists of the S1P2 Receptor Description: Sphingosine 1-phosphate (S1P) influences heart rate [1,2], coronary artery caliber, endothelial integrity, lung epithelial integrity [3] and lymphocyte recirculation [2,4] through five related high affinity G-protein coupled receptors [5]. Subtype-selective modulators of S1P receptors will be of broad utility in understanding cell functions in vitro and vascular physiology in vivo, as well as de-convoluting the role of individual subtypes in cellular processes. The S1P receptor 2 (S1P2), also known as endothelial differentiation sphingolipid G-pro Estrogen Receptor (alpha) binding: Dose Response of Primary Screen Assay University of New Mexico Assay Overview: Assay Support: 1X01 MH077627-01 Assay for Ligands of GPR30 and Classical Estrogen Receptors PI: Eric Prossnitz, PhD Assay Development: Megan Dennis Assay Implementation: Megan Dennis, Mark Haynes, PhD Target Team Leader for the Center: Eric Prossnitz, PhD (EProssnitz@salud.unm.edu) Assay Background and Significance: The physiological effects of estrogen are diverse and numerous, with roles in growth, development and homeostasis of numerous tissues. Roles for estrogen in mammalian female reproductive development are among the best defined, but estrogen also plays a part in regulation of skeletal cancer, (cardio)vascular function, the central nervous system as well as in the immune system. Stimulation with estrogen induces many signaling pathways, leading to an array of cellular responses including adhesion, migration, survival, proliferation and angiogenesis in both normal and neoplastic tissues [Edwards et al., 2005]. Effects of estrogen Estrogen Receptor (beta) binding: Dose Response of Primary Screen Assay University of New Mexico Assay Overview: Assay Support: 1X01 MH077627-01 Assay for Ligands of GPR30 and Classical Estrogen Receptors PI: Eric Prossnitz, PhD Assay Development: Megan Dennis Assay Implementation: Megan Dennis, Mark Haynes, PhD Target Team Leader for the Center: Eric Prossnitz, PhD (EProssnitz@salud.unm.edu) Assay Background and Significance: The physiological effects of estrogen are diverse and numerous, with roles in growth, development and homeostasis of numerous tissues. Roles for estrogen in mammalian female reproductive development are among the best defined, but estrogen also plays a part in regulation of skeletal cancer, (cardio)vascular function, the central nervous system as well as in the immune system. Stimulation with estrogen induces many signaling pathways, leading to an array of cellular responses including adhesion, migration, survival, proliferation and angiogenesis in both normal and neoplastic tissues [Edwards et al., 2005]. Effects of estrogen Androgen Receptor Binding Assay Androgen receptor (AR) binding affinities of test compounds were studied in cytosolic lysates obtained from ventral prostates of castrated rats by competition binding assay (Schilling K. and Liao S., The Prostate, 1984; 5(6):581-588). Cytosol preparations and 1 nM [3H]mibolerone were incubated with increasing concentrations of test compounds. To determine non-specific binding, parallel incubations were carried out using excess of unlabelled testosterone. After incubation, bound and free steroids were separated by treatment with dextran-coated charcoal suspension. Bound radioactivity was determined by counting of supernatant fraction in scintillation fluid. Radioactivity was measured using a Microbeta counter. All data points were done as quadrublicates. Dissociation constant of [3H]mibolerone for rat androgen receptor was determined by saturation binding assay obtained from ventral prostates of castrated rats essentially as described (Isomaa V. et al., Endocrinology, 1982; 111(3):833-843). Binding Assay Androgen receptor (AR) binding affinities of test compounds were studied in cytosolic lysates obtained from ventral prostates of castrated rats by competition binding assay (Schilling K. and Liao S., The Prostate, 1984; 5(6):581-588). Cytosol preparations and 1 nM [3H]mibolerone were incubated with increasing concentrations of test compounds. To determine non-specific binding, parallel incubations were carried out using excess of unlabelled testosterone. After incubation, bound and free steroids were separated by treatment with dextran-coated charcoal suspension. Bound radioactivity was determined by counting of supernatant fraction in scintillation fluid. Radioactivity was measured using a Microbeta counter. All data points were done as quadrublicates. Dissociation constant of [3H]mibolerone for rat androgen receptor was determined by saturation binding assay obtained from ventral prostates of castrated rats essentially as described (Isomaa V. et al., Endocrinology, 1982; 111(3):833-843). Biochemical Assay 2 Preparation of GST-tagged hK-RasGl2C loaded with fluorescent nucleotide was performed as follows: incubation of 11.5 μM hK-RasGl2C with 5-fold excess GDP-Dy647 nucleotide (54 μM) in 500 μl NLS-buffer (RAS activation Kit Jena Bioscience, Kat. #PR-950) for 10 min at 37° C. Addition of 20 μl 1 M MgCl2 (Sigma) to final 40 mM and store on ice. Purification into buffer (10 mM HEPES pH 7.4 (Applichem), 150 mM NaCl (Sigma), 5 mM MgCl2 (Sigma)) by use of a PD-Minitrap desalting column (GE Healthcare). Concentration of 1 ml purified hK-Ras-GDP-Dy647 is approx. 4-5 μM.The assay buffer contained 10 mM HEPES pH 7.4 (Applichem), 150 mM NaCl (Sigma), 5 mM MgCl2 (Sigma), 1 mM DTT (Thermofisher), 0.05% BSA Fraction V, pH 7.0, (ICN Biomedicals), 0.0025% (v/v) Igepal (Sigma). Enzymatic Assay HDAC5 reagents: N-terminal GST tagged HDAC5 was purchased from BPS Bioscience [catalog #50045]. Assays were performed with buffer containing 20 mM HEPES, pH 8.0 [Boston BioProducts, catalog #BB-104, 1M stock], 137 mM NaCl [Sigma, catalog #S5150, 5M stock], 2.7 mM KCl [BioChemika, catalog #87526, 4M stock], 1 mM MgCl2 [Fluka, catalog #63020, 1M stock], and 0.05% BSA (Fraction V) [Invitrogen, catalog #15260, 7.5% stock]. The HDAC5 enzyme was run at the final concentration of 0.447 nM. Boc-Lys(TFA)-AMC substrate [Bachem, catalog #1-1985.0050], used to evaluate enzyme activity, was added at the final concentration of 60 uM. To enable detection of the signal, Developer II [BioMol Research Laboratories, catalog #KI-176] was added at a 1:200 dilution to the stop solution, which also included 20 uM trichostatin A (TSA) [Sigma, catalog #T8552] to ensure complete termination of the reaction. Fluorescence-based biochemical high throughput dose response assay for activators of the calcium sensitivity of cardiac Regulated Thin Filaments (RTF) Source (MLPCN Center Name): The Scripps Research Institute Molecular Screening Center (SRIMSC) Affiliation: The Scripps Research Institute, TSRI Assay Provider: James D. Potter, University of Miami School of Medicine Network: Molecular Library Probe Production Centers Network (MLPCN) Grant Proposal Number: 1 R21 NS064821-01 Grant Proposal PI: James D. Potter, University of Miami School of Medicine External Assay ID: RTF_ACT_FLINT_1536_3XEC50 DRUN SENS Name: Fluorescence-based biochemical high throughput dose response assay for activators of the calcium sensitivity of cardiac Regulated Thin Filaments (RTF). Description: Cardiomyopathies are myocardial diseases that often lead to cardiac remodeling to compensate for deficiencies in cardiac output (1). Cardiomyopathies are characterized as having systolic dysfunctions (i.e. reduced ejection fraction) in dilated cardiomyopathy or diastolic dysfunctions (i.e. impaired relaxation) in hypertrophic and restrictive cardiomyopathies (2). The Fluorescence-based biochemical high throughput dose response assay for inhibitors of the calcium sensitivity of cardiac Regulated Thin Filaments (RTF) Source (MLPCN Center Name): The Scripps Research Institute Molecular Screening Center (SRIMSC) Affiliation: The Scripps Research Institute, TSRI Assay Provider: James D. Potter, University of Miami School of Medicine Network: Molecular Library Probe Production Centers Network (MLPCN) Grant Proposal Number 1 R21 NS064821-01 Grant Proposal PI: James D. Potter, University of Miami School of Medicine External Assay ID: RTF_INH_FLINT_1536_3XIC50 DRUN DESENS Name: Fluorescence-based biochemical high throughput dose response assay for inhibitors of the calcium sensitivity of cardiac Regulated Thin Filaments (RTF). Description: Cardiomyopathies are myocardial diseases that often lead to cardiac remodeling to compensate for deficiencies in cardiac output (1). Cardiomyopathies are characterized as having systolic dysfunctions (i.e. reduced ejection fraction) in dilated cardiomyopathy or diastolic dysfunctions (i.e. impaired relaxation) in hypertrophic and restrictive cardiomyopathies (2). Th Fluorescent Assay hABHD6:Initial Fluorescent Inhibition Assay (3-Point) In each well of a 96-well plate 8 μL of membrane fraction containing full-length hABHD6 (1 μg total protein) was mixed with 168 μL of assay buffer (50 mM Tris-HCl, pH 7.6), and 20 μL of diluted compound (diluted in a dilution buffer consisting of 50% DMSO/50% assay buffer v/v). Each plate was incubated at RT for 15 minutes before adding 4 μL of 1 mM AHMMCE substrate (final concentration of 20 μM AHMMCE and final volume of 200 μL). The reaction was allowed to proceed for 1 hr at RT before the fluorescence was read at λex 360 nm and λem 460 nm and the inhibition calculated. Each experiment can test 8 compounds at three concentrations (usually 10 μM, 1 μM, and 100 nM). Inhibition Assay Assay plates: 96-well MultiScreen 0.65 um filter plates (Millipore Cat. No.: MADVNOB10)Streptavidin coated beads: Streptavidin Sepharose, suspension 5.0 mL, in 50 mM EDTA/PBS diluted (1:100), (Amersham, Cat. No.: 17-5113-01)Compounds: 10 mM in 100% dimethylsulfoxide (DMSO), final conc.: compound 0.003-100 uM in 10% DMSOEnzyme: SYK RPA purified, truncated construct of Spleen Tyrosine Kinase aa 360-635, stock solution 1 mg/mL, MW: 31.2 KDa, final conc.:0.0005 uM. Peptide 1: biotinylated peptide is derived from a naturally occurring phosphor-acceptor consensus sequence (Biotin-EPEGDYEEVLE), special order from QCB, stock solution 20 mM, final conc.: 5.0 uM.ATP: Adenosine-5'-triphosphate 20 mM, (ROCHE Cat. No.: 93202720), final concentration: 20 uM Buffer: HEPES: 2-Hydroxyethyl piperazine-2-ethanesulfonic acid (Sigma, Cat. No.: H-3375)final concentration: 50 mM HEPES pH7.5BSA: Bovine Serum Albumin Fraction V, fatty acid free (Roche Diagnostics GmbH, Cat. No. 9100221). Probe Displacement Assay The probe displacement assay is conducted as follows: In a 384-well plate, test compound along with recombinantly expressed His-tagged protein corresponding to amino acids 556-871 of human TYK2 (sequence shown below) at 5 nM, 33 nM probe (BB, preparation described below) and 0.33 mg/mL Yttrium oxide (YOx) His-capture SPA beads (PerkinElmer, RPNQ0276) in IX PBS containing 10 mM MgCl2, 0.1 mg/mL BSA, 0.1 mM TCEP, 0.005% Tween-20, and 1% DMSO (final concentration) were incubated for 10 hours at room temperature. The amount of radiolabeled probe bound to TYK2 was then quantified using a ViewLux Microplate Imager, and the fraction bound by the test compound calculated by comparison to wells containing scale references for 0% and 100% probe displacement. The IC50 value is defined as the concentration of test compound required to inhibit radiolabeled probe binding by 50%. Proton Potassium-adenosine Triphosphatase (H+,K+-ATPase) Inhibitory Activity According to the method [Biochem. Biophys. Acta., 728, 31 (1983)] of Wallmark et al., a gastric mucous membrane microsomal fraction was prepared from the stomach of swine. First, the stomach was removed, washed with tap water, immersed in 3 mol/L brine, and the surface of the mucous membrane was wiped with a paper towel. The gastric mucous membrane was detached, chopped, and homogenized in a 0.25 mol/L saccharose solution (pH 6.8) containing 1 mmol/L EDTA and 10 mmol/L tris-hydrochloric acid using polytron (Kinematica). The obtained homogenate was centrifuged at 20,000×g for 30 min and the supernatant was centrifuged at 100,000×g for 90 min. The precipitate was suspended in 0.25 mol/L saccharose solution, superimposed on a 0.25 mol/L saccharose solution containing 7.5% Ficoll, and centrifuged at 100,000×g for 5 hr. The fraction containing the interface between the both layers was recovered, and centrifugally washed with 0.25 mol/L saccharose solution.The obtained microsomal fraction was used as a proton, potassium-adenosine triphosphatase standard product.To 40 μL of a 50 mmol/L HEPES-tris buffer (5 mmol/L magnesium chloride, 10 mmol/L potassium chloride, 10 μmol/L valinomycin, pH=6.5) containing 2.5 μg/mL (based on the protein concentration) of the enzyme standard product was added a test compound (5 μL) dissolved in a 10% aqueous dimethyl sulfoxide solution, and the mixture was incubated at 37° C. for 30 min. The enzyme reaction was started by adding 5 μL of a 2 mmol/L adenosine triphosphate tris salt solution (50 mmol/L HEPES-tris buffer (5 mmol/L magnesium chloride, pH 6.5)). The enzyme reaction was carried out at 37° C. for 20 min, and 15 μL of a malachite green solution (0.12% malachite green solution in sulfuric acid (2.5 mol/L), 7.5% ammonium molybdate and 11% Tween 20 were mixed at a ratio of 100:25:2) was added to quench the reaction. After allowing to stand at room temperature for 15 min, the resulting reaction product of inorganic phosphorus with malachite green was colorimetrically determined at a wavelength of 610 nm. In addition, the amount of the inorganic phosphoric acid in the reaction solution free of potassium chloride was measured in the same manner, which was subtracted from the inorganic phosphoric acid amount in the presence of potassium chloride to determine the proton, potassium-adenosine triphosphatase activity. The inhibitory rate (%) was determined from the activity value of the control and the activity values of various concentrations of the test compound, and the 50% inhibitory concentration (IC50) of the proton, potassium-adenosine triphosphatase was determined. Counter screen for S1P2 Agonists: Dose Response High Throughput Cell-Based Screen to Identify Activators of CRE-BLA Source (MLSCN Center Name): The Scripps Research Institute Molecular Screening Center Center Affiliation: The Scripps Research Institute (TSRI) Assay Provider: Hugh Rosen, TSRI Grant Proposal Number: 1 R03 MH076533-01 Grant Proposal PI: Germana Sanna External Assay ID: Counterscreen for S1P2 Agonists: Dose Response High Throughput Cell-Based Screen to Identify Activators of CRE-BLA Name: CRE_AG_BLA_384_EC50_%ACT Description: Sphingosine 1-phosphate (S1P) influences heart rate [1,2], coronary artery caliber, endothelial integrity, lung epithelial integrity [3] and lymphocyte recirculation [2,4] through five related high affinity G-protein coupled receptors [5]. Subtype-selective modulators of S1P receptors will be of broad utility in understanding cell functions in vitro and vascular physiology in vivo, as well as de-convoluting the role of individual subtypes in cellular processes. The S1P receptor 2 (S1P2), also known as endothelial differentiation sphingolipid G-protein-coup Counterscreen for S1P2 Agonists: Dose Response High Throughput Cell-Based Screen to Identify Activators of CRE-BLA: S1P2 Purchased Analogues Source (MLSCN Center Name): The Scripps Research Institute Molecular Screening Center Center Affiliation: The Scripps Research Institute (TSRI) Assay Provider: Hugh Rosen, TSRI Grant Proposal Number: 1 R03 MH076533-01 Grant Proposal PI: Germana Sanna External Assay ID: CRE_AG_BLA_384_EC50_S1P2_Purchased_Analogues Name: Counterscreen for S1P2 Agonists: Dose Response High Throughput Cell-Based Screen to Identify Activators of CRE-BLA: S1P2 Purchased Analogues Description: Sphingosine 1-phosphate (S1P) influences heart rate [1,2], coronary artery caliber, endothelial integrity, lung epithelial integrity [3] and lymphocyte recirculation [2,4] through five related high affinity G-protein coupled receptors [5]. Subtype-selective modulators of S1P receptors will be of broad utility in understanding cell functions in vitro and vascular physiology in vivo, as well as de-convoluting the role of individual subtypes in cellular processes. The S1P receptor 2 (S1P2), also known as endothelial Dose Response Cell Based Assay for Agonists of the S1P2 Receptor Source (MLSCN Center Name): The Scripps Research Institute Molecular Screening Center Center Affiliation: The Scripps Research Institute (TSRI) Assay Provider: Hugh Rosen, TSRI Grant Proposal Number: 1 R03 MH076533-01 Grant Proposal PI: Germana Sanna External Assay ID: S1P2_AG_BLA_384_EC50 Name: Dose Response Cell Based Assay for Agonists of the S1P2 Receptor Description: Sphingosine 1-phosphate (S1P) influences heart rate [1, 2], coronary artery caliber, endothelial integrity, lung epithelial integrity [3] and lymphocyte recirculation [2, 4] through five related high affinity G-protein coupled receptors [5]. Subtype-selective modulators of S1P receptors will be of broad utility in understanding cell functions in vitro and vascular physiology in vivo, as well as de-convoluting the role of individual subtypes in cellular processes. The S1P receptor 2 (S1P2), also known as endothelial differentiation sphingolipid G-protein-coupled receptor 5 (EDG5), signals through Gi, Gq and G1 Dose Response Cell Based Assay for Agonists of the S1P2 Receptor of Purchased Analogues Source (MLSCN Center Name): The Scripps Research Institute Molecular Screening Center Center Affiliation: The Scripps Research Institute (TSRI) Assay Provider: Hugh Rosen, TSRI Grant Proposal Number: 1 R03 MH076533-01 Grant Proposal PI: Germana Sanna External Assay ID: S1P2_AG_BLA_384_EC50_Purchased_Analogues Name: Dose Response Cell Based Assay for Agonists of the S1P2 Receptor of Purchased Analogues Description: Sphingosine 1-phosphate (S1P) influences heart rate [1, 2], coronary artery caliber, endothelial integrity, lung epithelial integrity [3] and lymphocyte recirculation [2, 4] through five related high affinity G-protein coupled receptors [5]. Subtype-selective modulators of S1P receptors will be of broad utility in understanding cell functions in vitro and vascular physiology in vivo, as well as de-convoluting the role of individual subtypes in cellular processes. The S1P receptor 2 (S1P2), also known as endothelial differentiation sphingolipid G-protein-coupled recepto VEGF-R2 Binding Assay A competition binding assay (DiscoveRx KINOMEscan) was used to measure the ability of the compound to compete for binding of an immobilized adenosine triphosphosphate (ATP) site directed ligand using a DNA-tagged vascular endothelial growth receptor 2 (VEGFR2) as the target. The ability of the test compound to compete with the immobilized ligand was measured using quantitative polymerase chain reaction (qPCR) of the DNA tag (Fabian, et al, Nature Biotechnology (2005) 23, 329-336; Karaman, et al, Nature Biotechnology (2008) 26, 127-132).A VEGFR2 tagged T7 phage strain was prepared in an Escherichia coli (E. coli) derived from the BL21 strain. The E. coli were grown to log-phase, infected with VEGFR2 tagged T7 phage and then incubated with shaking at 32 ° C. until lysis. The lysate containing the kinase was then centrifuged and filtered to remove cell debris. Affinity resin for the VEGFR2 assay was prepared by treating Streptavidin-coated magnetic beads with a biotinylated small molecule. 5α-Reductase Activity Assay The reaction mixtures contained a final volume of 1 mL, 1 mM DTT, sodium phosphate buffer 40 mM, at pH 6.5, 2 mM, NADPH, 2 nM [1,2,6,7-3H]T or [1,2,3,7-3H(N)] 4-dione and the prostatic enzyme fractions. The amount of prostatic enzyme fractions was determined to adjust the rate of conversion of T to DHT or 4-dione to 5α-dione to around 28%. T, DHT, unlabeled 4-dione or 5α-dione concentrationswere adjusted to 25-250 nM in the incubating medium, by adding one of the following reagents cold T, DHT, unlabeled 4-dione or 5α-dione. The reactions in duplicate were started when added to the enzymatic fraction (400 μg protein in a volume of 80 μL) incubated at 37°C for 60 min13 and stopped by mixing with 1 mL ofdichloromethane. Incubation without tissue was used as a control. All reactions were carried out in two different times by duplicate. AO Enzyme Assay Guinea pig AO activity was assayed spectrophotometrically using phenanthridine as a substrate at 322 nm. All spectrophotometric determinations were carried out at 25 °C using a PClinked perkin Elemer Lambda 25 UV/Vis spectrophotometer. The varying substrate concentrations (4-60 µM) were separately added into Sorenson's phosphate buffer (final concentration of 50 mM, pH 7.0) containing 0.1 mM of EDTA. Then, the reaction was started by addition of the enzyme and monitored for up to 2 min. The enzyme was also assayed in the presence of different concentrations of the compounds as potential inhibitors (1-200 µM) and constant concentration of substrate (40 µM) to calculation of IC50 values. The results were compared with the inhibitory effects of 1-100 µM menadione, a specific AO inhibitor. Before enzymatic reaction, all of the solutions were equilibrated at 25 °C, except the enzyme fraction which was kept on ice bath prior to addition to the incubation solution. All compounds were dis Binding Activity Assay Binding affinity of the present compound for human 5-HT1A receptor, human 5-HT2A receptor, and human D2 receptor was measured by the following procedures.CHO cell membrane fraction in which human 5-HT1A receptor, human 5-HT2A receptor, and human D2 receptor were expressed was purchased from PerkinElmer, Inc. In a test for evaluating binding affinity, a test compound dissolved in dimethylsulfoxide (DMSO) and each receptor membrane sample diluted in buffer were mixed with [3H]8-OH-DPAT, [3H]Ketanserin, or [3H]Spiperone (all purchased from PerkinElmer, Inc.) for 5-HT1A receptor, 5-HT2A receptor, and D2 receptor, respectively. Each mixture was incubated at room temperature for 60 minutes. Then, the mixture was added quickly on a glassfiber filter plate (Multiscreen FB, Millipore, Inc.) coated with 0.3% aqueous polyethylenimine, and vacuum-filtered. Radioactivity remaining on the filter was measured with a liquid scintillation counter (PerkinElmer, Inc.). Binding inhibition rate was calculated from the following formula. Binding Affinity Assay Present compound for human 5-HT1A receptor, human 5-HT2A receptor, and human D2 receptor was measured by the following procedures. CHO cell membrane fraction in which human 5-HT1A receptor, human 5-HT2A receptor, and human D2 receptor were expressed was purchased from PerkinElmer, Inc. In a test for evaluating binding affinity, a test compound dissolved in dimethylsulfoxide (DMSO) and each receptor membrane sample diluted in buffer were mixed with [3H]8-OH-DPAT, [3H]Ketanserin, or [3H]Spiperone (all purchased from PerkinElmer, Inc.) for 5-HT1A receptor, 5-HT2A receptor, and D2 receptor, respectively. Each mixture was incubated at room temperature for 60 minutes. Then, the mixture was added quickly on a glassfiber filter plate (Multiscreen FB, Millipore, Inc.) coated with 0.3% aqueous polyethylenimine, and vacuum-filtered. Radioactivity remaining on the filter was measured with a liquid scintillation counter (PerkinElmer, Inc.). Binding inhibition rate was calculated from the following formula. Binding Assay Membranes from CHO/Galpha16 cells stably transfected with human hMCH-1R are resuspended using a syringe (needle 0.6x25 mm) and diluted in test buffer (50 mM HEPES, 10 mM MgCl2, 2 mM EGTA, pH 7.00; 0.1% bovine serum albumin (protease-free), 0.021% bacitracin, 1 ug/ml aprotinin, 1 ug/ml leupeptin and 1 uM phosphoramidone) to a concentration of 5 to 15 ug/ml. 200 microliters of this membrane fraction (contains 1 to 3 ug of protein) are incubated for 60 minutes at ambient temperature with 100 pM of 125I-tyrosyl melanin concentrating hormone (125I-MCH commercially obtainable from NEN) and increasing concentrations of the test compound in a final volume of 250 microliters. After the incubation the reaction is filtered using a cell harvester through 0.5% PEI treated fibreglass filters (GF/B, Unifilter Packard). The membrane-bound radioactivity retained on the filter is then determined after the addition of scintillator substance (Packard Microscint 20). DELFIA Assay This test uses active PTK2 enzyme (Invitrogen Code PV3832) and poly-Glu-Tyr (4:1, Sigma P-0275) as the kinase substrate. The kinase activity is detected by means of the phosphorylation of the substrate in a DELFIA assay. The phosphorylated substrate is detected with the Europium-labelled phosphotyrosine antibody PY20 (Perkin Elmer, No.: AD0038). In order to determine concentration-activity curves with PTK2-inhibitors the compounds are serially diluted in 10% DMSO/H2O and 10 L of each dilution are dispensed per well in a 96-well microtitre plate (clear U-shaped base plate, Greiner No. 650101) (the inhibitors are tested in duplicates) and mixed with 10 L/well of PTK2 kinase (0.01 ug/well). PTK2 kinase is diluted accordingly beforehand with kinase dilution buffer (20 mM TRIS/HCl pH 7.5, 0.1 mM EDTA, 0.1 mM EGTA, 0.286 mM sodium orthovanadate, 10% glycerol with the addition of freshly prepared BSA (fraction V 1 mg/mL) and DTT (1 mM)). Determination of Affinity for Human 5-HT2A Receptors in Competitive Binding Assay Receptor membranes were prepared from the CHO-K1 recombinant AequoScreen® cell line stably expressing the human 5-HT2A receptor (PerkinElmer, Waltham, MA, USA). Cells were suspended in 4× volume in buffer A (15 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 0.3 mM EDTA, 1 mM EGTA) (1 g cell-4 mL buffer) and homogenized in a Dounce homogenizer. The crude membrane fraction was collected following two consecutive centrifugation steps at 40,000×g for 25 minutes separated by a washing step in buffer A. The final pellet was resuspended in buffer B (75 mM Tris-HCl, pH 7.5, 12.5 mM MgCl2, 0.3 mM EDTA, 1 mM EGTA, 250 mM sucrose) in a concentration of 80 mg wet cell weight in 0.5 mL buffer, aliquoted and flash frozen on dry ice. Protein content was determined using the bicinchoninic acid assay in the presence of sulfhydryl reagents with bovine serum albumin (BSA) as a standard. Enzymatic Assay HDAC8 reagents: HDAC8 was purchased from Enzo Life Sciences [catalog #BML-SE145]. Assays were performed with buffer containing 20 mM HEPES, pH 8.0 [Boston BioProducts, catalog #BB-104, 1M stock], 100 mM NaCl [Sigma, catalog #S5150, 5M stock], 20 mM KCl [BioChemika, catalog #87526, 4M stock], 1 mM MgCl2 [Fluka, catalog #63020, 1M stock], 0.05% BSA (Fraction V) [Invitrogen, catalog #15260, 7.5% stock], and 0.1% n-Octyl-β-D-glucopyranoside (N-OG) [Anatrace, catalog #O311, 10% stock]. The HDAC8 enzyme was run at the final concentration of 1.333 nM. Fluor-de-Lys substrate [BioMol Research Laboratories, catalog #KI-178], used to evaluate enzyme activity, was added at the final concentration of 200 uM. To enable detection of the signal, Developer II [BioMol Research Laboratories, catalog #KI-176] was added at a 1:200 dilution to the stop solution, which also included 20 uM SAHA [Sigma, catalog #SML0061] to ensure complete termination of the reaction. Enzyme Assay This test uses active PTK2 enzyme (Invitrogen Code PV3832) and poly-Glu-Tyr (4:1, Sigma P-0275) as the kinase substrate. The kinase activity is detected by means of the phosphorylation of the substrate in a DELFIA assay. The phosphorylated substrate is detected with the europium-labelled phosphotyrosine antibody PY20 (Perkin Elmer, No.: AD0038).In order to determine concentration-activity curves with PTK2-inhibitors the compounds are serially diluted in 10% DMSO/H2O and 10 uL of each dilution are dispensed per well in a 96-well microtitre plate (clear U-shaped base plate, Greiner No. 650101) (the inhibitors are tested in duplicates) and mixed with 10 uL/well of PTK2 kinase (0.01 ug/well). PTK2 kinase is diluted accordingly beforehand with kinase dilution buffer (20 mM TRIS/HCl pH 7.5, 0.1 mM EDTA, 0.1 mM EGTA, 0.286 mM sodium orthovanadate, 10% glycerol with the addition of freshly prepared BSA (fraction V 1 mg/mL) and DTT (1 mM)). Fluorescence Polarization Assay An in vitro competition fluorescence polarization assay in which a test compound competes with a fluorescent probe for binding to the binding domain of human recombinant HSP90. The reaction can be followed kinetically using fluorescence (excitation lamda==485 nm; emission lamda==538 nm). The binding affinity of the test compound to HSP90 is determined by the changes in the polarized fluorescence; the intensity of the polarized fluorescence is proportional to the fraction of bound probe. To each test well, an aliquot of buffer, 2 ul of test compound in 10% DMSO, 4 ul of 6.25 nM of TSD FP probe, 4 ul of 12.5 nM of purified HSP90a protein were added. For positive control, 1 uM geldanamycin (GM) was used instead of the test compound (GM is a natural benzoquinone ansamycin that is known to bind to the N-terminal ATP-binding pocket of HSP90 and inhibits ATP binding and ATP-dependent chaperone activities). For negative control, no inhibitor was added. GOAT Activity Assay Assays were performed with ~100 μg of membrane protein, as determined by a Bradford assay. The membrane fraction was preincubated with 1 μM methyl arachidonyl fluorophosphonate (MAFP) and inhibitor or vehicle as indicated in 50 mM HEPES (pH 7.0) for 30 min at room temperature [McGovern-Gooch et al., Endocrinology, 157:4330-4338]. Reactions were initiated via the addition of 500 μM octanoyl CoA and 1.5 μM fluorescently labeled ghrelin mimetic, GSSFLCAcrylodan, for a total volume of 50 μL, and were incubated for 3 h at room temperature in the dark. Reactions were stopped with the addition of 50 μL of 20% acetic acid in isopropanol, and solutions were clarified by proteinprecipitation with 16.7 μL of 20% trichloroacetic acid, followed by centrifugation (1000g for 1 min). The supernatant was then analyzed by reverse phase HPLC, as previously described [Darling et al., Anal. Biochem., 437:68-76]. In Vitro Competitive Activity-Based Protein Assay Proteomes (mouse brain membrane fraction or cell lysates) (50 uL, 1.0 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37 C. After 30 min, FP-Rh (1.0 uL, 50 uM in DMSO) was added and the mixture was incubated for another 30 min at 37 C. Reactions were quenched with SDS loading buffer (50 uL-4x) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL, ABHD6 and FAAH using ImageJ 1.43u software.Preparation of Mouse Brain Proteomes from Inhibitor Treated Mice. Inhibitors were administered to wild-type C57Bl/6J by oral gavage in a vehicle of polyethylene glycol. Each animal was sacrificed 4 h following administration and brain proteomes were prepared and analyzed according to previously established methods (See Niphakis, M. J., et al. (2011) ACS Chem. Neurosci. and Long, J. Z., et al. Nat. Chem. Biol. 5:37-44). Inhibition Assay The PDE 7 (phosphodiesterase VII) inhibiting effect of the compounds of the present invention was performed by the following method, which was modified assay method described in Biochemical. Pharmacol. 48(6), 1219-1223 (1994). (1) The active fraction of PDE 7 (phosphodiesterase VII) was obtained. That is, MOLT-4 (obtainable from ATCC as ATCC No. CRL-1582), which was cell line of human acute lymphoblastic lymphoma T cells, was incubated in RPMI1640 culture medium containing 10% fetal bovine serum to obtain 5x108 MOLT-4 cells. The cells were collected by centrifugation and suspended with 10 mL of buffer solution A [25 mM of tris-HCl, 5 mM of 2-mercaptoethnol, 2 mM of benzamidine, 2 mM of EDTA, 0.1 mM of 4-(2-aminoethyl)benzensulfonyl hydrochloride; pH 7.5], then homogenized by Polytron homogenizer. The homogenate were centrifuged under 25,000xG for 10 minutes at 4 C. The supernatant was separated and thus obtained supernatant was further centrifuged under 100,000xG for 60 minutes. Inhibitory Assay For the measurement of CH24H inhibitory activity, using the human CH24H lysate prepared in Experimental Example 2, the amount of 24-HC produced from cholesterol by catalytic activity of CH24H was measured in the presence of a test compound, and compared with that measured in the absence of the test compound. That is, a test compound solution at various concentrations was mixed with a reaction buffer (50 mM potassium phosphate containing 0.1% BSA and Complete, EDTA-free, pH 7.4) and human CH24H lysate. Then, [14C] cholesterol (53 mCi/mmol specific activity, 15 μM) was added, and CH24H reaction was performed at 37° C. for 5 hr. After completion of the reaction, a quenching solution consisting of chloroform/methanol/distilled water (2:2:1 v/v) was added, and the resulting 24-HC was extracted by shaking. The extract was applied to silica gel thin layer chromatography (ethyl acetate:toluene=4:6), and the obtained 14C-24HC fraction was measured with BAS2500 (Fujifilm Corporation). JAK1 and TYK2 Inhibition Assay To determine inhibition constants (Ki), compounds were diluted serially in DMSO and added to 50 uL kinase reactions containing 1.5 nM JAK1, 0.2 nM purified JAK2 or 1 nM purified TYK2 enzyme, 100 mM Hepes pH7.2, 0.015% Brij-35, 1.5 uM peptide substrate, 25 uM ATP, 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22 ° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 25 uL of an EDTA containing solution (100 mM Hepes pH 7.2, 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip 3000 according to the manufacturer's specifications. Ki values were then determined using the Morrison tight binding model (Morrison, J. F., Biochim. Biophys. Acta. 185:269-296 (1969). Radioligand Binding Assay The detailed experimental protocols for the radioligand and functional receptor assays are available on the NIMH PDSP website at http://pdsp.med.unc.edu/UNC-CH %20Protocol %20Book.pdf. A. Serotonin receptors: 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT5A, 5-HT6 and 5-HT7. Assay Buffer: Standard Binding Buffer (50 mM Tris-HCl, 10 mM MgCl2, 1 mM EDTA, pH 7.4) Membrane Fraction Source: Transiently or stably transfected cell lines (e.g., HEK293, COS, CHO, NIH3T3). Protocol adapted from Roth et al. (1986), J. Pharmacol. Exp. Ther., 238(2): 480-485; and Roth et al. (1994), J. Pharmacol. Exp. Ther., 268(3): 1403-1410. Adrenergic Receptors: alpha1A, alpha1B, alpha2A, alpha2B, alpha2C, beta1, beta2, beta3 Assay Buffers: For alpha1 receptors, alpha1 Binding Buffer (20 mM Tris-HCl, 145 mM NaCl, pH 7.4); for alpha2 receptors, alpha2 Binding Buffer (50 mM Tris-HCl, 5 mM MgCl2, pH 7.7); for beta receptors. Scintillation Assay The test compounds (2.5 μL) dissolved in DMSO were added to wells containing 37.5 μL of the reaction solution (25 mM HEPES (pH 7.5), 10 mM magnesium acetate, 1 mM DTT) including 30 ng of active ASK1 protein and 1 μg of myelin basic protein (Wako), and incubated at room temperature for 5 min. To start the reaction, 10 μL of ATP solution (2.5 μM ATP, 0.1 μCi [γ-32P]ATP) was added to wells. After incubating at room temperature for 30 min, the reaction was terminated by adding 50 μL of 20% TCA solution. The reaction solution was incubated at 4° C. for 30 min and an acid-insoluble fraction was transferred onto a GF/C filter (Packard) with Cell Harvester (Packard), and washed with 250 mM phosphoric acid. After drying at 45° C. for 60 min., 40 μL of Microscint 0 (Packard) was added and the radioactivity was measured with TopCount (Packard). Binding Activity Assay Human 5-HT2A Receptor, Human 5-HT7 Receptor, and Human D2 Receptor: Binding affinity of the present compound for human 5-HT2A receptor, human 5-HT7 receptor, and human D2 receptor was measured by the following procedures.CHO cell membrane fraction in which human 5-HT2A receptor, human 5-HT7 receptor, or human D2 receptor was expressed was purchased from PerkinElmer, Inc. In a test for evaluating binding affinity, a test compound dissolved in dimethylsulfoxide (DMSO) and each receptor membrane sample diluted in buffer were mixed with [3H]Ketanserin, [3H]SB-269970, or [3H]Spiperone (all purchased from PerkinElmer, Inc.) for 5-HT2A receptor, 5-HT7 receptor, or D2 receptor, respectively. Each mixture was incubated at room temperature for 60 minutes. Non specific binding to receptors was obtained from a competitive binding test in the presence of 10 μmol/L 8-OH-DPAT, 10 μmol/L Mianserin, or 10 μmol/L Spiperone, respectively. Radioactivity caused by binding to receptors was measured with a liquid scintillation counter (PerkinElmer, Inc.), and 50% inhibition concentration was calculated. Biological Assay Endothelial lipase (EL) and hepatic lipase (HL) activities were measured using a fluorescent substrate, A10070, (Invitrogen, CA) doped into an artificial vesicle containing DMPG (Avanti Polar Lipids) as the excipient. Vesicles were prepared by combining 571 uL of 29 mM DMPG in a 1:1 mixture of MeOH and CHCl3 with 2000 uL of 1 mM A10070 in a 1:1 mixture of MeOH and CHCl3. The mixture was dried under nitrogen in multiple vials then resuspended in 20 mL total volume of 50 mM HEPES pH 8.0 buffer containing 50 mM NaCl and 0.2 mM EDTA. The sample was allowed to sit at room temperature for 15 min and then was sonicated 3x4 mins on ice with a Branson Sonicator using duty cycle 1. This preparation provides vesicles with a mole fraction of 0.11 for the FRET substrate.The enzymatic assay was measured using 384-well white Optiplates. Each well contained 20 uL of assay buffer (50 mM HEPES pH 8.0, 50 mM NaCl and 1 mM CaCl2) and 0.25 uL of a DMSO solution containing a compound. Biological Assay Endothelial lipase activity was measured using a fluorescent substrate, A10070, (Invitrogen, CA) doped into an artificial vesicle containing DMPG (Avanti Polar Lipids) as the excipient. Vesicles were prepared by combining 285 uL, of 1 mM DMPG in a 1:1 mixture of MeOH and CHCl3 with 15 uL, of 1 mM A10070 in a 1:1 mixture of MeOH and CHCl3. The mixture was dried under nitrogen and resuspended in 150 uL, of 50 mM HEPES pH 8.0 buffer containing 100 mM NaCl and 0.2 mM EDTA. The sample was allowed to sit at rt for 15 min and then was sonicated 3x4 mins on ice with a Branson Sonicator using duty cycle 1. This preparation provides vesicles with a mole fraction of 0.05 for the FRET substrate.The enzymatic assay was measured using white, opaque 96-well half area plates. Each well contained 60 uL, of assay buffer (50 mM HEPES pH 8.0, 50 mM NaCl and 1 mM CaCl2) and 2 ul of a DMSO solution containing compound of interest. Conditioned media obtained from HT-1080 cells. Competitive Inhibition Assay A competitive inhibition binding assay is performed using membrane homogenates prepared from HEK-293 cells that express recombinant hMC4-R, hMC3-R, or hMC5-R, and from B-16 mouse melanoma cells (containing endogenous MC1-R). In some instances, HEK-293 cells that express recombinant hMC1-R were employed. In the examples that follow, all MC3-R, MC4-R and MC5-R values are for human recombinant receptors. MC1-R values are for B-16 mouse melanoma cells, unless the heading is hMC1-R, in which case the value is for human recombinant MC1-R. Assays were performed in 96 well GF/B Millipore multiscreen filtration plates (MAFB NOB10) pre-coated with 0.5% bovine serum albumin (Fraction V). Membrane homogenates were incubated with 0.2 nM (for hMC4-R) 0.4 nM (for MC3-R and MC5-R) or 0.1 nM (for mouse B16 MC1-R or hMC1-R) [I125]-NDP-alpha -MSH (Perkin Elmer) and increasing concentrations of test peptides of the present invention in buffer containing 25 mM HEPES buffer (pH 7.5) with 100 mM NaCl. Competitive Inhibition Assay A competitive inhibition binding assay was performed for exemplified peptides according to the invention using membrane homogenates prepared from HEK-293 cells that express recombinant hMC4-R, hMC3-R, or hMC5-R, and from B-16 mouse melanoma cells (containing endogenous MC1-R). In some instances, HEK-293 cells that express recombinant hMC1-R were employed. Assays were performed in 96 well GF/B Millipore multiscreen filtration plates (MAFB NOB10) pre-coated with 0.5% bovine serum albumin (Fraction V). Membrane homogenates were incubated with 0.2 nM (for hMC4-R) 0.4 nM (for MC3-R and MC5-R) or 0.1 nM (for mouse B16 MC1-R or hMC1-R) [I125]-NDP-alpha -MSH (Perkin Elmer) and increasing concentrations of test peptides of the present invention in buffer containing 25 mM HEPES buffer (pH 7.5) with 100 mM NaCl, 2 mM CaCl2, 2 mM MgCl2, 0.3 mM 1,10-phenanthroline, and 0.2% bovine serum albumin. After incubation for 60 minutes at 37 C., the assay mixture was filtered and the membranes washed. Enzyme Inhibition Assay A reaction was performed on a 384-well plate (Greiner Bio One) using a reaction volume of 24 µL, and all the samples were diluted with an assay buffer (50 mM tris buffer, pH 7.4, 10% glycerol). 0.8 mM NADPH, 6 mM glucose 6-phosphate, 0.35 units/mL glucose 6-phosphate dehydrogenase (Sigma Chemical), and 3 mM magnesium chloride and a microsome fraction as an enzyme source were added to the plate, and a solution containing a test compound dissolved in a dimethyl sulfoxide/methanol solution was added at a final concentration of 0.1%. After the addition of the test compound, cortisone at a final concentration of 160 nM was added to start the reaction, and the reaction was performed at room temperature for 3 h. 25 µL of 100 mM carbenoxolone (Sigma Chemical) was added to terminate the reaction, and the amount of cortisol produced was measured with RUBYstar (BMG LABTECH JAPAN Ltd.) using a cortisol prototype kit (Cisbio International) according to the package insert. Enzyme Inhibition Assay Human SCD-1 enzyme activity using HepG2 cell microsomes after treating with inhibitory compounds (% inhibition):Human hepatocarcinoma HepG2 cells (ATCC, HB-8065) are cultured to confluence and trypsinised. The cell pellet is taken up with 10 mM Tris (pH 7.4) sucrose (250 mM) DTT (1 mM) buffer and the cells are lysed by sonication. The microsomes are obtained after centrifugation at 10,000 g for 20 minutes at 4 C. followed by centrifugation of the supernatant at 100,000 g for 60 minutes at 4 C. The pellet is taken up with 10 mM Tris (pH 7.4) sucrose (250 mM) buffer at 4 C. and the microsomal proteins are assayed and stored at -196 C. (liquid nitrogen).The enzyme reaction measures the conversion of stearic acid (C18:0 fatty acid) to oleic acid (C18:1 fatty acid) by SCD-1. The enzyme reaction is started by adding 125 ug of HepG2 cell microsomal fraction to tubes (total reaction volume of 500 ul) containing 62 uM of stearic acid. In Vitro Acyltransferase Assays ALCAT1 enzymatic activity was determined by measuring the conversion of monolysocardiolipin (MLCL) to cardiolipin or lysophosphatidylglycerol (LPG) to phosphatidylglycerol (PG) in an enzymatic reaction mixture that contained 50 mm Tris/HCl, pH 7.0, 100 μm lysophospholipids, 25 μm [14C]acyl-CoA (50 mCi/mmol, American Radiolabeled Chemicals, Inc), and membrane fraction (0.5-2.5 μg) in a total volume of 200 μl. The reaction was incubated at room temperature for 30 min. The lipids were extracted will chloroform, dried, and separated by thin layer chromatography (TLC) with chloroform:hexane:methanol:acetic acid (50:30:10:5, v/v) or chloroform:methanol:water (65:25:4, v/v). After separation, TLC plates were exposed to a Phosphorlmager screen to visualize the radiolabeled products with a Molecular Dynamics Typhoon Scanner (Sunnyvale, Calif.). In some experiments, NBD-CoA was used at 100 μM to replace the [14C]acyl-CoA in the enzymatic reaction. All quantitative data were expressed as mean±S.E. Statistical analyses for differences between two groups were carried out using a Student's t test. In Vitro Competitive Activity-Based Protein Profiling Proteomes (mouse brain membrane fraction or cell lysates for mouse assays; human prefrontal cortex or cell membrane fractions for human assays) (50 μL, 1.0 mg/mL total protein concentration) were preincubated with varying concentrations of inhibitors at 37° C. After 30 min, FP Rh or HT-01 (1.0 μL, 50 μM in DMSO) was added and the mixture was incubated for another 30 min at 37° C. Reactions were quenched with SDS loading buffer (15 μL-4×) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL and FAAH using ImageJ 1.43u software.Preparation of Mouse Brain Proteomes from Inhibitor Treated Mice.Inhibitors were administered to wild-type C57Bl/6J by oral gavage in a vehicle of polyethylene glycol. Each animal was sacrificed 4 h following administration and brain proteomes were prepared and analyzed according to previously established methods (See Niphakis, M. J., et al. (2011) ACS Chem. Neurosci. and Long, J. Z., et al. Nat. Chem. Biol. 5:37-44). Inhibition Assay Rat brain tissue (hippocampus or whole brain) is homogenized in aqueous homogenization buffer (10% w/v, 0.32 M sucrose, 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 0.01% (w/v) NaN3, pH 7.4, 4C.) at 600 rpm in a glass homogenizer. The homogenate is centrifuged (1000xg, 4C., 10 min) and the supernatant is removed. The pellet is resuspended (20% w/v) and the suspension is centrifuged (1000xg, 4C., 10 min). The two supernatants are combined and centrifuged (15 000xg, 4C., 30 min). The pellet obtained in this way is referred to as the P2 fraction. The P2 pellet is suspended in binding buffer (50 mM Tris-HCl, 1 mM MgCl2, 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, pH 7.4), and the suspension is centrifuged (15 000xg 4C., 30 min), twice. The residue is resuspended in binding buffer to a concentration of 4 mg/ml and incubated in a volume of 250 ul (amount of membrane protein 0.4 mg) in the presence of 2 nM [3H]-methyllyeaconitine, 0.1% (w/v) BSA (bovine serum albumin). Inhibitory Activity Assay The inhibition of CYP17A1 by the test compounds was evaluated using a recombinant enzyme. Human CYP17A1 was expressed in E. coli (Ehmer, P. B. et al.; J. Steroid Biochem. Mol. Biol., 75, 57-63 (2000)). The microsomal fraction and 140 μL of phosphate buffer (50 mM Na phosphate, 1 mM MgCl2, 0.1 mM EDTA, 0.1 mM dithiothreitol, pH 7.4) were preincubated separately with a mixture of progesterone (24.95 μM) and 3H-progesterone (0.05 μM, 101.3 Ci/mmol), 50 μM of an NADPH regeneration system (in phosphate buffer with 10 mM NADP+, 100 mM glucose 6-phosphate and 2.5 U of glucose 6-phosphate dehydrogenase) and the appropriate test substances (in 5 μl of DMSO) at 37° C. for 5 minutes. The reaction was started by addition of the enzyme and, after 30 minutes of incubation at 37° C., stopped by addition of 50 μl of 1N hydrochloric acid.The steroids were extracted with ethyl acetate. After evaporation of the organic phase, the steroids were taken up in acetonitrile. JAK2 Inhibition Assay To determine the inhibition constants (Ki), compounds were diluted serially in DMSO and added to 50 kinase reactions containing 0.2 nM purified JAK2 enzyme, 100 mM Hepes pH7.2, 0.015% Brij-35, 1.5 μM peptide substrate, 25 μM ATP, 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 250 μL of an EDTA containing solution (100 mM Hepes pH 7.2, 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip 3000 according to the manufacturer's specifications. Ki values were then determined using the Morrison tight binding model. Morrison, J. F., Biochim. Biophys. Acta. 185:269-296 (1969); William, J. W. and Morrison, J. F., Meth. Enzymol., 63:437-467 (1979). JAK3 Inhibition Assay To determine inhibition constants (Ki), compounds were diluted serially in DMSO and added to 50 uL kinase reactions containing 5 nM purified JAK3 enzyme, 100 mM Hepes pH7.2, 0.015% Brij-35, 1.5 uM peptide substrate, 5 uM ATP, 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 25 uL of an EDTA containing solution (100 mM Hepes pH 7.2, 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip 3000 according to the manufacturer's specifications. Ki values were then determined using the Morrison tight binding model (Morrison, J. F., Biochim. Biophys. Acta. 185:269-296 (1969); William, J. W. and Morrison, J. F., Meth. Enzymol., 63:437-467 (1979)). MGAT SPA Assay MGAT2 enzyme was assayed using membranes isolated from Sf9 cells expressing the recombinant human MGAT2 cDNA with 2-monooleoylglycerol and [3H]-oleoyl-CoA as substrates as described by Seethala et al. [Anal. Biochem., 383(2):144-150 (Dec. 15, 2008)]. Briefly, the assays were conducted in 384-well plates in a total volume of 30 uL at 25° C. In each assay, 200 ng of recombinant human MGAT2 membrane was incubated with 10 uM of 2-monooleoylglycerol and 15 uM of [3H]-oleoyl-CoA in 100 mM potassium phosphate (pH 7.4) for 20 min with various concentrations of compounds delivered in DMSO. The assay was terminated by the addition of 20 ul of Stopping Solution (7.5 mg/ml Yttrium Oxide Polylysine beads, 3.3 mg/ml Fraction V BSA and 200 uM Mercuric chloride in 50 mM HEPES, pH 7.4). The signal was measured 1 h after quenching the reaction using LEADSEEKERSM for 5 minutes. To calculate the degree of inhibition, the zero level of enzyme activity (blank) was defined. Radioligand Binding Assay The cell homogenate was centrifuged at 19000 r.p.m. for 20 min at 4° C. using a Beckman Ti-60 rotor. The resultant pellet was resuspended in TME buffer to give a final 1 mg/ml protein concentration, as determined by Biorad assay. Radioligand binding competition assays vs. [3H-]17-phenyl PGF2a , (5 nM) were performed in a 100 μl volume for 60 min. Binding reactions were started by adding plasma membrane fraction. The reaction was terminated by the addition of 4 ml ice-cold TRIS-HCl buffer and rapid filtration through glass fiber GF/B filters using a Brandel cell harvester. The filters were washed 3 times with ice-cold buffer and oven dried for one hour. [3H-] PGE2 (specific activity 180 Ci mmol) was used as the radioligand for EP receptors. [3H] 17-phenyl PGF2a, was employed for FP receptor binding studies. Binding studies employing EP1, EP2, EP4 and FP receptors were performed in duplicate in at least three separate experiments. A 200 μl assay volume was used. Incubations w Competitive ABPP Assays in Proteomes with FP-alkyne For in vitro experiments, proteomes were diluted to 1 mg/mL in PBS (pH 7.5, 50 μL total reaction volume), doped with 1 μM recombinant PAFAH1b2 and PAFAH1b3, and incubated with compound at the indicated concentrations (1 μL of a 50x stock in DMSO) for 30 min at 37 °C, followed by labeling with 1 μM FP-alkyne (1 μL of a 50x stock in DMSO) for 10 min at 25 °C. Total protein concentrations for each fraction were adjusted to 1 mg/mL in PBS (50 μL total reaction volume). Samples were then subjected to click chemistry by the addition of 1 mM CuSO4 (1 μL of a 50x stock in DMSO), followed by TBTA (3 μL of a 17x stock in 4:1 t-butanol:DMSO), then 25 μM Rh-N3 (1 μL of a 50x stock in DMSO) and 1 mM TCEP (1 μL of a 50x stock in DMSO). Reactions were vortexed, incubated for 1 h at 25 °C, and quenched with 4x SDS-PAGE loading buffer and analyzed by in-gel fluorescence scanning. Enzymatic Assay Endothelial lipase activity was measured using a fluorescent substrate, A10070, (Invitrogen, CA) doped into an artificial vesicle containing DMPG (Avanti Polar Lipids) as the excipient. Vesicles were prepared by combining 285 uL of 1 mM DMPG in a 1:1 mixture of MeOH and CHCl3 with 15 uL of 1 mM A10070 in a 1:1 mixture of MeOH and CHCl3. The mixture was dried under nitrogen and resuspended in 150 uL of 50 mM HEPES pH 8.0 buffer containing 100 mM NaCl and 0.2 mM EDTA. The sample was allowed to sit at rt for 15 min and then was sonicated 3~4 mins on ice with a Branson Sonicator using duty cycle 1. This preparation provides vesicles with a mole fraction of 0.05 for the FRET substrate.The enzymatic assay was measured using white, opaque 96-well half area plates. Each well contained 60 uL of assay buffer (50 mM HEPES pH 8.0, 50 mM NaCl and 1 mM CaCl2) and 2 ul of a DMSO solution containing compound of interest. Conditioned media obtained from HT-1080 cells, which were transformed by RAGE technology. Enzyme Assay The activity of the isolated recombinant JAK1 and JAK2 kinase domain was measured by monitoring phosphorylation of a peptide derived from JAK3 (Val-Ala-Leu-Val-Asp-Gly-Tyr-Phe-Arg-Leu-Thr-Thr, fluorescently labeled on the N-terminus with 5-carboxyfluorescein) using the Caliper LabChip R technology (Caliper Life Sciences, Hopkinton, Mass.). To determine inhibition constants (Ki), compounds were diluted serially in DMSO and added to 50 μL kinase reactions containing purified enzyme (1.5 nM JAK1, or 0.2 nM JAK2), 100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 1.5 μM peptide substrate, ATP (25 μM), 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 25 μL of an EDTA containing solution (100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip R 3000 according to the manufacturer's specifications. Enzyme Assay The activity of the isolated recombinant JAK1 and JAK2 kinase domain was measured by monitoring phosphorylation of a peptide derived from JAK3 (Val-Ala-Leu-Val-Asp-Gly-Tyr-Phe-Arg-Leu-Thr-Thr, fluorescently labeled on the N-terminus with 5-carboxyfluorescein) using the Caliper LabChip technology (Caliper Life Sciences, Hopkinton, Mass.). To determine inhibition constants (Ki), compounds were diluted serially in DMSO and added to 50 μL kinase reactions containing purified enzyme (1.5 nM JAK1, or 0.2 nM JAK2), 100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 1.5 μM peptide substrate, ATP (25 μM), 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 25 μL of an EDTA containing solution (100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip 3000 according to the manufacturer's specifications. HTRF assay The HTRF assay was carried out using a two-step protocol essentially according to the kit manufacturer's instructions, in 20 μL total volume per well in 384-well plate format (ProxiPlates; PerkinElmer, Fremont, Calif.; catalog #6008280). To each of the experimental wells was transferred 3000 recombinant CHO-K1 cells in 5 μL assay buffer (phosphate buffered saline containing calcium chloride and magnesium chloride (Invitrogen, Carlsbad, Calif.; catalog #14040) supplemented with IBMX (100 gμM) and rolipram (10 gμM) (phosphodiesterase inhibitors; Sigma-Aldrich, St. Louis, Mo.; catalog #15879 and catalog # R6520, respectively) and 0.1% bovine serum albumin (BSA) fraction V (Sigma-Aldrich; catalog # A3059)), followed by test compound in 5 μL assay buffer or 5 μL assay buffer. The plate was then incubated at room temperature for 1 h. To each well was then added 5 μL cAMP-d2 conjugate in lysis buffer and 5 μL Cryptate conjugate in lysis buffer according to the kit manufacturer's instructions. The plate was then further incubated at room temperature for 1 h, after which the assay plate was read. Homogeneous Time-Resolved Fluorescence (HTRF) Assay for Direct cAMP Measurement The HTRF assay was carried out using a two-step protocol essentially according to the kit manufacturer's instructions, in 20 μL total volume per well in 384-well plate format (ProxiPlates; PerkinElmer, Fremont, Calif.; catalog #6008280). To each of the experimental wells was transferred 3000 recombinant CHO-K1 cells in 5 μL assay buffer (phosphate buffered saline containing calcium chloride and magnesium chloride (Invitrogen, Carlsbad, Calif.; catalog #14040) supplemented with IBMX (100 μM) and rolipram (10 μM) (phosphodiesterase inhibitors; Sigma-Aldrich, St. Louis, Mo.; catalog #15879 and catalog #R6520, respectively) and 0.1% bovine serum albumin (BSA) fraction V (Sigma-Aldrich; catalog # A3059)), followed by test compound in 5 μL assay buffer or 5 μL assay buffer. The plate was then incubated at room temperature for 1 h. To each well was then added 5 μL cAMP-d2 conjugate in lysis buffer and 5 μL Cryptate conjugate in lysis buffer according to the kit manufacturer's instructions. The plate was then further incubated at room temperature for 1 h, after which the assay plate was read. JAK Enzyme Assay The activity of the isolated recombinant JAK1 and JAK2 kinase domain was measured by monitoring phosphorylation of a peptide derived from JAK3 (Val-Ala-Leu-Val-Asp-Gly-Tyr-Phe-Arg-Leu-Thr-Thr, fluorescently labeled on the N-terminus with 5-carboxyfluorescein) using the Caliper LabChip® technology (Caliper Life Sciences, Hopkinton, Mass.). To determine inhibition constants (Ki), compounds were diluted serially in DMSO and added to 50 μL kinase reactions containing purified enzyme (1.5 nM JAK1, or 0.2 nM JAK2), 100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 1.5 μM peptide substrate, ATP (25 μM), 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 25 μL of an EDTA containing solution (100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip® 3000 according to the manufacturer's specifications. Radioligand Binding HEK-293 cells stably expressing the human or feline FP receptor, or EP1, EP2, EP3, or EP4 receptors were washed with TME buffer, scraped from the bottom of the flasks, and homogenized for 30 sec using a Brinkman PT 10/35 polytron. TME buffer was added to achieve a final 40 ml volume in the centrifuge tubes (the composition of TME is 100 mM TRIS base, 20 mM MgCl2, 2M EDTA; 10 N HCl is added to achieve a pH of 7.4).The cell homogenate was centrifuged at 19000 r.p.m. for 20 min at 4 C. using a Beckman Ti-60 rotor. The resultant pellet was resuspended in TME buffer to give a final 1 mg/ml protein concentration, as determined by Biorad assay. Radioligand binding competition assays vs. [3H-]17-phenyl PGF2 (5 nM) were performed in a 100 ul volume for 60 min. Binding reactions were started by adding plasma membrane fraction. The reaction was terminated by the addition of 4 ml ice-cold TRIS-HCl buffer and rapid filtration through glass fiber GF/B filters using a Brandel cell. Ribonuclease Activity Assay The Ribonuclease Activity assay used was adapted from the procedure described by Anfinsen et al. and Slifman et al. Determination of ribonuclease activity of RNase A and EDN proceeded as follows: yeast tRNA (Sigma-Aldrich) was solubilized in 50 mM sodium phosphate, pH = 7.4, at a concentration of 60 uM. Various tRNA concentrations were used: 2 uM, 3 uM, 4 uM, 5 uM and 6 uM. The final volume of the reaction was 300 ul. tRNA solutions were incubated at 30 C for 10 min. 10 nM of enzyme final concentration from a 0.6 uM stock solution added to the reactionmixture. The reaction was stopped one minute and five minutes later in the case of RNase A and EDN respectively, by the addition of 300 ul of fresh ice-cold solution of 40 mM lanthanum nitrate, 6% v/v perchloric acid. Stopped reactions were maintained on ice for 15 min, and insoluble tRNA was removed by centrifugation at 4 C for 15 min at 14.500 rpm on a bench centrifuge. The amountof cleaved tRNA was determined from the ultraviolet absorbance at 260 nm (A260) of the supernatant fraction. Nedd8 Conjugation Inhibitory Activity A purified NAE (heterodimer of APPBP1 and UBA3) solution was prepared in the following manner. The human APPBP1 gene (NCBI Reference Sequence number: NM_003905) region corresponding to amino acids 1 to 534 of human APPBP1 protein (NCBI Reference Sequence number: NP_003896, full length: 534 amino acids) was inserted in pBacPAK9 (produced by Clontech) to construct a plasmid pBacPAK9-APPBP1 for expressing APPBP1 full-length protein having a His tag and a TEV protease-recognition sequence at the N-terminus. Next, the human UBA3 gene (NCBI Reference Sequence number: NM_003968) region corresponding to amino acids 1 to 463 of human UBA3 protein (NCBI Reference Sequence number: NP_003959, full length: 463 amino acids) was inserted in pBacPAK9 to construct a plasmid pBacPAK9-UBA3 for expressing UBA3 full-length protein. The pBacPAK9-APPBP1 or pBacPAK9-UBA3, and BacPAK6 DNA were cotransfected into insect cells (Sf9, produced by Clontech) to produce a recombinant baculovirus containing APPBP1 or UBA3 gene. The APPBP1 gene recombinant baculovirus was mixed with the UBA3 gene recombinant baculovirus, and the resulting mixture was used to infect Sf9 cells. The baculovirus-infected Sf9 cells were incubated at 28° C. with shaking for 72 hours in Grace's Insect Medium (produced by Gibco), and the collected cells were suspended in a lysis buffer (50 mM Tris-HCl, 200 mM NaCl, and 10% glycerol (pH 7.4)), followed by sonication. The sonicated cell solution was centrifuged (40,000×g, for 30 minutes) to obtain the supernatant as a crude extract. The crude extract was fractionated on a HisTrap HP column (produced by GE Healthcare) and a TALON Superflow column (produced by Clontech), followed by the addition of a TEV protease. Then, a His-tag cleavage reaction was performed at 4° C. overnight. The resulting solution was subjected to TALON Superflow column chromatography, and the unadsorbed fraction was collected. This fraction was applied to a HiLoad 16/60 Superdex 75 prep grade column equilibrated with 50 mM Tris-HCl, 200 mM NaCl, and 10% glycerol (pH 7.4), and fractionated. A fraction containing an APPBP1/UBA3 complex was concentrated to obtain a purified NAE solution. The purification above was performed entirely at 4° C. The purified NAE solution was stored at −80° C. until use.A purified GST-UBC12 solution was prepared in the following manner. The human UBC12 gene (NCBI Reference Sequence number: NM_003969) region corresponding to amino acids 1 to 183 of human UBC12 protein (NCBI Reference Sequence number: NP_003960, full length: 183 amino acids) was inserted in pGEX-4T-2 (produced by GE Healthcare) to construct a plasmid pGEX-UBC12 for expressing UBC12 full-length protein having a GST tag at the N-terminus. The pGEX-UBC12 was introduced into Escherichia coli (BL21 (DE3), produced by Stratagene), followed by culture at 37° C. for 2 hours in the presence of 1 mM isopropyl-beta-D-thiogalactopyranoside (produced by Sigma-Aldrich). The collected Escherichia coli was suspended in PBS, followed by sonication. The sonicated cell solution was centrifuged (40,000×g, for 5 minutes) to obtain the supernatant as a crude extract. A Glutathione Sepharose 4B carrier (produced by GE Healthcare) was added to the crude extract, and eluted with 50 mM Tris-HCl (pH 7.9), 150 mM NaCl, and a 10 mM reduced glutathione solution, followed by dialysis with 50 mM HEPES (pH 7.5) and a 0.05% BSA solution to obtain a purified GST-UBC12 solution. The purified GST-UBC12 solution was divided and stored at −80° C. until use.The Nedd8 conjugation inhibitory activity was measured using an AlphaScreen assay system. Each of the purified NAE solution and GST-UBC12 solution was diluted with an assay buffer (50 mM HEPES (pH 7.5), 5 mM MgCl2, 1 mM DTT, 0.05% BSA) and added to a 384-well plate (#3673, produced by Corning) containing the test compound. After reaction at room temperature for 30 minutes, a solution obtained by diluting ATP and Biotin-Nedd8 Binding Activity Assay The binding affinity of the present compounds to human 5-HT1A receptor, human D4 receptor, and human D2 receptor was measured in a manner mentioned below.CHO cell membrane fraction in which human 5-HT1A receptor, human D4 receptor, and human D2 receptor were expressed was purchased from PerkinElmer Co., Ltd. In the evaluation test of binding, the test compound dissolved in DMSO, each receptor membrane preparation diluted with buffer solution, and [3H] 8-OH-DPAT (for 5-HT1A receptor), [3H] dopamine (for D4 receptor), or [3H] spiperone (for D2 receptor) (all were obtained from PerkinElmer Co., Ltd.) were mixed, and each mixture was incubated at room temperature for 30 or 60 minutes. The nonspecific binding to each receptor was evaluated by a competition binding experiment in the presence of 10 μmol/L 8-OH-DPAT, 10 μmol/L dopamine, or 10 μmol/L spiperone, respectively. The radioactivity of each receptor-binding sample was measured with a liquid scintillation counter (PerkinElmer Co., Ltd.), the 50% inhibitory concentration was calculated, and Ki value was evaluated based on the dissociation constant and the substrate concentration caluculated in the saturated bond test, which was used as binding affinity. Enzymatic Assay HDAC1 and 6 reagents: FLAG-tagged HDACs 1 and 6 were prepared in-house by protein expression in HEK293F cells followed by anti-FLAG affinity purification. Assays were performed with buffer containing 20 mM HEPES, pH 8.0 [Boston BioProducts, catalog #BB-104, 1M stock], 137 mM NaCl [Sigma, catalog #S5150, 5M stock], 2.7 mM KCl [BioChemika, catalog #87526, 4M stock], 1 mM MgCl2 [Fluka, catalog #63020, 1M stock], and 0.05% BSA (Fraction V) [Invitrogen, catalog #15260, 7.5% stock]. In addition to the above buffer ingredients, TCEP [CalBiochem, catalog #580561, 500 mM stock] was added at a final concentration of 0.5 mM to the buffer for the HDAC6 assays. HDAC1, 2, 3, and 6 enzymes were run at the final concentrations of 0.3 nM, 1.5 nM, 0.3 nM, and 1.333 nM, respectively. Fluor-de-Lys substrate [BioMol Research Laboratories, catalog #KI-104], used to evaluate enzyme activity, was added at the final concentrations of 20 uM, 40 uM, 20 uM, and 2.5 uM for HDACs 1, 2, 3, and 6. To enable detection of the signal, Developer [BioMol Research Laboratories, catalog #KI-105] was added at a 1:250 dilution to the stop solution, which also included 10 uM SAHA [Sigma, catalog #SML0061] to ensure complete termination of the reaction. Evaluation of Binding Activity to Human 5-HT1A Receptor, Human D4 Receptor, and Human D2 Receptor The binding affinity of the present compounds to human 5-HT1A receptor, human D4 receptor, and human D2 receptor was measured in a manner mentioned below.CHO cell membrane fraction in which human 5-HT1A receptor, human D4 receptor, and human D2 receptor were expressed was purchased from PerkinElmer Co., Ltd. In the evaluation test of binding, the test compound dissolved in DMSO, each receptor membrane preparation diluted with buffer solution, and [3H] 8-OH-DPAT (for 5-HT1A receptor), [3H] dopamine (for D4 receptor), or [3H] spiperone (for D2 receptor) (all were obtained from PerkinElmer Co., Ltd.) were mixed, and each mixture was incubated at room temperature for 30 or 60 minutes. The nonspecific binding to each receptor was evaluated by a competition binding experiment in the presence of 10 μmol/L 8-OH-DPAT, 10 μmol/L dopamine, or 10 μmol/L spiperone, respectively. The radioactivity of each receptor-binding sample was measured with a liquid scintillation counter (PerkinElmer Co., Ltd.), the 50% inhibitory concentration was calculated, and Ki value was evaluated based on the dissociation constant and the substrate concentration calculated in the saturated bond test, which was used as binding affinity. JAK Enzyme Assay The activity of the isolated recombinant JAK1 and JAK2 kinase domain was measured by monitoring phosphorylation of a peptide derived from JAK3 (Val-Ala-Leu-Val-Asp-Gly-Tyr-Phe-Arg-Leu-Thr-Thr, fluorescently labeled on the N-terminus with 5-carboxyfluorescein) using the Caliper LabChip technology (Caliper Life Sciences, Hopkinton, Mass.). To determine inhibition constants (Ki), compounds were diluted serially in DMSO and added to 50 μL kinase reactions containing purified enzyme (1.5 nM JAK1, or 0.2 nM JAK2), 100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 1.5 μM peptide substrate, ATP (25 μM), 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 25 μL of an EDTA containing solution (100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip 3000 according to the manufacturer's specifications. Ki values were then determined using the Morrison tight binding model. EP3 Binding Activity Measurement To each well of a 96 well plate, 10 μL of a medium (dimethyl sulfoxide; DMSO) which had been 10-fold diluted with an assay buffer solution (10 mmol/L KH2PO4 KOH containing 1 mmol/L EDTA, 10 mmol/L Mg2+ and 100 mmol/L NaCl, pH6.0) or a DMSO solution of a test compound (final concentration of DMSO: 0.5%), 90 μL of assay buffer solution, 50 μL of 10 nmol/L [3H]-PGE2 (final concentration: 2.5 nmol/L), and 50 μL of human EP3 receptor expressing cell membrane fraction (manufactured by Millipore) (membrane protein mass: 2.5 μg) were placed, and subjected to incubation at room temperature. In nonspecific binding group, instead of the medium, 2 mmol/L of PGE2 was added (final concentration of PGE2: 10 μmol/L). After 60 minutes, a membrane fraction was subjected to suction filtration using a cell harvester, and collected onto a glass fiber (GF/B) plate (hereinafter, filter plate ) which had been wetted with a washing buffer solution (10 mmol/L KH2PO4 KOH containing 100 mmol/L NaCl, pH6.0) in advance. Furthermore, an operation of adding about 0.2 mL of the washing buffer solution to the 96 well plate after suction filtration, and carrying out suction filtration was repeated twice, and the remaining membrane fractions were collected. The filter plate was washed with 150 mL of washing buffer solution twice, and then dried at 50° C. to 60° C. for about 60 minutes. After an accessary back seal was attached to the bottom surface of the filter plate, about 50 μL per well of liquid scintillation cocktail was added to the filter plate, and a scaling film sheet was attached to the upper surface of the filter plate. The filter plate was shaken, and then radioactivity (cpm) of the filter plate was measured using a microplate scintillation counter. A specific binding amount of [3H]-PGE2 to EP3 receptor was calculated by subtracting the radioactivity of the nonspecific binding group from the radioactivity other than the radioactivity of the nonspecific binding group. The inhibition rate by the test compound was calculated from the specific binding amount of [3H]-PGE2 in a medium group and the compound of the present invention group, a Ki value (dissociation constant of the test compound) was calculated from estimated IC50 value (the concentration of the test compound required for inhibiting 50% with respect to the specific binding amount of the medium group) according to the following formula. CETP Inhibition Assay A recombinant human CETP protein (manufactured by Roar Biomedical Inc.; 4.5 ng), the acceptor lipoprotein described in (2) above (32.5 μg) and 5,5'-dithio-bis-(2-nitrobenzoic acid) (7 mM, 15 μl) were taken in a 96 well plate and the total amount of the mixture was adjusted to 48.5 μl with the PBS solution. The test compound [DMSO solution (concentration: 0.15, 0.5, 1.5, 5, 15, 50, 150 and 500 μM); 1.5 μl] was added to each well and the mixture was incubated in a thermostatic bath at 37 °C. for 60 minutes. The reconstituted HDL (50 μl) described in (1) above was added to each well and the mixture was reacted in a thermostatic bath at 37 °C. for 60 minutes. The 96 well plate was moved onto ice and a precipitation reagent [a mixed solution of magnesium chloride (60 mM) and 0.1% dextran sulfate[1/1(v/v)]; 15 μl] was added to each well, and then the mixture was allowed to stand on ice for 15 minutes. The reaction solution (80 μl) in each well was moved to a filter plate and centrifuged at 1500 rpm for 1 minute, and the filtrate which passed through the filter was designated as the HDL fraction and the radioactivity thereof was measured with a scintillation counter. Fluorescence Polarization (FP) Assay In a flat black bottom 96 well plate (Corning), the buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 20% Glycerol, 0.5 mM TCEP, 0.01% Tween 20) is added—200 μL to column 1 (blank), 295 μL to column 2, 150 μL to columns 3-12. 5 μL of protein (179.0 μM JAK2-JH2-WT, 154.7 μM JAK2-JH2-V617F, 126.3 μM JAK2-JH1) were added to column 2. 150 μL was transferred, using a multichannel pipette, from column 2 to 3, 3 to 4, 4 to 5, until reaching the last column to make a serial dilutions (1:2). 50 μ1 of 24.0 nM tracer were added from columns 2-12 and fluorescence polarization was measured at λexc=485±20 nm, λem=535±25 nm using an Infinite F500 plate reader until no FP variation was observed. From the lowest and highest FP values (tracer free and tracer fully bound to JAK) fraction of ligand bound to the protein to ligand total (Lb/Lt) was calculated for each concentration of the JAK2-JH2-WT, JAK2-JH2-V617F, and JAK2-JH1 (FIGS. 8A-8B). Experiments were carried out by quadruplicates in three independent experiments. The data provided a typical saturation-binding curve and Kd was calculated fitting the results to the Hill equation using Prism 7. JAK Enzyme Assay The activity of the isolated recombinant JAK1 and JAK2 kinase domain was measured by monitoring phosphorylation of a peptide derived from JAK3 (Val-Ala-Leu-Val-Asp-Gly-Tyr-Phe-Arg-Leu-Thr-Thr, fluorescently labeled on the N-terminus with 5-carboxyfluorescein) using the Caliper LabChip technology (Caliper Life Sciences, Hopkinton, Mass.). To determine inhibition constants (Ki), compounds were diluted serially in DMSO and added to 50 μL kinase reactions containing purified enzyme (1.5 nM JAK1, or 0.2 nM JAK2), 100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 1.5 μM peptide substrate, ATP (25 μM), 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 25 μL of an EDTA containing solution (100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip 3000 according to the manufacturer's specifications. Ki values were then determined using the Morrison tight binding model (Morrison, J. F., Biochim. Biophys. Acta. 185:269-296 (1969); William, J. W. and Morrison, J. F., Meth. RTX Competition Binding Assay. Binding studies with [3H]RTX were carried out as follows. The binding assay mixtures were prepared in 1.5 ml centrifuge tubes and consisted of a fixed concentration (approximately 2 nM) of [3H]RTX (37 Ci/mmol specific activity, PerkinElmer Life Sciences), various concentrations of competing ligands, and 100 g protein of membranes from induced CHO-hTRPV1 expressing cells (approximately 1-3×106 cells) in Dulbecco's phosphate buffered saline (DPBS, with Ca2+ & Mg2+) for a total volume of 350 μl. The assay mix contained bovine serum albumin at a final concentration of 0.25 mg/ml (Cohn fraction V; Sigma-Aldrich, St. Louis, Mo.). In each set of experiments, nonspecific binding were determined in the presence of 200 nM non-radioactive RTX. The binding reaction was initiated by placing the assay mixture in a 37° C. shaking water bath for 60 minutes (−30 rpm). The assay mixture was then chilled on ice for 2-3 min before adding 100 μl of α1-acid glycoprotein (2 mg/ml; Sigma-Aldrich) and mixed thoroughly. The tubes were kept on ice for an additional 10 min. The bound and free ligands were then separated by centrifugation (12,200 rpm for 15 minutes) in a Beckman Coulter centrifuge Allegra 21R. 200 μl of supernatant was collected for determination of free ligand. The remainder was removed by aspiration. VEGFR2 and RET9 Binding Assay A competition binding assay, was used to measure the ability of a compound to compete for binding of an immobilized adenosine triphosphate (ATP) site directed ligand using a DNA-tagged vascular endothelial growth receptor 2 (VEGFR2) as the target. A VEGFR2 tagged T7 phage strain was prepared in an Escherichia coli (E. coli) derived from the BL21 strain. The E. coli were grown to log-phase, infected with VEGFR2 tagged T7 phage and then incubated with shaking at 32° C. until lysis. The lysate containing the kinase was then centrifuged and filtered to remove cell debris. Affinity resin for the VEGFR2 assay was prepared by treating Streptavidin-coated magnetic beads with a biotinylated small molecule ligand for 30 minutes at room temperature. The beads were blocked with excess biotin and then washed with blocking buffer (SEABLOCK (PIERCE), 1% bovine serum albumin, 0.17% phosphate buffered saline, 0.05% TWEEN 20, 6 mM dithiothreitol). The binding reaction was initiated by combining in a well of a polystyrene 96-well plate, DNA tagged VEGFR2, liganded affinity beads and the serial diluted test compound in 1× binding buffer (20% SEABLOCK, 0.17× phosphate buffered saline, 0.05% TWEEN 20, 6 mM dithiothreitol) in a final volume of 0.135 ml. The assay plates were incubated at room temperature with shaking for 1 hour and then the beads were washed with wash buffer (1× phosphate buffered saline, 0.05% TWEEN 20). Human Recombinant Btk Dissociation Dialysis Assay A test compound was incubated with human recombinant Btk (100 nM) for 1.5 h at a concentration of 25 times the IC50 of Btk inhibition or 200 nM (whichever was greater). The incubation was performed in assay buffer (20 mM HEPES pH 7.5, 10 mM MgCl2, 2 mM dithiothreitol, 50 μg/mL bovine serum albumen and 0.015% Brij 35). The reaction mixture was then dialyzed twice for 6 h each time against 1 L of assay buffer. The dialyzed reaction mixture (0.5 μL) was then diluted into a solution (100 μL) of ATP (2 mM) and substrate peptide (5 μM Src-tide, AnaSpec) such that the final Btk concentration was 1 nM (along with any inhibitor still bound). The assay was performed in matrix polypropylene 384-well plates. The reaction progress curve was monitored on the Caliper LABCHIP by electrophoretic separation of the substrate and phosphorylated product (pressure −1.2 psi, downstream voltage −500 V, upstream voltage −2300 V). Reaction velocity was measured over the linear phase and percent recovery of Btk activity was assessed at 2 h by comparing the fraction of phosphorylated peptide product relative to a DMSO-treated Btk control reaction containing no Example inhibitor. A control reaction with no Btk was also used to measure the background signal. A reversible inhibitor would show nearly complete recovery of Btk activity, while an irreversible inhibitor, would show little or no recovery of Btk activity. In Vitro DGAT2 Assay For determination of IC50 values, the reactions were carried out in 384-well white Polyplates (Perkin Elmer) in a total volume of 20 μL. To 14 of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 20 minutes. To this mixture, 10 μL of hDGAT2 membrane fraction (0.01 mg/mL) diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock) was added. After this mixture was preincubated at room temperature for 2 hours, DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). Inhibition of Recombinant PDE2A The DNA encoding PDE2A (NM002599) was cloned and the gene was inserted in the baculovirus and the enzyme-protein expressed in SF21-cells. The enzyme was isolated from these cells by harvesting the cells by an centrifugation at 200 g to collect the cells. The cells were resuspended in 50 mM Tris-HCl/5 mM MgCl2 buffer (pH=7.4)(Sigma, Deisenhofen, Germany; Merck, Darmstadt, Germany) and lysed by a sonication of the cells (three times for 15 seconds, Labsonic U, Fa. Braun, Degersheim, Switzerland, level high). The membrane fraction of PDE2A was obtained by a centrifugation at 48 000 g for 1 h, resuspended in buffer and stored at −70° C.PDE2A activity was determined in a one step procedure in microtiterplates. The reaction mixture of 100 μl contained 50 mM Tris-HCl/5 mM MgCl2 buffer (pH=7.4)(Sigma, Deisenhofen, Germany; Merck, Darmstadt, Germany), 0.5 OA [3H]-cAMP (PerkinElmer, Shelton, USA), 1000 nM cGMP and the enzyme. Non-specific enzyme activity was tested in the absence of cGMP. The reaction was initiated by addition of the substrate solution and was carried out at 37° C. for 30 minutes. Enzymatic activity then was stopped by addition of 25 μl SPA-beads (PerkinElmer, Shelton, USA). One hour later the mixture was measured in a liquid scintillation counter for microtiterplates (Microbeta Trilux). For pipetting of the incubation mixture we routinely use the robot Biomek (Fa. Beckman). JAK Enzyme Assay The activity of the isolated recombinant JAK1 and JAK2 kinase domain was measured by monitoring phosphorylation of a peptide derived from JAK3 (Val-Ala-Leu-Val-Asp-Gly-Tyr-Phe-Arg-Leu-Thr-Thr, fluorescently labeled on the N-terminus with 5-carboxyfluorescein) using the Caliper LabChip technology (Caliper Life Sciences, Hopkinton, Mass.). To determine inhibition constants (Ki) compounds were diluted serially in DMSO and added to 50 μL kinase reactions containing purified enzyme (1.5 nM JAK1, or 0.2 nM JAK2), 100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 1.5 μM peptide substrate, ATP (25 μM), 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 25 μL of an EDTA containing solution (100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip 3000 according to the manufacturer's specifications. Ki values were then determined using the Morrison tight binding model (Morrison, J. F., Biochim. Biophys. Acta. 185:269-296 (1969); William, J. W. and Morrison, J. F., Meth. Enzymol., 63:437-467 (1979)) modified for ATP-competitive inhibition [Ki=Ki,app/(1+[ATP]/Km,app)]. JAK Enzyme Assays The activity of the isolated recombinant JAK1 and JAK2 kinase domain was measured by monitoring phosphorylation of a peptide derived from JAK3 (Val-Ala-Leu-Val-Asp-Gly-Tyr-Phe-Arg-Leu-Thr-Thr, fluorescently labeled on the N-terminus with 5-carboxyfluorescein) using the Caliper LabChip technology (Caliper Life Sciences, Hopkinton, Mass.). To determine inhibition constants (Ki), compounds were diluted serially in DMSO and added to 50 μL kinase reactions containing purified enzyme (1.5 nM JAK1, or 0.2 nM JAK2), 100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 1.5 μM peptide substrate, ATP (25 μM), 10 mM MgCl2, 4 mM DTT at a final DMSO concentration of 2%. Reactions were incubated at 22° C. in 384-well polypropylene microtiter plates for 30 minutes and then stopped by addition of 25 μL of an EDTA containing solution (100 mM HEPES buffer (pH 7.2), 0.015% Brij-35, 150 mM EDTA), resulting in a final EDTA concentration of 50 mM. After termination of the kinase reaction, the proportion of phosphorylated product was determined as a fraction of total peptide substrate using the Caliper LabChip 3000 according to the manufacturer's specifications. Ki values were then determined using the Morrison tight binding model (Morrison, J. F., Biochim. Biophys. Acta. 185:269-296 (1969); William, J. W. and Morrison, J. F., Meth. Enzymol., 63:437-467 (1979)) modified for ATP-competitive inhibition [Ki=Ki,app/(1+[ATP]/Km,app)]. Metalloenzyme Activity V79-4 cells expressing recombinant andrenodoxin and andrenodoxin reductase with either recombinant human CYP11B2 or CYP11B1 were prepared according to methods previously described (LaSala et al 2009 Anal Bioch 394:56-61). An enzyme enriched microsomal fraction was prepared from cellular lysates and subsequently used as the enzyme source for determining inhibitor IC50s. The substrate Km values were experimentally determined for 11-deoxycorticosterone (CYP11B2 substrate) and 11-deoxycortisol (CYP11B1 substrate). Enzyme assays for inhibitor screening employed CYP11B2 and CYP11B1 enzyme enriched microsomes and were run at the Km of the respective substrates. Products of the enzyme reactions, aldosterone for CYP11B2 or cortisol for CYP11B1, were measured by LC-MS. Assays were run under conditions of less than 20% substrate turnover. Inhibitor IC50s were generated by determining the product formation in the absence or presence of inhibitor at various concentrations. In the absence of the test compound, the product formed (Pt) in each data set was defined as 100% activity. In the absence of enzyme, the product formed (Pb) in each data set was defined as 0% activity. The percent activity in the presence of each inhibitor was calculated according to the following equation: % activity=(P−Pb)/(Pt−Pb), where P=the product formed in the presence of the inhibitor. The IC50 value was defined as the inhibitor concentration causing a 50% decrease in activity relative to the no inhibitor control reaction. DNA Helicase Assay Helicase assay was demonstrated using the purified fraction of PfUDN. The specially designed partial duplex substrate consisted of a 32P-labelled 47-mer DNA oligodeoxynucleotide annealed to M13mp19 phage ssDNA. This oligodeoxynucleotide of the nucleotide sequence 5'-(T)15GTTTTCCCAGTCACGAC(T)15-3' contains 15 base-pairs of non-complementary region (T)15 at both the 5' and 3' ends. Oligodeoxynucleotide was labeled at 5'-end with T4 polynucleotide kinase (PNK) (5U) (New England Biolabs) and 1.85 MBq of [γ-32P] ATP (specific activity 222 TBq/mmol) at 37°C for one hour and then annealed using standard annealing buffer (20 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 100 mM NaCl, 1 mM DTT) with 0.5 μg of single-stranded circular M13mp19 (+)phage DNA by heating at 95°C for 1 min and then transferring immediately to 65°C for 2 min and then slow cooling to room temperature. Using gel filtration through a Sepharose 4B column (Pharmacia, Sweden) the nonhybridized oligodeoxynucleotide was removed [Ahmad et al., PLoS One, 7(11):e49385]. The reaction volume of 10 μl containing the 32P-labeled helicase substrate (1000 cpm/10 μl) in appropriate buffer (20 mM Tris-HCl, pH 8.0, 8 mM DTT, 1.0 mM MgCl2, 20 mMKCl and 16 μg/ml BSA) and PfUDN was incubated at 37°C for 60 min. The substrate and products were separated by electrophoresis on a nondenaturing 12% PAGE and the gel was exposed to hyper film for autoradiography or scanned on phosphoimager. Protective Effects of Compounds on Primary Cerebellum Granule Cells of Rats Isolated primary cerebellum granule cells of infant rats were inoculated in 96-well plates with 1.2×105/well by using 10% FBS+25 mM KCl+2 mM Glutamine+1% of double-antibody BME medium. After 24 hours, cytarabine with a final concentration of 10 iM was added to inhibit the proliferation of neurogliocyte cells. After the day 4, glucose with the final concentration of 5 mM was added every four days to complement energy metabolism and water evaporation of cells. The materials were placed in a cell incubator (37° C., 5% CO2) to be cultured for 10 days. A 200 iM of glutamate was used to induce the excitotoxic injury of the primary cerebellum granule cells, with test groups of normal control group, glutamate group, pretreatment groups with different memantine nitrate compounds, and pretreatment control group with memantine. In the testing groups, the compounds of NM-001, NM-002, NM-003, NM-004, NM-005, NM-008, NM-009, NM-011, NM-012 and memantine were respectively added. After pre-protection for 2 h, 200 iM of glutamate was added to induce cell damage for 24 h, and then MTT was added to culture for 4 h. The supernatant fraction was sucked, and 150 iL of DMSO was added to each well for dissolving. After blending with shaking, the light absorption values under 570 nm wavelength was measured with a microplate reader, and the viability of cells was calculated. Cell viability (%)=absorbance of different groups/absorbance of the normal control group×100%. Biological Assay Receptor competition binding assays are run in a buffer made up of DPBS (1 L) (Hyclone #SH30028.03), 2.2 g BSA Fraction v (Roche #9048-46-8), 100 mL glycerol (Fischer #56-81-5) and 40 mL DMSO (reagent grade). The final wells contain 20 μg/mL aprotinin and 20 μg/mL leupeptin and 10 μM Pefabloc. Typically, receptor binding assays include radio-labeled ligands, such as 7 nM [3H]-25-hydroxycholesterol for alpha binding, 20 nM [3H]-3-[[4-[[3-(2,6-dichlorophenyl)-5-isopropyl-isoxazol-4-yl]methoxy]-N,2-dimethyl-anilino]methyl]benzoic acid for beta binding, and 6 nM [3H]-25-hydroxycholesterol for gamma binding, and 0.5 μg RORa receptor, 0.03 μg RORb receptor, or 0.13 μg RORg receptor per well. Assays are typically run in 96-well format. Competing test compounds are added at various concentrations ranging from about 0.4 nM to 25 μM. Non-specific binding is determined in the presence of 250 nM 25-hydroxycholesterol for RORa and RORg binding, 250 nM 3-[[4-[[3-(2,6-dichlorophenyl)-5-isopropyl-isoxazol-4-yl]methoxy]-N,2-dimethyl-anilino]methyl]benzoic acid for RORb binding. The sample, label and receptor solutions are combined in a 96 well assay plate (Costar 3632) and incubated overnight at room temperature, then 25 μl beads (Amersham YSi (2-5 micron) copper His-tag Spa Beads, #RPNQ0096) for a final bead concentration of 1 mg/well is added to each reaction. Plates are mixed for 30 minutes on an orbital shaker at room temperature. After an incubation of 4 hours, plates are read in a Wallac MICROBETA counter. Binding Assay A competition binding assay was used to measure the ability of a compound to compete for binding of an immobilized adenosine triphosphosphate (ATP) site directed ligand using a DNA-tagged vascular endothelial growth receptor 2 (VEGFR2) as the target. The ability of the test compound to compete with the immobilized ligand was measured using quantitative polymerase chain reaction (qPCR) of the DNA tag. A VEGFR2 tagged T7 phage strain was prepared in an Escherichia coli (E. coli) derived from the BL21 strain. The E. coli were grown to log-phase, infected with VEGFR2 tagged T7 phage and then incubated with shaking at 32° C. until lysis. The lysate containing the kinase was then centrifuged and filtered to remove cell debris. Affinity resin for the VEGFR2 assay was prepared by treating Streptavidin-coated magnetic beads with a biotinylated small molecule ligand for 30 minutes at room temperature. The beads were blocked with excess biotin and then washed with blocking buffer SEABLOCK(PIERCE), 1% bovine serum albumin, 0.17% phosphate buffered saline, 0.05% TWEEN 20, 6 mM dithiothreitol). The binding reaction was initiated by combining in a well of a polystyrene 96-well plate, DNA tagged VEGFR2, liganded affinity beads and the serial diluted test compound in 1× binding buffer (20% SEABLOCK, 0.17× phosphate buffered saline, 0.05% TWEEN 20, 6 mM dithiothreitol) in a final volume of 0.135 ml. The assay plates were incubated at room temperature with shaking for 1 hour and then the beads were washed with wash buffer (1× phosphate buffered saline, 0.05% TWEEN 20). The beads were re-suspended in elution buffer (1× phosphate buffered saline, 0.05% TWEEN 20, 0.05 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The VEGFR2 concentration in the eluate was measured using qPCR. Alpha-Synuclein Competitive Binding Assay (A) Expression and purification of recombinant wild-type human α-synuclein: 0.5 mM IPTG was used to induce production of wild type human α-synuclein by bacteria transformed with a full-length α-synuclein expression plasmid. After shaking at 16° C. for 20 hours, cell pellet was resuspended in lysis buffer (10 mM Tris, 1 mM EGTA, 0.75 mM NaCl, 1 mM PMSF, pH 7.5) and lysed by sonication followed by centrifugation (6000 g, 30 min, 4° C.). The supernatant was then boiled at 95° C. for 15 min with manual agitation by every 5 minutes, followed by centrifugation (6000 g, 30 min, 4° C.). Supernatant was dialyzed with (10 mM Tris, 1 mM EGTA, 50 mM NaCl, pH 7.5) buffer and concentrated with concentrator. The collected sample was loaded onto a Superdex200 column, the flow-through fraction was then loaded onto a Q-HP column. Collected fractions with α-synuclein eluates were pooled, concentrated and stored at −80° C.(B) Preparation of aggregated α-synuclein: 5 mg/mL of α-synuclein in PBS buffer (pH 7.4) was incubated in tube with at 37° C. with shaking (700 rpm) for 5-10 days.(C) In vitro fluorometric α-synuclein binding assays: 2 μM α-synuclein was incubated with serially diluted compound (three-fold serial dilutions, from 10 to 0.001 μM) in a 96-well plate at 37° C. for 1 hour. Fluorescence intensity was read by microplate spectrometer. Compound Kd values were calculated using the following equation: Y=Bmax*X/(Kd+X), where X is the concentration of compound; Y is the fluorescence signal of (compound+α-synuclein)−(compound+DMSO); and Bmax is the maximum signal. Biochemical Assay: hK-RasG12C Interaction Assay with hSOS1 The assay buffer containes 5 mM HEPES pH 7.4 (Applichem), 150 mM NaCl (Sigma), 10 mM EDTA (Promega), 1 mM DTT (Thermofisher), 0.05% BSA Fraction V, pH 7.0, (ICN Biomedicals), 0.0025% (v/v) Igepal (Sigma) and 100 mM KF (FLUKA). The expression and purification of N-terminal GST-tagged K-RasG12C and N-terminal His-tagged SOS1 is described below. Concentrations of protein batches used are optimized to be within the linear range of the HTRF signal. A Ras working solution is prepared in assay buffer containing typically 10 nM GST-hK-RasG12C and 2 nM antiGST-Eu(K) (Cisbio, France). A SOS1 working solution is prepared in assay buffer containing typically 20 nM His-hSOS1 and 10 nM anti-6His-XL665 (Cisbio, France). An inhibitor control solution is prepared in assay buffer containing 10 nM anti-6His-XL665 without SOS1. Fifty nl of a 100-fold concentrated solution of the test compound in DMSO are transferred into a black microtiter test plate (384 or 1536, Greiner Bio-One, Germany). For this, either a Hummingbird liquid handler (Digilab, MA, USA) or an Echo acoustic system (Labcyte, CA, USA) is used. All steps of the assay are performed at 20° C. A volume of 2.5 μl of the Ras working solution is added to all wells of the test plate using a Multidrop dispenser (Thermo Labsystems). After 2 min preincubation, 2.5 μl of the SOS1 working solution are added to all wells except for those wells at the side of the test plate that are subsequently filled with 2.5 μl of the inhibitor control solution. After 60 min incubation the fluorescence is measured with a Pherastar (BMG, Germany) using the HTRF module (excitation 337 nm, emission 1: 620 nm, emission 2: 665 nm). Competitive Inhibition Assay A competitive inhibition binding assay is performed using membrane homogenates prepared from HEK-293 cells that express recombinant hMC4-R, hMC3-R, or hMC5-R, and from B-16 mouse melanoma cells (containing endogenous MC1-R). In some instances, HEK-293 cells that express recombinant hMC1-R were employed. In the examples that follow, all MC3-R, MC4-R and MC5-R values are for human recombinant receptors. MC1-R values are for B-16 mouse melanoma cells, unless the heading is hMC1-R, in which case the value is for human recombinant MC1-R. Assays were performed in 96 well GF/B Millipore multiscreen filtration plates (MAFB NOB10) pre-coated with 0.5% bovine serum albumin (Fraction V). Membrane homogenates were incubated with 0.2 nM (for hMC4-R) 0.4 nM (for MC3-R and MC5-R) or 0.1 nM (for mouse B16 MC1-R or hMC1-R) [I125]-NDP-α-MSH (Perkin Elmer) and increasing concentrations of test peptides of the present invention in buffer containing 25 mM HEPES buffer (pH 7.5) with 100 mM NaCl, 2 mM CaCl2, 2 mM MgCl2, 0.3 mM 1,10-phenanthroline, and 0.2% bovine serum albumin. After incubation for 60 minutes at 37° C., the assay mixture was filtered and the membranes washed three times with ice-cold buffer. Filters were dried and counted in a gamma counter for bound radioactivity. Non-specific binding was measured by inhibition of binding of [I125]-NDP-α-MSH in the presence of 1 μM NDP-α-MSH. Maximal specific binding (100%) was defined as the difference in radioactivity (cpm) bound to cell membranes in the absence and presence of 1 μM NDP-α-MSH. Radioactivity (cpm) obtained in the presence of test compounds was normalized with respect to 100% specific binding to determine the percent inhibition of [I125]-NDP-α-MSH binding. Enzymatic Assay Endothelial lipase activity was measured using a fluorescent substrate, A10070, (Invitrogen, CA) doped into an artificial vesicle containing DMPG (Avanti Polar Lipids) as the excipient. Vesicles were prepared by combining 285 uL of 1 mMDMPG in a 1:1 mixture of MeOH and CHCl3 with 15 uL of 1 mM A10070 in a 1:1 mixture of MeOH and CHCl3. The mixture was dried under nitrogen and resuspended in 150 uL of 50 mM HEPES pH 8.0 buffer containing 100 mM NaCl and 0.2 mM EDTA. The sample was allowed to sit at rt for 15 min and then was sonicated 3x4 mins on ice with a Branson Sonicator using duty cycle 1. This preparation provides vesicles with a mole fraction of 0.05 for the FRET substrate.The enzymatic assay was measured using white, opaque 96-well half area plates. Each well contained 60 uL of assay buffer (50 mM HEPES pH 8.0, 50 mM NaCl and 1 mM CaCl2) and 2 ul of a DMSO solution containing compound of interest. Conditioned media obtained from HT-1080 cells, which were transformed by RAGE technology (Athersys) to overexpress endogenous EL, was added and the reaction was allowed to incubate for 20 min at 37deg; C. with gentle agitation. The reaction was started by the addition of 20 uL of a 1:4 dilution of vesicles. The final total reaction volume was 100 uL. The reaction rates were measured on a Gemini plate reader with an excitation wavelength of 488 nm and a emission of 530 nm. Readings were taken every 20 seconds for 10 min with agitation between each reading. The slope of the linear portion of the readout was used to calculate the rate of the reaction. Fluorescence Polarization (FP) Assay In a typical experiment the PDE10 inhibitory activity of the compounds of the present invention was determined in accordance with the following experimental method. PDE10A2 was amplified from human fetal brain cDNA (Clontech, Mountain View, Calif.) using a forward primer corresponding to nucleotides 56-77 of human PDE10A2 (Accession No. AF127480, Genbank Identifier 4894716), containing a Kozak consensus sequence, and a reverse primer corresponding to nucleotides 2406-2413 of human PDE10A2 (Accession No. AF127480, Genbank Identifier 4894716). Amplification with Easy-A polymerase (Stratagene, La Jolla, Calif.) was 95° C. for 2 minutes followed by thirty three cycles of 95° C. for 40 seconds, 55° C. for 30 seconds, and 72° C. for 2 minutes 48 seconds. Final extension was 72° C. for 7 minutes. The PCR product was TA cloned into pcDNA3.2-TOPO (Invitrogen, Carlsbad, Calif.) according to standard protocol. AD293 cells with 70-80% confluency were transiently transfected with human PDE10A2/pcDNA3.2-TOPO using Lipofectamine 2000 according to manufacturer specifications (Invitrogen, Carlsbad, Calif.). Cells were harvested 48 hours post-transfection and lysed by sonication (setting 3, 10x5 sec pulses) in a buffer containing 20 mM HEPES, 1 mM EDTA and protease inhibitor cocktail (Roche). Lysate was collected by centrifugation at 75,000 xg for 20 minutes. Supernatant containing the cytoplasmic fraction was used for evaluation of PDE10A2 activity. The fluorescence polarization assay for cyclic nucleotide phosphodiesterases was performed using an IMAP FP kit supplied by Molecular Devices, Sunnyvale, Calif. (product #R8139). IMAP technology has been applied previously to phosphodiesterase assays (Huang, W., et al., J. Biomol Screen, 2002, 7: 215). Assays were performed at room temperature in 384-well microtiter plates with an incubation volume of 20.2 uL. Human Glucocorticoid Receptor (hGR) Ligand-Binding Assay The human lymphoblast cell line IM9 (ATCC, Bethesda, MD) was cultivated in RPMI 1640 media containing 10% fetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml), and 2 mM L-glutamine at 37° and 7% CO2 in a humidified incubator. Cells were centrifuged for 10 minutes at 1500 g and were washed in PBS and repelleted. Cell were then resuspended in homogenization buffer consisting of: 10 mM TES, 10 mM sodium molybdate, 1 mM EDTA, pH 7.4, 20 mM 2-mercaptoethanol, and 10% glycerol. Disruption of the cells was performed by nitrogen cavitation using 2×15 minutes at 600 to 750 psi nitrogen in a N2 cavitator at 0° C. The cell preparation was then centrifuged at 27,000 g for 15 minutes, and the resultant supernatant (=cytosol of IM9 cells) was centrifuged at 103,000 g for 60 minutes at 4° C. The amount of protein in the supernatant fraction was determined using a BCA assay kit and aliquots were snap frozen in a dry ice-acetone bath and stored at −70° C. Competitive binding assays were done in duplicate in homogenization buffer with a total volume of 200 μl. To this end, 1 mg of IM9 cytosol, 0.05 μCi (1.5 nM) of 3H-dexamethasone and unlabeled Example compounds as competitor compounds at 1 μM were mixed. The reaction was stopped after incubation at 0° C. for 16 to 18 hours by the addition of 100 μl of a charcoal-dextran mixture (2% activated charcoal, 0.5% dextran in 10 mM Tris, 1 mM EDTA, pH 7.4). Another incubation step at 0° C. for 10 minutes followed before the samples were centrifuged for 5 minutes at 8200 g. 100 μl of the supernatant) was finally assayed for radioactivity by liquid scintillationspectrometry, and percentage inhibition of 3H-dexamethasone binding was calculated. In Vitro DGAT2 Assay For determination of IC50 values, the reactions were carried out in 384-well white Polyplates (Perkin Elmer) in a total volume of 20 uL. To 1 uL of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 uL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 20 minutes. To this mixture, 10 uL of hDGAT2 membrane fraction (0.01 mg/mL) diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock) was added. After this mixture was preincubated at room temperature for 2 hours, DGAT2 reactions were initiated by the addition of 4 uL of substrates containing 30 uM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 uM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 uL of 1% H3PO4. After the addition of 45 uL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). Plates were centrifuged at 2,000×g for 1 min in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). DGAT2 activity was measured by quantifying the generated product [14C]tridecanoylglycerol in the upper organic phase. Myofibril Assays To evaluate the effect of compounds on the ATPase activity of full-length cardiac myosin in the context of the native sarcomere, skinned myofibril assays were performed. Bovine cardiac myofibrils were obtained by homogenizing bovine cardiac left ventricular tissue in the presence of a detergent such as triton X-100. Such treatment removes membranes and a majority of the soluble cytoplasmic proteins but leaves intact the cardiac sarcomeric acto-myosin apparatus. Myofibril preparations retain the ability to hydrolyze ATP in an Ca2+ regulated manner. ATPase activities of such myofibril preparations in the presence and absence of compounds were assayed at Ca2+ concentrations activating to a defined fraction of the maximal rate (i.e., 25%, 75%). Small molecule agents were assessed for their ability to inhibit the steady-state ATPase activity of bovine cardiac myofibrils using pyruvate kinase and lactate dehydrogenase (PK/LDH)-coupled enzyme system. This assay regenerates myosin-produced ADP into ATP by oxidizing NADH, producing an absorbance change at 340 nm. Prior to testing small molecule agents, the bovine cardiac myofibrils were assessed for their calcium responsiveness and the calcium concentration that achieves either a 50% (pCa50) or 75% (pCa75) activation of the myofibril system was chosen as the final condition for assessing the inhibitory activity of the small molecule agents. All enzymatic activity was measured in a buffered solution containing 12 mM PIPES (piperazine-N,N′-bis(2-ethanesulfonic acid), 2 mM magnesium chloride at pH 6.8 (PM 12 buffer). Final assay conditions were 1 mg/mL of bovine cardiac myofibrils, 4 U/mL pyruvate kinase, 6 U/mL lactate dehydrogenase, 50 μM ATP, 0.1 mg/mL BSA (bovine serum albumin), 10 ppm antifoam, 1 mM DTT, 0.5 mM NADH, 1.5 mM PEP, 0.6 mM EGTA, and an amount of CaCl2) sufficient to achieve either 50% or 75% activation of the myofibril ATPase activity. Nox2 Assay Cells: Human blood was purchased in buffy coat, prepared the same day for isolation of neutrophils, from Labjoy AB, Lund, Sweden. Blood components were separated by density gradient centrifugation using Ficoll-Paque Plus. Plasma, PBMCs and Ficoll were removed before erythrocytes were removed by dextran sedimentation. Remaining erythrocytes were lyses before neutrophils were washed and counted. Isolated neutrophils were kept on ice resuspended in HBSS without Mg and Ca until assayed.Buffers: The isoluminol buffer contained Isoluminol (0.175 mg/ml) and HRP fraction II (1.75 U/ml). The buffer was prepared by diluting these ingredients at 4× working concentration in HBSS.Procedures: Compounds (Nox inhibitors) were diluted at 4× working concentration and titrated from 100 μM to 0.006 μM in 1:4 steps. PMA was diluted in Isoluminol buffer at 4× working concentration for a final concentration of 30 ng/ml. Compounds had a final DMSO concentration of 1% in the wells; therefore a DMSO control of 1% was included on the plates. 25 μl diluted compound or control/well were added to a white 96-well plate. 25 μl/well of PMA diluted in Isoluminol buffer was added to each well. To non-stimulated control wells only Isoluminol buffer was added. Neutrophils were washed and resuspended at 2×106 cells/ml in HBSS with Mg and Ca just before adding 50 μl of the neutrophil cell suspension/well, which was followed by immediate initiation of luminescence measurement. Luminescence was measured using a FluoStar Optima (BMG Labtech). Graphs were performed using Prism 5 for Mac OS X (Prism 5.0 Software, San Diego Calif. USA). Inhibitors were evaluated at 50% inhibition (IC50) in comparison to cell control without inhibitor present. Radioligand Binding Assay HEK-293 cells stably expressing the human or feline FP receptor, or EP1, EP2, or EP4 receptors were washed with TME buffer, scraped from the bottom of the flasks, and homogenized for 30 sec using a Brinkman PT 10/35 polytron. TME buffer was added to achieve a final 40 ml volume in the centrifuge tubes (the composition of TME is 100 mM TRIS base, 20 mM MgCl2, 2M EDTA; 10N HCl is added to achieve a pH of 7.4).The cell homogenate was centrifuged at 19000 r.p.m. for 20 min at 4° C. using a Beckman Ti-60 rotor. The resultant pellet was resuspended in TME buffer to give a final 1 mg/ml protein concentration, as determined by Biorad assay. Radioligand binding competition assays vs. [3H-]17-phenyl PGF2a (5 nM) were performed in a 100 μl volume for 60 min. Binding reactions were started by adding plasma membrane fraction. The reaction was terminated by the addition of 4 ml ice-cold TRIS-HCl buffer and rapid filtration through glass fiber GF/B filters using a Brandel cell harvester. The filters were washed 3 times with ice-cold buffer and oven dried for one hour.[3H] PGE2 (specific activity 180 Ci mmol) was used as the radioligand for EP receptors. [3H] 17-phenyl PGF2a was employed for FP receptor binding studies. Binding studies employing EP1, EP2, EP4 and FP receptors were performed in duplicate in at least three separate experiments. A 200 μl assay volume was used. Incubations were for 60 min at 25° C. and were terminated by the addition of 4 ml of ice-cold 50 mM TRIS-HCl, followed by rapid filtration through Whatman GF/B filters and three additional 4 ml washes in a cell harvester (Brandel). Receptor Competition Binding Assay Receptor competition binding assays are run in a buffer made up of DPBS (1 L) (Hyclone #SH30028.03), 2.2 g BSA Fraction v (Roche #9048-46-8), 100 mL glycerol (Fischer #56-81-5) and 40 mL DMSO (reagent grade). The final wells contain 20 μg/mL aprotinin and 20 μg/mL leupeptin and 10 μM Pefabloc. Typically, receptor binding assays include radio-labeled ligands, such as 7 nM [3H]-25-hydroxycholesterol for alpha binding, 20 nM [3H]-3-[[4-[[3-(2,6-dichlorophenyl)-5-isopropyl-isoxazol-4-yl]methoxy]-N,2-dimethyl-anilino]methyl]benzoic acid for beta binding, and 6 nM [3H]-25-hydroxycholesterol for gamma binding, and 0.5 μg RORα receptor, 0.03 μg RORβ receptor, or 0.13 μg RORγ receptor per well. Assays are typically run in 96-well format. Competing test compounds are added at various concentrations ranging from about 0.4 nM to 25 μM. Non-specific binding is determined in the presence of 250 nM 25-hydroxycholesterol for RORα and RORγ binding, 250 nM 3-[[4-[[3-(2,6-dichlorophenyl)-5-isopropyl-isoxazol-4-yl]methoxy]-N,2-dimethyl-anilino]methyl]benzoic acid for RORβ binding. The sample, label and receptor solutions are combined in a 96 well assay plate (Costar 3632) and incubated overnight at room temperature, then 25 μl beads (Amersham YSi (2-5 micron) copper His-tag Spa Beads, #RPNQ0096) for a final bead concentration of 1 mg/well is added to each reaction. Plates are mixed for 30 minutes on an orbital shaker at room temperature. After an incubation of 4 hours, plates are read in a Wallac MICROBETA counter. Fluorometric Assay The capability of compounds described herein to inhibit the enzymatic activity of vascular adhesion protein-1 (VAP-1) was determined by fluorometric assay. The assay procedure provided by R&D Systems was modified slightly for use as an inhibitor screening assay. The assay measured the H2O2 generated from the oxidative deamination of benzylamine by rhVAP-1 by converting Amplex Red to the fluorescent product, resorufin, via horseradish peroxidase (HRP) and H2O2. Reaction buffer was 10 mM NaHCO3, pH 7.4. A standard curve was prepared by serially diluting H2O2 in reaction buffer. A 10 μM solution of resorufin was prepared in reaction buffer and was dispensed to 2 wells. These wells served as positive control for maximum fluorescence. Reaction buffer was added to the individual wells so that the final volume in each well was 100 μL. Amplex Red was diluted to 1 mM in reaction buffer. A 10 U stock of HRP was prepared in reaction buffer. A reaction cocktail was prepared by mixing equal parts 1 mM Amplex Red with 10 U HRP with 3 parts reaction buffers. The reaction cocktail was added to the appropriate wells, so that that final concentration in the well was 0.05 mM Amplex Red and 0.5 U HRP. A 200 μM solution of benzylamine was prepared in reaction buffer. Benzylamine was added to the appropriate wells so that the final concentration was 50 μM. Inhibitors were diluted in DMSO to 20 and distributed to the appropriate wells. UP-1207 served a positive control. rhVAP-1 was purchased from R&D Systems. The concentration used for each lot was determined so that the activity was equal to the first lot. Two wells were left blank as controls. The plate was incubated for 30 minutes at 37 C., protected from light under foil. The plate was read at 530/35 emission and 590/35 excitation. Data processing was performed using Excel and Prism software. VAP-1 Assay The capability of compounds described herein to inhibit the enzymatic activity of vascular adhesion protein-1 (VAP-1) was determined by fluorometric assay. The assay procedure provided by R&D Systems was modified slightly for use as an inhibitor screening assay. The assay measured the H2O2 generated from the oxidative deamination of benzylamine by rhVAP-1 by converting Amplex Red to the fluorescent product, resorufin, via horseradish peroxidase (HRP) and H2O2. Reaction buffer was 10 mM NaHCO3, pH 7.4. A standard curve was prepared by serially diluting H2O2 in reaction buffer. A 10 μM solution of resorufin was prepared in reaction buffer and was dispensed to 2 wells. These wells served as positive control for maximum fluorescence. Reaction buffer was added to the individual wells so that the final volume in each well was 100 μL. Amplex Red was diluted to 1 mM in reaction buffer. A 10 U stock of HRP was prepared in reaction buffer. A reaction cocktail was prepared by mixing equal parts 1 mM Amplex Red with 10 U HRP with 3 parts reaction buffers. The reaction cocktail was added to the appropriate wells, so that that final concentration in the well was 0.05 mM Amplex Red and 0.5 U HRP. A 200 μM solution of benzylamine was prepared in reaction buffer. Benzylamine was added to the appropriate wells so that the final concentration was 50 μM. Inhibitors were diluted in DMSO to 20 and distributed to the appropriate wells. LJP-1207 served a positive control. rhVAP-1 was purchased from R&D Systems. The concentration used for each lot was determined so that the activity was equal to the first lot. Two wells were left blank as controls. The plate was incubated for 30 minutes at 37 C., protected from light under foil. The plate was read at 530/35 emission and 590/35 excitation. Data processing was performed using Excel and Prism software. 5α-Reductase Activity Assay The reaction mixture contained a final volume of 1 mL: 1 mM dithiothreitol, sodium phosphate buffer 40 mM, at pH 6.5 for human prostate, 2 mM NADPH, 2 nM [1,2,6,7-3H]T [Cabeza et al., Steroids 60:630-5]. The reaction in duplicate was started when it was added to the enzymatic fraction (500 μg protein in a volume of 80 μL) incubated at 37°C for 60 min [Hirosumi et al., J. Steroid Biochem. Mol. Biol., 52:357-63] and stopped by mixing with 1 mL of dichloromethane; this was considered as the end point. Incubation without tissue was used as a control. The mixture (incubation medium/dichloromethane) was agitated on a vortex for 1 min and the dichloromethane phase was separated and placed in another tube. This procedure was repeated four more times. The dichloromethane extract was evaporated to dryness under a nitrogen stream and suspended in 50 mL of methanol that was spotted on high-performance thin layer chromatography (HPTLC) Keiselgel 60 F254 plates. T and DHT were used as carriers, and were applied in different lanes on both lateral sides of the plates (T, T + DHT, and DHT). The plates were developed in chloroform-acetone, 9:1, and were air-dried; the chromatography was repeated twice more. The steroid carriers were detected using phosphomolybdic acid reagent (DHT) and with an ultraviolet (UV) lamp (254 nm) (T). After the plates were segmented into pieces of 1 cm each, these were cut off and the strips soaked in 5 mL of Ultima Gold (Packard). The radioactivity was determined in a scintillation counter (Packard Tri-Carb 2100 TR). The radioactivity content in the segment corresponding to T and DHT carriers was identified. Radioactivity with identical chromatographic behavior to the DHT standard was considered as the DHT transformation. Control incubations, chromatography separations, and identifications were carried out in the same manner as described above, except that the tubes did not contain tissue. Biochemical Assay The assay buffer containes 5 mM HEPES pH 7.4 (Applichem), 150 mM NaCl (Sigma), 10 mM EDTA (Promega), 1 mM DTT (Thermofisher), 0.05% BSA Fraction V, pH 7.0, (ICN Biomedicals), 0.0025% (v/v) Igepal (Sigma) and 100 mM KF (FLUKA).The expression and purification of N-terminally GST-tagged human K-RasG12C (termed GST-hK-RasG12C) and N-terminally His-tagged human SOS1 (termed His10-hSOS1) is described in WO 2019/201848, page 220 line 12-34 and page 222, line 13-25 (expression) and page 222, line 26 to page 223, line 17 (purification). Concentrations of protein batches used are optimized to be within the linear range of the HTRF signal. A Ras working solution is prepared in assay buffer containing typically 10 nM GST-hK-RasG12C and 2 nM antiGST-Eu(K) (Cisbio, France). A SOS1 working solution is prepared in assay buffer containing typically 20 nM His-hSOS1 and 10 nM anti-6His-XL665 (Cisbio, France). An inhibitor control solution is prepared in assay buffer containing nM anti-6His-XL665 without SOS1.Fifty nl of a 100-fold concentrated solution of the test compound in DMSO are transferred into a black microtiter test plate (384 or 1536, Greiner Bio-One, Germany). For this, either a Hummingbird liquid handler (Digilab, MA, USA) or an Echo acoustic system (Labcyte, CA, USA) is used.All steps of the assay are performed at 20° C. A volume of 2.5 μl of the Ras working solution is added to all wells of the test plate using a Multidrop dispenser (Thermo Labsystems). After 2 min preincubation, 2.5 μl of the SOS1 working solution are added to all wells except for those wells at the side of the test plate that are subsequently filled with 2.5 μl of the inhibitor control solution. After 60 min incubation the fluorescence is measured with a Pherastar (BMG, Germany) using the HTRF module (excitation 337 nm, emission 1: 620 nm, emission 2: 665 nm). Biological Activity Endothelial lipase (EL) and hepatic lipase (HL) activities were measured using a fluorescent substrate, A10070, (Invitrogen, CA) doped into an artificial vesicle containing DMPG (Avanti Polar Lipids) as the excipient. Vesicles were prepared by combining 571 μL of 29 mM DMPG in a 1:1 mixture of MeOH and CHCl3 with 2000 μL of 1 mM A10070 in a 1:1 mixture of MeOH and CHCl3. The mixture was dried under nitrogen in multiple vials then resuspended in 20 mL total volume of 50 mM HEPES pH 8.0 buffer containing 50 mM NaCl and 0.2 mM EDTA. The sample was allowed to sit at room temperature for 15 min and then was sonicated 3×4 mins on ice with a Branson Sonicator using duty cycle 1. This preparation provides vesicles with a mole fraction of 0.11 for the FRET substrate.The enzymatic assay was measured using 384-well white Optiplates. Each well contained 20 μL of assay buffer (50 mM HEPES pH 8.0, 50 mM NaCl and 1 mM CaCl2) and 0.25 μL of a DMSO solution containing a compound of interest. EL or HL (10 μL) was added and allowed to incubate with the compound for 30 min at 37° C. The source of EL was conditioned media obtained from HT-1080 cells that were transformed using RAGE technology (Athersys) to overexpress endogenous EL, and HL was partially purified from conditioned media obtained from COS cells overexpressing HL. The reaction was started by the addition of 10 μL of a 1:10 dilution of vesicles. The final total reaction volume was 20.25 μL. The reaction rates were measured on a Gemini plate reader with an excitation wavelength of 490 nm and an emission wavelength of 530 nm. Readings were taken over a period of 60 minutes, and the slope between 300 and 900 secs of the readout was used to calculate the rate of the reaction. Competitive Binding Assay Table 11: Receptor membranes were prepared from the CHO-K1 recombinant AequoScreen® cell line stably expressing the human 5-HT2A receptor (PerkinElmer, Waltham, MA, USA). Cells were suspended in 4× volume in buffer A (15 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 0.3 mM EDTA, 1 mM EGTA) (1 g cell-4 mL buffer) and homogenized in a Dounce homogenizer. The crude membrane fraction was collected following two consecutive centrifugation steps at 40,000×g for 25 minutes separated by a washing step in buffer A. The final pellet was resuspended in buffer B (75 mM Tris-HCl, pH 7.5, 12.5 mM MgCl2, 0.3 mM EDTA, 1 mM EGTA, 250 mM sucrose) in a concentration of 80 mg wet cell weight in 0.5 mL buffer, aliquoted and flash frozen on dry ice. Protein content was determined using the bicinchoninic acid assay in the presence of sulfhydryl reagents with bovine serum albumin (BSA) as a standard.In binding experiments, 15 μg protein/well membrane preparation and 1 nM ketanserin hydrochloride, [ethylene-3H] (PerkinElmer) as radioligand were incubated with compounds or vehicle (DMSO, 1% (v/v) final concentration) in an incubation buffer (50 mM Tris, 0.3% BSA, pH 7.4). Non-specific binding (NSB) was determined in the presence of 1 μM mianserin hydrochloride (Tocris, Bristol, UK). Samples were incubated in a final volume of 250 μL for 15 minutes at 25° C. Binding reactions were terminated by rapid filtration through a Filtermate™ harvester (PerkinElmer) using UniFilter® GF/C plates pre-soaked for at least 1 hour in 0.5% (v/v) polyethylene imine (PEI, dissolved in distilled water). The filter plates were washed three times with 0.5 mL of ice-cold washing buffer (50 mM Tris, pH 7.4). Washed filter plates were dried at 40° C. for 60 minutes and 40 μL of Microscint™-20 scintillation cocktail (PerkinElmer) was added to each well. Radioactivity was determined with a MicroBeta2® microplate counter (PerkinElmer). IDO1 Enzyme Assay Compounds to be tested were serially diluted in ten 3-fold steps in DMSO starting from 10 mM DMSO stocks. Compound dilutions or DMSO alone were then dispensed from the dilution plate into a Greiner black 384-well assay plate (catalog #781086) using an Echo 555 acoustic liquid handler (Labcyte).HIS-tagged IDO1 protein was recombinantly expressed in Escherichia coli using ZYP5052 autoinduction media supplemented with 500 μM delta aminolevulinic acid for 48 hours at 16 degrees Celcius. IDO1 protein was purified using Ni2+-affinity resin and size exclusion chromatography. Purified protein was then diluted in assay buffer (50 mM Tris pH 7.0, 1% glycerol, 20 uM methylene blue, 0.05% Tween-20, 20 mM sodium ascorbate, 100 units/mL catalase to obtain a final IDOL concentration of 40 nM. IDOL solution (30 uM) or buffer alone (30 uM) were dispensed to wells of the assay plate using a BioRAPTR liquid dispenser (Beckman Coulter). Assay plates containing compound and IDO1 enzyme were incubated at room temperature for 30 minutes. Afterwards, 10 μL of 400 μM tryptophan in assay buffer were added to each well of the assay plate using a BioRAPTR liquid dispenser. Plates were incubated at room temperature for 60 minutes and reactions were quenched by addition of 10 μL of 0.5 M methyl isonipecotate in dimethyl sulfoxide. Plates were sealed and incubated at 37 degrees Celcius for 4 hours or 50 degrees Celcius for 2 hours. The plates are allowed to cool and then centrifuged for 1 minute at 1000×g. The resulting fluoresence was measured in an Envision plate reader (Perkin Elmer) with a 400/25 nm excitation filter and an 510/20 nm emission filter.The fluoresence intensity of each well was corrected for the background observed in wells that did not receive IDO1 and was expressed as a fraction of the intensity observed in wells that received IDO1 enzyme and DMSO only. Potencies were calculated by linear least squares fit to the four parameter logistic IC50 equation. PLpro Inhibition Assay The assays were performed in 40 μL total volume in black half area 96-well plates (Greiner PN 675076) at 25° C. The assay buffer contained 20 mM Tris-HCl pH 7.45, 0.1 mg/mL bovine serum albumin fraction V, and 2 mM reduced glutathione. The final DMSO concentration in all assays was 2.5% v/v. PLpro initial rates were measured using a previously established fluorogenic peptide substrate assay (e.g., K. Ratia et al., Proc. Natl. Acad. Sci. USA, 105(42), 16119-24, 2008). The substrates Z-LRGG-AMC and Z-RLRGG-AMC were dissolved to 10 mM in DMSO and stored in aliquots at −20° C. To determine Michaelis-Menten parameters, 20 μL enzyme solution was dispensed into wells (250 nM final concentration), and reactions were initiated by adding 20 μL substrate to 0-500 μM final concentration, in triplicate. Release of aminomethylcoumarin (AMC) was monitored by a fluorescence plate reader every 50 s with an excitation wavelength of 345 nm and an emission wavelength of 445 nm, 6.25 mm read height, and gain=60. After background subtraction of the average of no-enzyme negative controls, product formation was quantified using a 0.02-5 μM calibration curve of AMC. Initial rates were determined for time points in the initial linear range by linear regression in Excel, and GraphPad Prism 9 was used to perform nonlinear regression of the Michaelis-Menten equation to the initial rate vs. substrate concentration data to yield KM and Vmax.Inhibitors were characterized by dispensing 10 μL enzyme solution into wells (115 nM final concentration), followed by 10 μL inhibitor solution at 4× desired final concentrations in 5% v/v DMSO in at least duplicate, centrifuging briefly, and incubating for 30 min. Reactions were initiated by adding 20 μL substrate to 100 μM final concentration. Initial rates were determined as described above and % residual activities were determined by normalizing to the average of no inhibitor controls (100% activity). Radioligand Binding HEK-293 cells stably expressing the human or feline FP receptor, or EP1, EP2, or EP4 receptors were washed with TME buffer, scraped from the bottom of the flasks, and homogenized for 30 sec using a Brinkman PT 10/35 polytron. TME buffer was added to achieve a final 40 ml volume in the centrifuge tubes (the composition of TME is 100 mM TRIS base, 20 mM MgCl2, 2M EDTA; 10N HCl is added to achieve a pH of 7.4). The cell homogenate was centrifuged at 19000 r.p.m. for 20 min at 4 °C. using a Beckman Ti-60 rotor. The resultant pellet was resuspended in TME buffer to give a final 1 mg/ml protein concentration, as determined by Biorad assay. Radioligand binding competition assays vs. [3H-]17-phenyl PGF2a (5 nM) were performed in a 100 μl volume for 60 min. Binding reactions were started by adding plasma membrane fraction. The reaction was terminated by the addition of 4 ml ice-cold TRIS-HCl buffer and rapid filtration through glass fiber GF/B filters using a Brandel cell harvester. The filters were washed 3 times with ice-cold buffer and oven dried for one hour. [3H−] PGE2 (specific activity 180 Ci mmol) was used as the radioligand for EP receptors. [3H] 17-phenyl PGF2a was employed for FP receptor binding studies. Binding studies employing EP1, EP2, EP4 and FP receptors were performed in duplicate in at least three separate experiments. A 200 μl assay volume was used. Incubations were for 60 min at 25 °C. and were terminated by the addition of 4 ml of ice-cold 50 mM TRIS-HCl, followed by rapid filtration through Whatman GF/B filters and three additional 4 ml washes in a cell harvester (Brandel). Competition studies were performed using a final concentration of 5 nM [3H]-PGE2, or 5 nM [3H] 17-phenyl PGF2a and non-specific binding determined with 10^−5M of unlabeled PGE2, or 17-phenyl PGF2a, according to receptor subtype studied. Receptor Binding Assay The buffer solution for the receptor binding test was dispensed to the wells of a 96-well assay plate (Greiner) at 22.5 μL/well. DMSO solutions of a test compound, which were prepared at an 80-time higher concentration using 100% dimethyl sulfoxide (DMSO), were added to the wells at 2.5 μL/well (final concentrations of 1 nM to 100 nM), and the solutions were mixed. As a radiolabeled ligand, 125I-substance P (Substance P, [125I]Tyr8-, Perkin Elmer) was used. 125I-substance P was diluted with the buffer solution for the receptor binding test to a concentration resulting in 125 pmol/25 μL/well and added to the 96-well assay plate, and the solutions were mixed. The membrane fraction prepared from the human NK1 receptor-expressing cells was diluted with the buffer solution for the receptor binding test to a concentration resulting in 8 to 10 μg/well, suspended until the suspension became in such a homogenous state that the suspension could flow through a 27G injection needle smoothly and then added to the 96-well assay plate at 150 μL/well. Then, the plate was incubated at room temperature for 60 minutes while shaking the plate. The reaction solutions were suction-filtered through a multiscreen 96-well filter plate (Millipore) which had been pre-treated with 0.3% polyethyleneimine, and the reaction was terminated by washing with a washing solution (50 mM Tris and 0.02% bovine serum albumin, pH 7.4) four times. The bottom of the microplate was dried at 60° C., and then 100 μL/well of MicroScint 20 (PerkinElmer) was dispensed to the wells. The top of the plate was sealed with TopSeal A (PerkinElmer), and the plate was shaken for 5 to 10 minutes. Then, the radioactivities were measured with TopCount NXT (registered trademark) (PerkinElmer). The radioactivity of each well was calculated by subtracting the radioactivity of the well to which 10 μM aprepitant was added (non-specific binding). SYK Assa SYK kinase assay is a standard kinase assay adapted to a 96 well plate format. This assay is performed in 96-well format for IC50 determination with 8 samples which represented 10 half log dilutions and a 40 μL reaction volume. The assay measures the incorporation of radiolabeled 33P γATP into an N-terminally biotinylated peptide substrate, derived from naturally occurring phosphoacceptor consensus sequence (Biotin-11aa DY*E). Phosphorylated products were detected upon termination of reactions with EDTA and the addition of Streptavidin coated beads. Representative results are in Table II above.Assay plates: 96-well MultiScreen 0.65 um filter plates (Millipore Cat. No.: MADVNOB10) Streptavidin coated beads: Streptavidin Sepharose , suspension 5.0 mL, in 50 mM EDTA/PBS diluted (1:100), (Amersham, Cat. No.: 17-5113-01)Compounds: 10 mM in 100% dimethylsulfoxide (DMSO), final conc.: compound 0.003-100 uM in 10% DMSOEnzyme: SYK RPA purified, truncated construct of Spleen Tyrosine Kinase aa 360-635, stock solution 1 mg/mL, MW: 31.2 KDa, final conc.:0.0005 μM.Peptide 1: biotinylated peptide is derived from a naturally occurring phosphor-acceptor consensus sequence (Biotin-EPEGDYEEVLE), special order from QCB, stock solution 20 mM, final conc.: 5.0 μM.ATP: Adenosine-5′-triphosphate 20 mM, (ROCHE Cat. No.: 93202720), final concentration: 20 μMBuffer: HEPES: 2-Hydroxyethyl piperazine-2-ethanesulfonic acid (Sigma, Cat. No.: H-3375)final concentration: 50 mM HEPES pH7.5BSA: Bovine Serum Albumin Fraction V, fatty acid free (Roche Diagnostics GmbH, Cat. No. 9100221) diluted to a final concentration of 0.1%EDTA: EDTA stock solution 500 mM, (GIBCO, Cat. No.: 15575-038) final concentration: 0.1 mMDTT: 1,4-Dithiothreitol (Roche Diagnostics GmbH, Cat. No.: 197777), final conc.: 1 mMMgCl2×6H2O: MERCK, Cat. No.: 105833.1000, final concentration: 10 mMAssay Dilution Buffer (ADB): 50 mM HEPES, 0.1 mM EGTA, 0.1 mM Na Vanadate, 0.1 mM β-glycerophosphate, 10 mM MgCl2, 1 mM DTT, 0.1% BSA, pH 7.5Bead wash buffer: 10 g/L PBS (Phosphate buffered saline) with 2M NaCl+1% phosphoric acid. TR-FRET Assay TR-FRET assay for retinol-induced RBP4-TTR interaction Binding of a desired RBP4 antagonist displaces retinol and induces hindrance for RBP4-TTR interaction resulting in the decreased FRET signal (FIG. 7). Bacterially expressed MBP-RBP4 and untagged TTR were used in this assay. For the use in the TR-FRET assay the maltose binding protein (MBP)-tagged human RBP4 fragment (amino acids 19-201) was expressed in the Gold(DE3)pLysS E. coli strain (Stratagene) using the pMAL-c4x vector. Following cell lysis, recombinant RBP4 was purified from the soluble fraction using the ACTA FPLC system (GE Healthcare) equipped with the 5-ml the MBP Trap HP column. Human untagged TTR was purchased from Calbiochem. Untagged TTR was labeled directly with Eu3+ Cryptate-NHS using the HTRF Cryptate Labeling kit from CisBio following the manufacturer's recommendations. HTRF assay was performed in white low volume 384 well plates (Greiner-Bio) in a final assay volume of 16 ul per well. The reaction buffer contained 10 mM Tris-HCl pH 7.5, 1 mM DTT, 0.05% NP-40, 0.05% Prionex, 6% glycerol, and 400 mM KF. Each reaction contained 60 nM MBP-RBP4 and 2 nM TTR-Eu along with 26.7 nM of anti-MBP antibody conjugated with d2 (Cisbio). Titration of test compounds in this assay was conducted in the presence of 1 uM retinol. All reactions were assembled in the dark under dim red light and incubated overnight at +4° C. wrapped in aluminum foil. TR-FRET signal was measured in the SpectraMax M5e Multimode Plate Reader (Molecular Device). Fluorescence was excited at 337 nm and two readings per well were taken: Reading 1 for time-gated energy transfer from Eu(K) to d2 (337 nm excitation, 668 nm emission, counting delay 75 microseconds, counting window 100 microseconds) and Reading 2 for Eu(K) time-gated fluorescence (337 nm excitation, 620 nm emission, counting delay 400 microseconds, counting window 400 microseconds). TR-FRET Assay for Retinol-Induced RBP4-TTR Interaction Binding of a desired RBP4 antagonist displaces retinol and induces hindrance for RBP4-TTR interaction resulting in the decreased FRET signal (FIG. 7 ). Bacterially expressed MBP-RBP4 and untagged TTR were used in this assay. For the use in the TR-FRET assay the maltose binding protein (MBP)-tagged human RBP4 fragment (amino acids 19-201) was expressed in the Gold(DE3)pLysS E. coli strain (Stratagene) using the pMAL-c4x vector. Following cell lysis, recombinant RBP4 was purified from the soluble fraction using the ACTA FPLC system (GE Healthcare) equipped with the 5-ml the MBP Trap HP column. Human untagged TTR was purchased from Calbiochem. Untagged TTR was labeled directly with Eu3+ Cryptate-NHS using the HTRF Cryptate Labeling kit from CisBio following the manufacturer's recommendations. HTRF assay was performed in white low volume 384 well plates (Greiner-Bio) in a final assay volume of 16 μl per well. The reaction buffer contained 10 mM Tris-HCl pH 7.5, 1 mM DTT, 0.05% NP-40, 0.05% Prionex, 6% glycerol, and 400 mM KF. Each reaction contained 60 nM MBP-RBP4 and 2 nM TTR-Eu along with 26.7 nM of anti-MBP antibody conjugated with d2 (Cisbio). Titration of test compounds in this assay was conducted in the presence of 1 μM retinol. All reactions were assembled in the dark under dim red light and incubated overnight at +4° C. wrapped in aluminum foil. TR-FRET signal was measured in the SpectraMax M5e Multimode Plate Reader (Molecular Device). Fluorescence was excited at 337 nm and two readings per well were taken: Reading 1 for time-gated energy transfer from Eu(K) to d2 (337 nm excitation, 668 nm emission, counting delay 75 microseconds, counting window 100 microseconds) and Reading 2 for Eu(K) time-gated fluorescence (337 nm excitation, 620 nm emission, counting delay 400 microseconds, counting window 400 microseconds). The TR-FRET signal was expressed as the ratio of fluorescence intensity: Flu665/Flu620×10,000. TR-FRET Assay for Retinol-induced RBP4-TTR Interaction Binding of a desired RBP4 antagonist displaces retinol and induces hindrance for RBP4-TTR interaction resulting in the decreased FRET signal (FIG. 7). Bacterially expressed MBP-RBP4 and untagged TTR were used in this assay. For the use in the TR-FRET assay the maltose binding protein (MBP)-tagged human RBP4 fragment (amino acids 19-201) was expressed in the Gold(DE3)pLysS E. coli strain (Stratagene) using the pMAL-c4x vector. Following cell lysis, recombinant RBP4 was purified from the soluble fraction using the ACTA FPLC system (GE Healthcare) equipped with the 5-ml the MBP Trap HP column. Human untagged TTR was purchased from Calbiochem. Untagged TTR was labeled directly with Eu3+ Cryptate-NHS using the HTRF Cryptate Labeling kit from CisBio following the manufacturer's recommendations. HTRF assay was performed in white low volume 384 well plates (Greiner-Bio) in a final assay volume of 16 μl per well. The reaction buffer contained 10 mM Tris-HCl pH 7.5, 1 mM DTT, 0.05% NP-40, 0.05% Prionex, 6% glycerol, and 400 mM KF. Each reaction contained 60 nM MBP-RBP4 and 2 nM TTR-Eu along with 26.7 nM of anti-MBP antibody conjugated with d2 (Cisbio). Titration of test compounds in this assay was conducted in the presence of 1 μM retinol. All reactions were assembled in the dark under dim red light and incubated overnight at +4° C. wrapped in aluminum foil. TR-FRET signal was measured in the SpectraMax M5e Multimode Plate Reader (Molecular Device). Fluorescence was excited at 337 nm and two readings per well were taken: Reading 1 for time-gated energy transfer from Eu(K) to d2 (337 nm excitation, 668 nm emission, counting delay 75 microseconds, counting window 100 microseconds) and Reading for Eu(K) time-gated fluorescence (337 nm excitation, 620 nm emission, counting delay 400 microseconds, counting window 400 microseconds). The TR-FRET signal was expressed as the ratio of fluorescence intensity: Flu665/Flu620x10,000. Biochemical Assay The binding of compounds to ASK1 were determined using DiscoverX's proprietary technology. KINOMEscan is based on a competition binding assay that quantitatively measures the ability of a compound to compete with an immobilized, active-site directed ligand. The assay is performed by combining three components: DNA-tagged ASK1 kinase; immobilized ligand; and a test compound. The ability of the test compound to compete with the immobilized ligand is measured via quantitative PCR of the DNA tag. Streptavidin-coated magnetic beads were treated with biotinylated small molecule ligands for 30 minutes at room temperature to generate affinity resins for ASK1 kinase assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific binding. Binding reactions were assembled by combining DNA-tagged ASK1 kinases, liganded affinity beads, and test compounds in 1× binding buffer (20% SeaBlock, 0.17×PBS, 0.05% Tween 20, 6 mM DTT). Test compounds were prepared as 111× stocks in 100% DMSO. Kds were determined using an 11-point 3-fold compound dilution series with three DMSO control points. All compounds for Kd measurements are distributed by acoustic transfer (non-contact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.9%. All reactions were performed in polypropylene 384-well plate. Each fraction had a final volume of 0.02 ml. The assay plates were incubated at room temperature with shaking for 1 hour and the affinity beads were washed with wash buffer (1×PBS, 0.05% Tween 20). The beads were then re-suspended in elution buffer (1×PBS, 0.05% Tween 20, 0.504 non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The kinase concentration in the eluates was measured by qPCR. Most Kds were determined using a compound top concentration=30,000 nM. If the initial Kd determined was <0.5 nM (the lowest concentration tested), the measurement was repeated with a serial dilution starting at a lower top concentration. Competitive Inhibition Assay Using [I125]-NDP-a-MSH A competitive inhibition binding assay is performed using membrane homogenates prepared from HEK-293 cells that express recombinant hMC4-R, hMC3-R, or hMC5-R, and from B-16 mouse melanoma cells (containing endogenous MC1-R). In some instances, HEK-293 cells that express recombinant hMC1-R were employed. In the examples that follow, all MC3-R, MC4-R and MC5-R values are for human recombinant receptors. MC1-R values are for B-16 mouse melanoma cells, unless the heading is hMC1-R, in which case the value is for human recombinant MC1-R. Assays were performed in 96 well GF/B Millipore multiscreen filtration plates (MAFB NOB10) pre-coated with 0.5% bovine serum albumin (Fraction V). Membrane homogenates were incubated with 0.2 nM (for hMC4-R) 0.4 nM (for MC3-R and MC5-R) or 0.1 nM (for mouse B16 MC1-R or hMC1-R) [I125]-NDP-α-MSH (Perkin Elmer) and increasing concentrations of test peptides of the present invention in buffer containing 25 mM HEPES buffer (pH 7.5) with 100 mM NaCl, 2 mM CaCl2, 2 mM MgCl2, 0.3 mM 1,10-phenanthroline, and 0.2% bovine serum albumin. After incubation for 60 minutes at 37° C., the assay mixture was filtered and the membranes washed three times with ice-cold buffer. Filters were dried and counted in a gamma counter for bound radioactivity. Non-specific binding was measured by inhibition of binding of [I125]-NDP-α-MSH in the presence of 1 uM NDP-α-MSH. Maximal specific binding (100%) was defined as the difference in radioactivity (cpm) bound to cell membranes in the absence and presence of 1 uM NDP-α-MSH. Radioactivity (cpm) obtained in the presence of test compounds was normalized with respect to 100% specific binding to determine the percent inhibition of [I125]-NDP-α-MSH binding. Each assay was conducted in triplicate and the actual mean values are described, with results less than 0% reported as 0%. Ki values for test peptides of the present invention were determined using Graph-Pad Prism curve-fitting software. BCA Protein Assay The buffer solution for the receptor binding test was dispensed to the wells of a 96-well assay plate (Greiner) at 22.5 uL/well. DMSO solutions of a test compound, which were prepared at an 80-time higher concentration using 100% dimethyl sulfoxide (DMSO), were added to the wells at 2.5 uL/well (final concentrations of 1 nM to 100 nM), and the solutions were mixed. As a radiolabeled ligand, 125I-substance P (Substance P, [125I]Tyr8-, PerkinElmer) was used. 125I-substance P was diluted with the buffer solution for the receptor binding test to a concentration resulting in 125 pmol/25 uL/well and added to the 96-well assay plate, and the solutions were mixed. The membrane fraction prepared from the human NK1 receptor-expressing cells was diluted with the buffer solution for the receptor binding test to a concentration resulting in 8 to 10 ug/well, suspended until the suspension became in such a homogenous state that the suspension could flow through as 27G injection needle smoothly and then added to the 96-well assay plate at 150 uL/well. Then, the plate was incubated at room temperature for 60 minutes while shaking the plate. The reaction solutions were suction-filtered. through a multiscreen 96-well filter plate (Millipore) which had been pre-treated with 0.3% polyethyleneimine, and the reaction was terminated by washing with a washing solution (50 mM Tris and 0.02% bovine serum albumin, pH 7.4) four times. The bottom of the microplate was dried at 60° C., and then 100 uL/well of MicroScint 20 (PerkinElmer) was dispensed to the wells. The top of the plate was sealed with TopSeal A (PerkinElmer), and the plate was shaken for 5 to 10 minutes. Then, the radioactivities were measured with TopCount NXT (registered trademark) (PerkinElmer). The radioactivity of each well was calculated by subtracting the radioactivity of the well to which 10 uM aprepitant was added (non-specific binding). The binding rate (%) of 125I-substance P=(the radioactivity of the group to which the test compound was added)/(the radioactivity of the group to which the vehicle was added)x100 was calculated. Using analysis software, GraphPad Prism (GraphPad Software), the binding rate (%) was plotted against the concentration of the test compound and linearly approximated, and the concentration required for 50% inhibition, IC50, was calculated. Human NK1 Receptor Binding Assay IM-9 cells (2×10^5 cells/mL) were cultured for 3 days after inoculation, and centrifuged at 500×g for 10 min to give cell pellets. The obtained pellets were washed with PBS (Phosphate-Buffered saline) (GIBCO), disrupted in buffer A (50 mM Tris-hydrochloric acid buffer (Tris-HCl) (pH 7.4) containing 120 mM sodium chloride, 5 mM potassium chloride, 2 μg/mL chymostatin, 40 μg/mL bacitracin, 40 μg/mL APMSF (p-amidinophenylmethanesulfonyl fluoride hydrochloride) and 1 mM ethylenediaminetetraacetic acid (EDTA)) using a Polytron homogenizer (Kinematika, Germany), and centrifuged at 100,000×g for 40 min. The precipitation fraction was suspended in buffer B (50 mM Tris-HCl (pH 7.4), 0.02% bovine serum albumin, 2 μg/mL chymostatin, 40 μg/mL bacitracin, 40 μg/mL APMSF, 3 mM manganese dichloride (MnCl2)) and cryopreserved (−80° C.) as a receptor reference standard. Buffer B (50 μL) was added to a 96-well microassay plate (Corning Incorporated). The membrane reference standard suspended in buffer B at 250 μg/mL was added by 50 μL. A measurement buffer containing 2% dimethyl sulfoxide was added by 50 μL to examine the total binding, 4 μM non-labeled SP diluted with a measurement buffer containing 2% dimethyl sulfoxide was added by 50 μL to examine the non-specific binding, and a test compound diluted with a measurement buffer (containing 2% dimethyl sulfoxide) was added by 50 μL to examine the binding inhibitory activity of the test compound. Furthermore, 400 μM 125I-Bolton-Hunter-SP (BH-SP) solution was added to each well by 50 μL. After reaction at room temperature for 30 min, the reaction was quenched using a cell harvester (PerkinElmer) by rapid filtration on a GF/C filter plate (PerkinElmer), and the cells were washed 10 times with 250 μL of a 50 mM Tris-hydrochloric acid buffer (pH 7.4) containing 0.02% bovine serum albumin. The GF/C filter plate was dried, MicroScinti-0 (PerkinElmer) was added by 20 μL, and the radioactivity was measured on a TopCount (PerkinElmer). The GF/C filter plate was immersed in 0.3% polyethyleneimine for one day before use. Human NK2 Receptor Binding Assay CHO cells expressing hNK2 receptor were cultured in a HAM-F12 medium containing 400 ug/mL geneticin, 100 U/mL penicillin, 100 ug/mL streptomycin and 10% inactivated serum. The medium was removed, the adhered cells were washed with PBS, and PBS containing 5 mM EDTA was added to detach the cells from the flask. The cells were collected by centrifugation, suspended in suspension buffer A (15 mM Tris-HCl (pH 7.5), 2 mM MgCl2, 0.3 mM ethylenediaminetetraacetic acid (EDTA), 1 mM O,O'-bis(2-aminoethyl)ethyleneglycol-N,N,N',N'-tetraacetic acid (EGTA)), disrupted by a Polytron homogenizer (Kinematika), and centrifuged at 800xg for 10 min. The supernatant was recovered and ultracentrifuged at 100,000xg for 25 min. The precipitation fraction was suspended in suspension buffer B (7.5 mM Tris-HCl (pH 7.5), 12.5 mM MgCl2, 0.3 mM EDTA, 1 mM EGTA, 250 mM sucrose), and cryopreserved (-80° C.) as a receptor reference standard. Measurement buffer (50 mM Tris-HCl (pH 7.4), 0.02% bovine serum albumin, 2 ug/mL chymostatin, 40 ug/mL bacitracin, 40 ug/mL APMSF, 3 mM MnCl2) (50 uL) was added to a 96-well microassay plate. The membrane reference standard (20 ug/mL) suspended in a measurement buffer was added by 50 uL. A measurement buffer containing 2% dimethyl sulfoxide was added by 50 uL to examine the total binding level, 4 uM non-labeled NK-A (PEPTIDE INSTITUTE, INC.) solution diluted with a measurement buffer containing 2% dimethyl sulfoxide was added by 50 uL to examine the non-specific binding level, and a test compound diluted with a measurement buffer (containing 2% dimethyl sulfoxide) was added by 50 uL to examine the binding inhibitory activity of the test compound. Furthermore, 400 uM [125I]-NK-A (GE Healthcare Bio-Sciences KK) solution was added to each well by 50 uL. After reaction at 25° C. for 30 min, the reaction was quenched using a cell harvester (PerkinElmer) by rapid filtration on a GF/C filter plate, and the cells were washed 5 times with 250 uL of a 50 mM Tris-HCl buffer (pH 7.4) containing 0.02% bovine serum albumin. The GF/C filter plate was dried, MicroScinti-0 (PerkinElmer) was added by 20 uL, and the radioactivity was measured on a TopCount (PerkinElmer). The GF/C filter plate used had been immersed in 0.3% polyethyleneimine for one day. Ki Determination for Genotypes 1b and 3a NS3 Protease Purified NS3 protease domain (amino acids 1-181) of the genotype 1b and 3a virus were generated as above. The internally quenched fluorogenic depsipeptide substrate Ac-DED(Edans)-EEAbuΨ[COO]ASK(Dabcyl)-NH2 and a synthetic peptide containing the hydrophobic core residues of the NS4A protein cofactor (KKGSVVIVGRIILSGRKK; NS4A peptide) were obtained from Anaspec, Inc. (San Jose, Calif.). Other chemicals and biochemicals were of reagent grade or better and were purchased from standard suppliers.Reactions were run at room temperature in buffer consisting of 50 mM HEPES, 40% glycerol, 0.05% Triton X-100, 10 mM DTT, and 10% DMSO. The final assay solutions contained 50 pM NS3 genotype 1 b protease or 200 pM genotype 3a protease, 20 μM NS4A peptide, and 4 μM substrate (genotype 1b) or 2 μM substrate (genotype 3a). Inhibitor concentrations varied from 100 nM to 5 pM in 3-fold dilutions, and no-inhibitor controls were included.Compound dilutions were made in DMSO at 20× final concentration. Reaction mixtures were prepared in 96-well assay plates. A solution of enzyme and NS4A peptide in assay buffer (25 μL volume with both reagents at 4× final concentration) was mixed with 45 μL assay buffer and 5 μL of either inhibitor or DMSO, and pre-incubated at room temperature for 1 hour. The reaction was started by addition of 25 μL substrate solution at 4× final concentration. Plates were mixed vigorously for 5-10 seconds and reactions were allowed to proceed for 90 minutes, fluorescence was measured every 30 s between 90 and 120 minutes reaction time using a Tecan InfiniTe M1000 or PerkinElmer Envision multimode plate reader with an excitation wavelength of 340 nm and an emission wavelength of 490 nm.Rates were calculated from the progress curves at steady state, in the time frame of 90-120 minutes after addition of substrate. To determine the Ki, rates were plotted as a function of inhibitor concentration, and the data were fit with equation 1 (Morrison, J. F., Biochimica et Biophysica Acta 1969, 185, 269-286) to calculate Ki app using GraphPad Prism 5. Active fraction of enzyme was determined by active site titration with known potent inhibitors. Ki was calculated from Ki app/(1+[[S]/Km]). RORa, b, and g Binding Inhibitors His-tagged human RAR-related orphan receptor alpha (hRORa), human RAR-related orphan receptor beta (hRORb), and human RAR-related orphan receptor gamma (hRORg) are used for receptor-ligand competition binding assays to determine Ki values. Typical procedures are provided below.Receptor competition binding assays are run in a buffer made up of DPBS (1 L) (Hyclone #SH30028.03), 2.2 g BSA Fraction v (Roche #9048-46-8), 100 mL glycerol (Fischer #56-81-5) and 40 mL DMSO (reagent grade). The final wells contain 20 μg/mL aprotinin and 20 μg/mL leupeptin and 10 μM Pefabloc. Typically, receptor binding assays include radio-labeled ligands, such as 7 nM [3H]-25-hydroxycholesterol for alpha binding, 20 nM [3H]-3-[[4-[[3-(2,6-dichlorophenyl)-5-isopropyl-isoxazol-4-yl]methoxy]-N,2-dimethyl-anilino]methyl]benzoic acid for beta binding, and 6 nM [3H]-25-hydroxycholesterol for gamma binding, and 0.5 μg RORa receptor, 0.03 μg RORb receptor, or 0.13 μg RORg receptor per well. Assays are typically run in 96-well format. Competing test compounds are added at various concentrations ranging from about 0.4 nM to 25 μM. Non-specific binding is determined in the presence of 250 nM 25-hydroxycholesterol for RORa and RORg binding, 250 nM 3-[[4-[[3-(2,6-dichlorophenyl)-5-isopropyl-isoxazol-4-yl]methoxy]-N,2-dimethyl-anilino]methyl]benzoic acid for RORb binding. The sample, label and receptor solutions are combined in a 96 well assay plate (Costar 3632) and incubated overnight at room temperature, then 25 μl beads (Amersham YSi (2-5 micron) copper His-tag Spa Beads, #RPNQ0096) for a final bead concentration of 1 mg/well is added to each reaction. Plates are mixed for 30 minutes on an orbital shaker at room temperature. After an incubation of 4 hours, plates are read in a Wallac MICROBETA counter.The data are used to calculate an estimated IC50 using a four parameter logistic fit. The Kd for [3H]-25-hydroxycholesterol for RORa and RORg, and [3H]-3-[[4-[[3-(2,6-dichlorophenyl)-5-isopropyl-isoxazol-4-yl]methoxy]-N,2-dimethyl-anilino]methyl]benzoic acid for RORb binding, is determined by saturation binding. The IC50 values for compounds are converted to Ki using the Cheng-Prushoff equation. Receptor Binding Assay The buffer solution for the receptor binding test was dispensed to the wells of a 96-well assay plate (Greiner) at 22.5 μL/well. DMSO solutions of a test compound, which were prepared at an 80-time higher concentration using 100% dimethyl sulfoxide (DMSO), were added to the wells at 2.5 μL/well (final concentrations of 1 nM to 100 nM), and the solutions were mixed. As a radiolabeled ligand, 125I-substance P (Substance P, [125I]Tyr8-, PerkinElmer) was used. 125I-substance P was diluted with the buffer solution for the receptor binding test to a concentration resulting in 125 pmol/25 μL/well and added to the 96-well assay plate, and the solutions were mixed. The membrane fraction prepared from the human NK1 receptor-expressing cells was diluted with the buffer solution for the receptor binding test to a concentration resulting in 8 to 10 μg/well, suspended until the suspension became in such a homogenous state that the suspension could flow through a 27 G injection needle smoothly and then added to the 96-well assay plate at 150 μL/well. Then, the plate was incubated at room temperature for 60 minutes while shaking the plate. The reaction solutions were suction-filtered through a multiscreen 96-well filter plate (Millipore) which had been pre-treated with 0.3% polyethyleneimine, and the reaction was terminated by washing with a washing solution (50 mM Tris and 0.02% bovine serum albumin, pH 7.4) four times. The bottom of the microplate was dried at 60° C., and then 100 μL/well of MicroScint 20 (PerkinElmer) was dispensed to the wells. The top of the plate was sealed with TopSeal A (PerkinElmer), and the plate was shaken for 5 to 10 minutes. Then, the radioactivities were measured with TopCount NXT (registered trademark) (PerkinElmer). The radioactivity of each well was calculated by subtracting the radioactivity of the well to which 10 μM aprepitant was added (non-specific binding). The binding rate (%) of 125I-substance P=(the radioactivity of the group to which the test compound was added)/(the radioactivity of the group to which the vehicle was added)×100 was calculated. Using analysis software, GraphPad Prism (GraphPad Software), the binding rate (%) was plotted against the concentration of the test compound and linearly approximated, and the concentration required for 50% inhibition, IC50, was calculated. VEGF-R2 Binding Assay A competition binding assay (DiscoveRx KINOMEscan ) was used to measure the ability of the compound to compete for binding of an immobilized adenosine triphosphosphate (ATP) site directed ligand using a DNA-tagged vascular endothelial growth receptor 2 (VEGFR2) as the target. The ability of the test compound to compete with the immobilized ligand was measured using quantitative polymerase chain reaction (qPCR) of the DNA tag (Fabian, et al, Nature Biotechnology (2005) 23, 329-336; Karaman, et al, Nature Biotechnology (2008) 26, 127-132).A VEGFR2 tagged T7 phage strain was prepared in an Escherichia coli (E. coli) derived from the BL21 strain. The E. coli were grown to log-phase, infected with VEGFR2 tagged T7 phage and then incubated with shaking at 32° C. until lysis. The lysate containing the kinase was then centrifuged and filtered to remove cell debris. Affinity resin for the VEGFR2 assay was prepared by treating Streptavidin-coated magnetic beads with a biotinylated small molecule ligand for 30 minutes at room temperature. The beads were blocked with excess biotin and then washed with blocking buffer (SeaBlock (Pierce), 1% bovine serum albumin, 0.17% phosphate buffered saline, 0.05% Tween 20, 6 mM dithiothreitol). The binding reaction was initiated by combining in a well of a polystyrene 96-well plate, DNA tagged VEGFR2, liganded affinity beads and the serial diluted test compound in 1× binding buffer (20% SeaBlock, 0.17× phosphate buffered saline, 0.05% Tween 20, 6 mM dithiothreitol) in a final volume of 0.135 ml. The assay plates were incubated at room temperature with shaking for 1 hour and then the beads were washed with wash buffer (1× phosphate buffered saline, 0.05% Tween 20). The beads were re-suspended in elution buffer (1× phosphate buffered saline, 0.05% Tween 20, 0.05 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The VEGFR2 concentration in the eluate was measured using qPCR.An 11-point dose response curve of 3-fold serial diluted test compound starting at 1 μM was used to determine the VEGFR2 binding constant (Kd). The compounds were prepared in 100% DMSO at 100× the final test concentration and then diluted to 1× in the assay for final DMSO concentration of 1%. Binding constants were calculated with standard dose-response curve using the Hill equation with Hill slope set to −1. Curves were fit using a non-linear least square fit with the Levenberg-Marquardt algorithm. VEGFR2 Binding Assay A competition binding assay (DISCOVERX KINOMESCAN ) was used to measure the ability of a compound to compete for binding of an immobilized adenosine triphosphosphate (ATP) site directed ligand using a DNA-tagged vascular endothelial growth receptor 2 (VEGFR2) as the target. The ability of the test compound to compete with the immobilized ligand was measured using quantitative polymerase chain reaction (qPCR) of the DNA tag (Fabian, M. A. et al., 23 Nature Biotechnology 329-336 (2005); Karaman, M. W. et al., 26 Nature Biotechnology 127-132 (2008)).A VEGFR2 tagged T7 phage strain was prepared in an Escherichia coli (E. coli) derived from the BL21 strain. The E. coli were grown to log-phase, infected with VEGFR2 tagged T7 phage and then incubated with shaking at 32° C. until lysis. The lysate containing the kinase was then centrifuged and filtered to remove cell debris. Affinity resin for the VEGFR2 assay was prepared by treating Streptavidin-coated magnetic beads with a biotinylated small molecule ligand for 30 minutes at room temperature. The beads were blocked with excess biotin and then washed with blocking buffer (SEABLOCK (PIERCE), 1% bovine serum albumin, 0.17% phosphate buffered saline, 0.05% TWEEN 20, 6 mM dithiothreitol). The binding reaction was initiated by combining in a well of a polystyrene 96-well plate, DNA tagged VEGFR2, liganded affinity beads and the serial diluted test compound in 1× binding buffer (20% SEABLOCK, 0.17× phosphate buffered saline, 0.05% TWEEN 20, 6 mM dithiothreitol) in a final volume of 0.135 ml. The assay plates were incubated at room temperature with shaking for 1 hour and then the beads were washed with wash buffer (1× phosphate buffered saline, 0.05% TWEEN 20). The beads were re-suspended in elution buffer (1× phosphate buffered saline, 0.05% TWEEN 20, 0.05 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The VEGFR2 concentration in the eluate was measured using qPCR.An 11-point dose response curve of 3-fold serial diluted test compound starting at 1 μM was used to determine the VEGFR2 binding constant (Kd). The compounds were prepared in 100% DMSO at 100× the final test concentration and the diluted to 1× in the assay for final DMSO concentration of 1%. Binding constants were calculated with standard dose-response curve using the Hill equation with Hill slope set to −1. Curves were fit using a non-linear least square fit with the Levenberg-Marquardt algorithm. Biochemical Assay Against EGFR Exon 19 Deletion/C797S Protein (EGFR d19/CS) The inhibitory activity of the compounds disclosed herein were measured in a biochemical assay utilizing a recombinant human EGFR exon 19 deletion/C797S double mutant protein, utilizing detection of chelation-enhanced fluorescence using the sulfonamido-oxine (Sox) chromophore covalently attached to an optimized kinase substrate.The following materials were used: EGFR d19-C797S (Carna Biosciences, Cat #08-564) phosphosens peptide substrate (AssayQuant, Cat #AQT0794); adenosine triphosphate (ThermoFisher, Cat #R0441); assay Buffer: 50 mM HEPES, pH=7.5, 10 mM MgCl2, 1 mM EGTA, 0.01% BSA, 0.01% Brij-35, 1 mM DTT; 384-well Polypropylene Plates (Greiner Cat #781201); 384-well Black Low Volume Plates (Greiner Cat #784076); TopSeal-A Plus (PerkinElmer Cat #6050185); Dimethyl Sulfoxide (Sigma, Cat #D2650); 1 M HEPES (VWR Chemicals, Cat #J848); 2M Magnesium Chloride (Quality Biological, Cat #340-034-721); 0.5M Ethylene Glycol Tetra Acetic Acid (Alfa Aesar, Cat #J60767); 30% Bovine Albumin, Fraction V (Alfa Aesar, Cat #165569); 10% Brij-35, (EMD Millipore, Cat #203728); 1M Dithiothreitol, (Invitrogen, Cat #P2325)In Row A of a 384-well polypropylene plate, was added 48 μL of DMSO. In wells A1 and A24, were added 12 μL of DMSO. Column 1 (16 replicates) served as enzyme+DMSO total signal (0% inhibition). Column 24 (16 replicates) served as no enzyme+DMSO background (100% inhibition). To a well in Row A were added 12 μL of each 10 mM test compound stock (100% DMSO). The master compound plate was transferred to the Bravo and run the “PPAR FRET cmpd serial dilution and intermediate dilution.pro” protocol to generate 16-point serial dilutions (3.16× or 0.5 log dilutions) of each compound. The final intermediate dilution plate contained 4×16-point compound serial dilutions in assay buffer. The DMSO concentration was 10%. The intermediate compound dilution plate was transferred to the Bravo and the “5 uL dispense into ARP.pro” protocol was run. To the black low volume assay ready plates, were added 5 μL of 40 uM Phosphosens peptide sensor/4 mM ATP working solution to each well. The plates were spun for 1 minute at 1200 rpm in an Eppendorf 5810R table-top centrifuge. Next, 10 μl of 2.0 nM EGFR d19/C797S working solution was added to all wells in columns 1-23. To column 24, was added 10 μL of assay buffer. The final DMSO concentration was 2.5%. The plates were spun for 1 minute at 1200 rpm in an Eppendorf 5810R table-top centrifuge. The final concentrations of EGFR d19/C797S, Phosphosens peptide substrate and ATP were 0.25 nM, 10 μM and 1 mM respectively. Biochemical Assay Against EGFR L858R/C797S Protein (EGFR LR/CS) The inhibitory activity of the compounds disclosed herein were measured in a biochemical assay utilizing a recombinant human EGFR L858R/C797S double mutant protein, utilizing detection of chelation-enhanced fluorescence using the sulfonamido-oxine (Sox) chromophore covalently attached to an optimized kinase substrate.The following materials were used: EGFR L858R C797S (Carna Biosciences, Cat #08-563); phosphosens peptide substrate (AssayQuant, Cat #AQT0794); adenosine triphosphate (ThermoFisher, Cat #R0441); assay Buffer: 50 mM HEPES, pH=7.5, 10 mM MgCl2, 1 mM EGTA, 0.01% BSA, 0.01% Brij-35, 1 mM DTT; 384-well Polypropylene Plates (Greiner Cat #781201); 384-well Black Low Volume Plates (Greiner Cat #784076); TopSeal-A Plus (PerkinElmer Cat #6050185); Dimethyl Sulfoxide (Sigma, Cat #D2650); 1 M HEPES (VWR Chemicals, Cat #J848); 2M Magnesium Chloride (Quality Biological, Cat #340-034-721); 0.5M Ethylene Glycol Tetra Acetic Acid (Alfa Aesar, Cat #J60767); 30% Bovine Albumin, Fraction V (Alfa Aesar, Cat #165569); 10% Brij-35, (EMD Millipore, Cat #203728); 1M Dithiothreitol, (Invitrogen, Cat #P2325)In Row A of a 384-well polypropylene plate, was added 48 μL of DMSO. In wells A1 and A24, were added 12 μL of DMSO. Column 1 (16 replicates) served as enzyme+DMSO total signal (0% inhibition). Column 24 (16 replicates) served as no enzyme+DMSO background (100% inhibition). To a well in Row A were added 12 μL of each 10 mM test compound stock (100% DMSO). The master compound plate was transferred to the Bravo and run the “PPAR FRET cmpd serial dilution and intermediate dilution.pro” protocol to generate 16-point serial dilutions (3.16× or 0.5 log dilutions) of each compound. The final intermediate dilution plate contained 4×16-point compound serial dilutions in assay buffer. The DMSO concentration was 10%. The intermediate compound dilution plate was transferred to the Bravo and the “5 uL dispense into ARP.pro” protocol was run. To the black low volume assay ready plates, were added 5 μL of 40 uM Phosphosens peptide sensor/4 mM ATP working solution to each well. The plates were spun for 1 minute at 1200 rpm in an Eppendorf 5810R table-top centrifuge. Next, 10 μl of 2.0 nM EGFR L858R/C797S working solution was added to all wells in columns 1-23. To column 24, was added 10 μL of assay buffer. The final DMSO concentration was 2.5%. The plates were spun for 1 minute at 1200 rpm in an Eppendorf 5810R table-top centrifuge. Human NK3 Receptor Binding Assay CHO cells expressing hNK3 receptor were cultured in a MEMalpha medium (Nikken Seibutsu Igaku Kenkyusho, K.K.) containing 100 U/mL penicillin, 100 ug/mL streptomycin and 10% inactivated dialyzed serum. The medium was removed, the adhered cells were washed with PBS, and PBS containing 5 mM EDTA was added to detach the cells from the flask. The cells were collected by centrifugation, suspended in suspension buffer A (50 mM Tris-HCl (pH 7.4) containing 120 mM NaCl, 5 mM KCl, 2 ug/mL chymostatin, 40 ug/mL bacitracin, 40 ug/mL APMSF, 1 mM EDTA), disrupted by a Polytron homogenizer (Kinematika), and centrifuged at 800xg for 10 min. The supernatant was recovered and ultracentrifuged at 100,000xg for 25 min. The precipitation fraction was suspended in suspension buffer B (50 mM Tris-HCl (pH 7.4), 0.02% bovine serum albumin, 2 ug/mL chymostatin, 40 ug/mL bacitracin, 40 ug/mL APMSF, 3 mM MnCl2), and cryopreserved (-80° C.) as a receptor reference standard. Measurement buffer (50 mM Tris-HCl (pH 7.4), 0.02% bovine serum albumin, 2 ug/mL chymostatin, 40 ug/mL bacitracin, 40 ug/mL APMSF, 3 mM MnCl2) (50 uL) was added to a 96-well microassay plate. The membrane reference standard (300 ug/mL) suspended in a measurement buffer was added by 50 uL. A measurement buffer containing 2% dimethyl sulfoxide was added by 50 uL to examine the total binding level, 16 uM non-labeled NK-B (PEPTIDE INSTITUTE, INC.) solution diluted with a measurement buffer containing 2% dimethyl sulfoxide was added by 50 uL to examine the non-specific binding level, and a test compound diluted with a measurement buffer (containing 2% dimethyl sulfoxide) was added by 50 uL to examine the binding inhibitory activity of the test compound. Furthermore, 400 uM [125I]-NK-B (Neurokinin-B (N-Me-Phe7), [125I]His3-) (PerkinElmer) solution was added to each well by 50 uL. After reaction at 25° C. for 30 min, the reaction was quenched using a cell harvester (PerkinElmer) by rapid filtration on a GF/C filter plate, and the cells were washed 5 times with 250 uL of a 50 mM Tris-HCl buffer (pH 7.4) containing 0.02% bovine serum albumin. The GF/C filter plate was dried, MicroScinti-0 (Perkin Elmer) was added by 20 uL, and the radioactivity was measured on a TopCount (PerkinElmer). The GF/C filter plate used had been immersed in 0.3% polyethyleneimine for one day. In Vitro Fluorescence-Based Recombinant Tau Binding Assay (A) Expression and Purification of Human Tau: 1 mM IPTG (Sangon Biotech, Cat. No A100487) was used to induce production of tau by bacteria (BL21, Invitrogen, Cat. No C600003) transformed with a full-length 2N4R tau expression plasmid. After 3 hours, cell pellet was resuspended in lysis buffer (100 mM PIPES, 1 mM EGTA, 1 mM MgSO4, pH 6.8) and lysed by sonication followed by centrifugation (15,000 rpm, 15 min, 4° C.). The supernatant was then placed in a boiling water bath for 20 min, followed by centrifugation (15,000 rpm, 15 min, 4° C.). Supernatant was loaded onto a Q-Sepharose Fast Flow column (GE healthcare, Cat. No 17-0510-01), the flow-through fraction was loaded onto an SP-Sepharose Fast Flow column (GE healthcare, Cat. No 17-0729-01), and tau protein was eluted with elution buffer (100 mM PIPES, 1 M NaCl, 1 mM EGTA, 1 mM MgSO4, 0.2 M NaCl, pH 6.8). Collected fractions of tau-containing eluates were pooled, concentrated and dialyzed against HEPES buffer (25 mM HEPES, 0.1 mM EDTA, 0.5 mM DTT, 100 mM NaCl, pH 7.2) and stored at −80° C. in small aliquots until use. Protein concentration was determined by UV absorption.(B) Preparation of Heparin-Induced Aggregated Tau (aTau):>2 μM of tau prepared in 30 mM Tris-HCl, pH 7.5 buffer was incubated in tube with 15 μM heparin (Sigma, Cat. No H3149) at 37° C. for 24 hours. (C) Compound Fluorescent Spectra Scanning Assay: Compounds were dissolved in 100% DMSO, and 40 nM aTau was incubated with 10 μM of each compound in 2% DMSO in 96 well plate (Corning, Cat. No 3573) at 37° C. for 1 hour. Emission and excitation of the compound was scanned by microplate spectrometer (EnSpire 2300, PerkinElmer).(D) In Vitro Fluorometric aTau Binding Assays: 2 μM aTau was diluted to 0.04 μM with 30 mM Tris-HCl (pH 6.8), and then incubated with serially diluted compound (three-fold serial dilutions, from 10 to 0.00017 μM) in a 96-well plate (Corning, Cat. No 3573) at 37° C. for 1 hour. Fluorescence intensity (excitation/emission=370/500 nm) of APN-0729 was read by microplate spectrometer (EnSpire 2300, PerkinElmer). Compound Kd values were calculated using the following equation: Y=B max *X/(Kd+X), where X is the concentration of compound; Y is the fluorescence signal of (compound+aTau)−(compound+DMSO); and B max is the maximum signal. Homogeneous Time-Resolved Fluorescence (HTRF) Assay for Direct cAMP Measurement Compounds were screened for agonists of the human prostacyclin (PGI2) receptor using the HTRF assay for direct cAMP measurement (Gabriel et al., ASSAY and Drug Development Technologies, 1:291-303, 2003) and recombinant CHO-K1 cells stably transfected with human prostacyclin receptor. CHO-K1 cells were obtained from ATCC (Manassas, Va.; Catalog # CCL-61). An agonist of the prostacyclin receptor was detected in HTRF assay for direct cAMP measurement as a compound which increased cAMP concentration. HTRF assay also was used to determine EC50 values for prostacyclin receptor agonists.Principle of the Assay:The HTRF assay kit was purchased from Cisbio-US, Inc. (Bedford, Mass.; Catalog #62AM4PEC). The HTRF assay supported by the kit is a competitive immunoassay between endogenous cAMP produced by the CHO-K1 cells and tracer cAMP labeled with the dye d2. The tracer binding is visualized by a monoclonal anti-cAMP antibody labeled with Cryptate. The specific signal (i.e., fluorescence resonance energy transfer, FRET) is inversely proportional to the concentration of unlabeled cAMP in the standard or sample.Standard Curve:The fluorescence ratio (665 nm/620 nm) of the standards (0.17 to 712 nM cAMP) included in the assay was calculated and used to generate a cAMP standard curve according to the kit manufacturer's instructions. The fluorescence ratio of the samples (test compound or compound buffer) was calculated and used to deduce respective cAMP concentrations by reference to the cAMP standard curve.Setup of the Assay:The HTRF assay was carried out using a two-step protocol essentially according to the kit manufacturer's instructions, in 20 μL total volume per well in 384-well plate format (ProxiPlates; PerkinElmer, Fremont, Calif.; catalog #6008280). To each of the experimental wells was transferred 3000 recombinant CHO-K1 cells in 5 μL assay buffer (phosphate buffered saline containing calcium chloride and magnesium chloride (Invitrogen, Carlsbad, Calif.; catalog #14040) supplemented with IBMX (100 μM) and rolipram (10 μM) (phosphodiesterase inhibitors; Sigma-Aldrich, St. Louis, Mo.; catalog #15879 and catalog # R6520, respectively) and 0.1% bovine serum albumin (BSA) fraction V (Sigma-Aldrich; catalog # A3059)), followed by test compound in 5 μL assay buffer or 5 μL assay buffer. The plate was then incubated at room temperature for 1 h. To each well was then added 5 μL cAMP-d2 conjugate in lysis buffer and 5 μL Cryptate conjugate in lysis buffer according to the kit manufacturer's instructions. The plate was then further incubated at room temperature for 1 h, after which the assay plate was read.Assay Readout:The HTRF readout was accomplished using a PHERAstar (BMG LABTECH Inc., Durham, N.C.) or EnVision (PerkinElmer, Fremont Calif.) microplate reader. Receptor [35S] GTPgammaS Binding Assays: (S1P1 GTPgammaS/S1P3 GTPgammaS) Compounds were loaded in a 384 Falcon v-bottom plate (0.5 μl/well in a 11 point, 3-fold dilution). Membranes prepared from S1P1/CHO cells or EDG3-Gal5-bla HEK293T cells (EDG3 equivalent S1P3) were added to the compound plate (40 l/well, final protein 3 μg/well) with MULTIDROP. [35S]GTP (1250 Ci/mmol, Perkin Elmer) was diluted in assay buffer: 20 mM HEPES, pH7.5, 10 mM MgCl2, 150 mM NaCl, 1 mM EGTA (ethylene glycol tetraacetic acid), 1 mM DTT (Dithiothreitol), 10 μM GDP, 0.1% fatty acid free BSA, and 10 μg/ml Saponin to 0.4 nM. 40 μl of the [35S] GTP solution was added to the compound plate with a final concentration of 0.2 nM. The reaction was kept at room temperature for 45 min. At the end of incubation, all the mixtures in the compound plate were transferred to Millipore 384-well FB filter plates via the VELOCITY11 Vprep liquid handler. The filter plate was washed with water 4 times by using the manifold Embla plate washer and dried at 60° C. for 45 min. MicroScint 20 scintillation fluid (30 μl) was added to each well for counting on the Packard TOPCOUNT . EC50 is defined as the agonist concentration that corresponds to 50% of the Ymax (maximal response) obtained for each individual compound tested. The EC50 for Example 689 was determined to be 5.7 nM in the assay utilizing membranes prepared from S1P1/CHO cells. The EC50 for Example 689 was determined to be >2000 nM in the assay utilizing membranes prepared from EDG3-Gal5-bla HEK293T cells.A smaller value for GTPγS S1P1 EC50 value indicated greater activity for the compound in the GTPγS S1P1 binding assay. A larger value for the GTPγS S1P3 EC50 value indicated less activity in the GTPγS S1P3 binding assay. Example 689, which is the phosphate ester of Example 672, possessed activity as an agonist of S1P1 and is selective over S1P3. Example 697, which is the phosphate ester of Example 681, possessed activity as an agonist of S1P1 and is selective over S1P3. Thus the compounds of the present invention may be used in treating, preventing, or curing various S1P1 receptor-related conditions while reducing or minimizing the side effects due to S1P3 activity. The selectivity of the compounds of the present invention indicate their potential use in treating, preventing, or curing autoimmune and inflammatory diseases such as multiple sclerosis, rheumatoid arthritis, inflammatory bowel disease, lupus, psoriasis, or vascular diseases, while reducing or minimizing possible side effects due to S1P3 activity. Other potential uses of the compounds of the present invention include minimizing or reducing rejection of transplanted organs, while reducing or minimizing side effects due to S1P3 activity. Receptor [35S] GTPgammaS Binding Assays: (S1P1 GTPgammaS/S1P3 GTPgammaS) Compounds were loaded in a 384 Falcon v-bottom plate (0.5 l/well in a 11 point, 3-fold dilution). Membranes prepared from S1P1/CHO cells or EDG3-Gal5-bla HEK293T cells (EDG3 equivalent S1P3) were added to the compound plate (40 l/well, final protein 3 μg/well) with MULTIDROP . [35S]GTP (1250 Ci/mmol, Perkin Elmer) was diluted in assay buffer: 20 mM HEPES, pH7.5, 10 mM MgCl2, 150 mM NaCl, 1 mM EGTA (ethylene glycol tetraacetic acid), 1 mM DTT (Dithiothreitol), 10 μM GDP, 0.1% fatty acid free BSA, and 10 μg/ml Saponin to 0.4 nM. 40 μl of the [35S] GTP solution was added to the compound plate with a final concentration of 0.2 nM. The reaction was kept at room temperature for 45 min. At the end of incubation, all the mixtures in the compound plate were transferred to Millipore 384-well FB filter plates via the VELOCITY11 Vprep liquid handler. The filter plate was washed with water 4 times by using the manifold Embla plate washer and dried at 60° C. for 45 min. MicroScint 20 scintillation fluid (30 μl) was added to each well for counting on the Packard TOPCOUNT . EC50 is defined as the agonist concentration that corresponds to 50% of the Ymax (maximal response) obtained for each individual compound tested. The EC50 for Example 689 was determined to be 5.7 nM in the assay utilizing membranes prepared from S1P1/CHO cells. The EC50 for Example 689 was determined to be >2000 nM in the assay utilizing membranes prepared from EDG3-Gal5-bla HEK293T cells.A smaller value for GTPγS S1P1 EC50 value indicated greater activity for the compound in the GTPγS S1P1 binding assay. A larger value for the GTPγS S1P3 EC50 value indicated less activity in the GTPγS S1P3 binding assay. Example 689, which is the phosphate ester of Example 672, possessed activity as an agonist of S1P1 and is selective over S1P3. Example 697, which is the phosphate ester of Example 681, possessed activity as an agonist of S1P1 and is selective over S1P3. Thus the compounds of the present invention may be used in treating, preventing, or curing various S1P1 receptor-related conditions while reducing or minimizing the side effects due to S1P3 activity. The selectivity of the compounds of the present invention indicate their potential use in treating, preventing, or curing autoimmune and inflammatory diseases such as multiple sclerosis, rheumatoid arthritis, inflammatory bowel disease, lupus, psoriasis, or vascular diseases, while reducing or minimizing possible side effects due to S1P3 activity. Other potential uses of the compounds of the present invention include minimizing or reducing rejection of transplanted organs, while reducing or minimizing side effects due to S1P3 activity. In Vitro DGAT2 Assay For determination of IC50 values, the reactions were carried out in 384-well white Polyplates (Perkin Elmer) in a total volume of 20 μL. To 14 of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 20 minutes. To this mixture, 10 μL of hDGAT2 membrane fraction (0.01 mg/mL) diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock) was added. After this mixture was preincubated at room temperature for 2 hours, DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). Plates were centrifuged at 2,000×g for 1 min in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). DGAT2 activity was measured by quantifying the generated product [14C]tridecanoylglycerol in the upper organic phase. Background activity obtained using 50 μM of (1R, 2R)-2-({3′-Fluoro-4′-[(6-fluoro-1, 3-benzothiazol-2-yl)amino]-1,1′-biphenyl-4-yl}carbonyl)cyclopentanecarboxylic acid (US 20040224997, Example 26) or (R)-1-(2-((S)-1-(4-Chloro-1H-pyrazol-1-yl)ethyl)-3H-imidazo[4,5-b]pyridin-5-yl)piperidin-3-yl)(pyrrolidin-1-yl)methanone (WO 2013150416, Example 196-A) for complete inhibition of DGAT2 was subtracted from all reactions. Inhibitors were tested at eleven different concentrations to generate IC50 values for each compound. The eleven inhibitor concentrations employed typically included 50, 15.8, 5, 1.58, 0.50, 0.16, 0.05, 0.016, 0.005, 0.0016, and 0.0005 μM. The data were plotted as percentage of inhibition versus inhibitor concentration and fit to the equation, y=100/[1+(x/IC50)z], where IC50 is the inhibitor concentration at 50% inhibition and z is the Hill slope (the slope of the curve at its inflection point). Table 3 below provides the IC50 values of the Examples for inhibition of DGAT2 in accordance with the above-described assay. Results are reported as geometric mean IC50 values. Measurement of PDE2 Inhibition In Vitro r-hPDE2A (Accession No. NM_002599, Homo sapiens phosphodiesterase 2A, cGMP-stimulated, transcript variant 1) A mammalian expression cloning vector with recombinant cDNA copy of the gene is purchased from Origene. Protein is expressed via transient transfection of HEK293 cells. The cells are harvested at 48 hours after transfection, washed once with TBS buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl), then lysed by sonication in cold homogenization buffer (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 1× protease inhibitor cocktail). The homogenate is centrifuged for 30 min at 15,000 g at 4° C. to obtain the soluble cytosolic fraction. The protein concentration of the cytosol is determined using BCA Protein Assay Kit (Pierce) with bovine serum albumin as a standard.Assay:PDE2A is assayed with FL-cAMP as substrate. An enzyme titration is first performed to determine the working concentration of PDE. The concentration of the enzyme giving activity of 100 ΔmP in the absence of inhibitor is deemed an appropriate working concentration for PDE.PDE enzyme is diluted in a standard reaction buffer buffer (10 mM Tris-HCl pH 7.2, 10 mM MgCl2, 0.1% BSA, 0.05% NaN3) according to the titration curve. For PDE2 assay the reaction buffer is supplemented with 1 μM cGMP to fully activate the enzyme. 99 μl of diluted enzyme solution is added into each well in a flat bottom 96-well polystyrene plate and then 1 μl of test compound dissolved in 100% DMSO is added. The compounds are mixed and pre-incubated with the enzyme for 10 min at room temperature.The FL-cNMP conversion reaction is initiated by addition of substrate (45 nM final). Enzyme and inhibitor mix (16 μl) and substrate solution (4 μl of 0.225 μM) are combined in a 384-well microtiter plate. The reaction is incubated in the dark at room temperature for 15 min. The reaction is halted by addition of 60 μl of binding reagent (1:400 dilution of IMAP beads in binding buffer supplemented with 1:1800 dilution of antifoam) to each well of the 384-well plate. The plate is incubated at room temperature for 1 hour to allow IMAP binding to proceed to completion, and then placed in an Envision multimode microplate reader (PerkinElmer, Shelton, Conn.) to measure the fluorescence polarization (Δmp).A decrease in cAMP concentration, measured as decreased Δmp, is indicative of inhibition of PDE activity. IC50 values are determined by measuring enzyme activity in the presence of 8 to 16 concentrations of compound ranging from 0.00037 nM to 80,000 nM and then plotting drug concentration versus ΔmP Test well values are normalized to control reactions run on the same plate (values converted to % of control). IC50 values are estimated using nonlinear regression software, fitting a four-parameter one-site dose-response model (XLFit; IDBS, Cambridge, Mass.). Bottom of curve is fixed at 0% of control. Fluorescence polarization assay Rhesus PDE9A2 was amplified from rhesus whole brain cDNA (Biochain Institute) essentially as described in Hutson, et al. Neuropharmacology (2011) 61(4):665-676. HEK 293 or CHO cells over-expressing rhesus or rat PDE9A2 (created by DiscoverX from mRNA Genbank accession # NM_138543) respectively were lysed in 20 mM HEPES, 1 mM EDTA buffer with protease inhibitors (Roche, Indianapolis, Ind.). After brief homogenization, cells were pelleted via centrifugation at 75,000×g for 20 min at 4° C. The pellets were re-suspended, centrifuged and re-pelleted again in the same manner. The membrane fraction was collected in 20 mM HEPES, 1 mM MgCl2 with protease inhibitors. Human PDE9 (PDE9A2, GenBank Accession No. NM_001001567), full length with N-terminal GST tag, was purchased from BPS Bioscience. The fluorescence polarization assay for cyclic nucleotide phosphodiesterases was performed using an IMAP FP kit supplied by Molecular Devices, Sunnyvale, Calif. (product # R8139). IMAP technology has been applied previously to phosphodiesterase assays (Huang, W., et al., J. Biomol Screen, 2002, 7: 215). Assays were performed at room temperature in 384-well microtiter plates with an incubation volume of 20.2 μL. Solutions of test compounds were prepared in DMSO and serially diluted with DMSO to yield 8 μL of each of 10 solutions differing by 3-fold in concentration, at 32 serial dilutions per plate. 100% inhibition is determined using a known PDE9 inhibitor, such as 1-(2-chlorophenyl)-6-[(2R)-3,3,3-trifluoro-2-methylpropyl]-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidine-4-one (BAY 73-6691) (Wunder et al, Mol. Pharmacol., 2005, 68(6): 1775-81), (6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (PF-04447943) (Wager et al., ACS Chemical Neuroscience, 2010, 1:435-449). 0% of inhibition is determined by using DMSO (1% final concentrations). A Labcyte Echo 555 (Labcyte, Sunnyvale, Calif.) is used to dispense 200 nL from each well of the titration plate to the 384 well assay plate. Rhesus membrane preps were diluted to 4 ng/ml, rat membrane preps diluted to 13 ng/ml, and human PDE9A2 diluted to 1 ng/ml. FAM-labeled cGMP substrate (Molecular Devices, Sunnyvale, Calif.) was at a concentration of 100 nM (Km of PDE9 for cGMP is 70-170 nM) in the assay buffer (10 mM Tris HCl, pH 7.2, 10 mM MgCl2, 0.05% NaN3 0.01% Tween-20, and 1 mM DTT). PDE9 enzyme mix and compounds were mixed and incubated at room temperature for 30 min. Following which, FAMcGMP substrate was added, shaken and incubated for an additional 60 min at room temperature. The final concentrations of rhesus and rat membrane preparations were 2 ng/ml and 6.5 ng/ml, respectively, while the human PDE9 was used at a final concentration of 0.5 ng/ml. The final concentration of FAM-cGMP was 50 nM. After the incubation period, the enzymatic reaction was stopped by addition of binding solution (IMAP-FP, Molecular Devices, comprised of 80% Solution A, 20% Solution B and a 1:600 dilution of binding reagent) to each well. The plates were shaken then incubated at room temperature for 1 h prior to determining the fluorescence polarization (mP) using a Perkin Elmer EnVision plate reader (Waltham, Mass.).
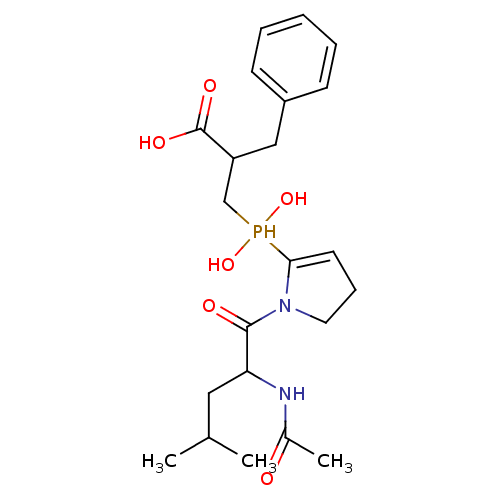 Phosphinic Peptide Inhibitor, 28 FII 2-benzyl-3-{[1-(2-acetamido-4-methylpentanoyl)pyrrolidin-2-yl](hydroxy)phosphoryl}propanoic acid The second fraction of the HPLC chromatogram. BDBM21464
Phosphinic Peptide Inhibitor, 28 FII 2-benzyl-3-{[1-(2-acetamido-4-methylpentanoyl)pyrrolidin-2-yl](hydroxy)phosphoryl}propanoic acid The second fraction of the HPLC chromatogram. BDBM21464 2-benzyl-3-({1-[2-acetamido-3-(1H-imidazol-4-yl)propanoyl]pyrrolidin-2-yl}(hydroxy)phosphoryl)propanoic acid Phosphinic Peptide Inhibitor, 34 FII The second fraction of the HPLC chromatogram. BDBM21470
2-benzyl-3-({1-[2-acetamido-3-(1H-imidazol-4-yl)propanoyl]pyrrolidin-2-yl}(hydroxy)phosphoryl)propanoic acid Phosphinic Peptide Inhibitor, 34 FII The second fraction of the HPLC chromatogram. BDBM21470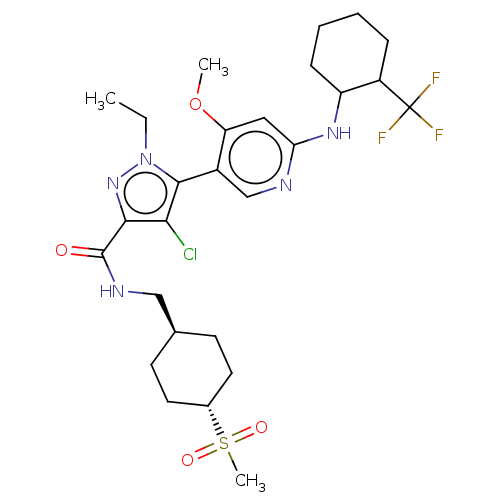 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclohexyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction A US10975057, Example 59 BDBM491775
4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclohexyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction A US10975057, Example 59 BDBM491775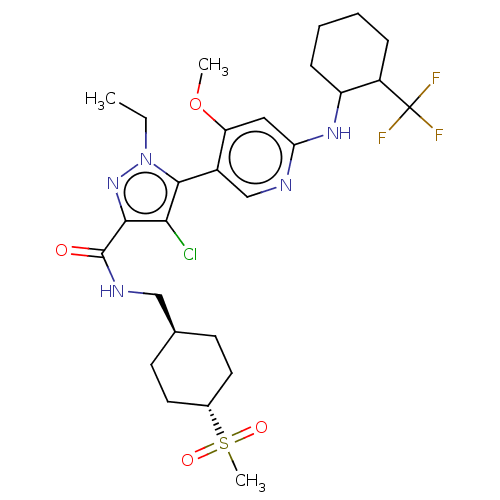 BDBM491776 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclohexyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction B US10975057, Example 60
BDBM491776 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclohexyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction B US10975057, Example 60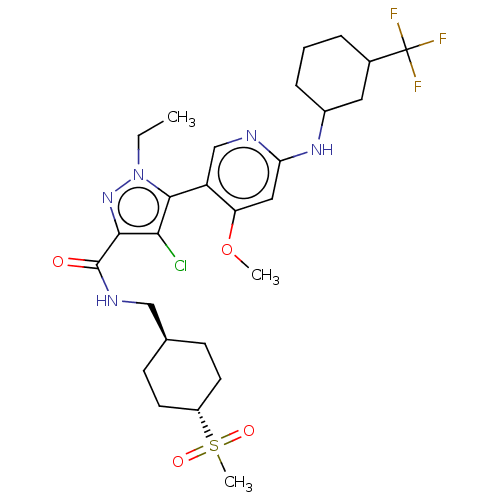 BDBM491777 US10975057, Example 61 4-Chloro-1-ethyl-5-(4-methoxy-6-((3-(trifluoromethyl)cyclohexyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction A
BDBM491777 US10975057, Example 61 4-Chloro-1-ethyl-5-(4-methoxy-6-((3-(trifluoromethyl)cyclohexyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction A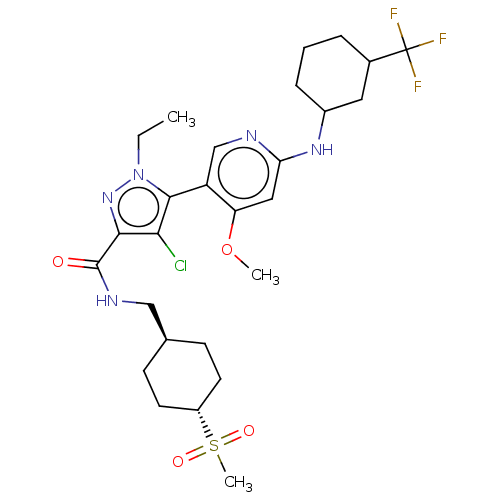 BDBM491778 US10975057, Example 62 4-Chloro-1-ethyl-5-(4-methoxy-6-((3-(trifluoromethyl)cyclohexyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction B
BDBM491778 US10975057, Example 62 4-Chloro-1-ethyl-5-(4-methoxy-6-((3-(trifluoromethyl)cyclohexyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction B BDBM491786 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclobutyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction B US10975057, Example 72
BDBM491786 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclobutyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction B US10975057, Example 72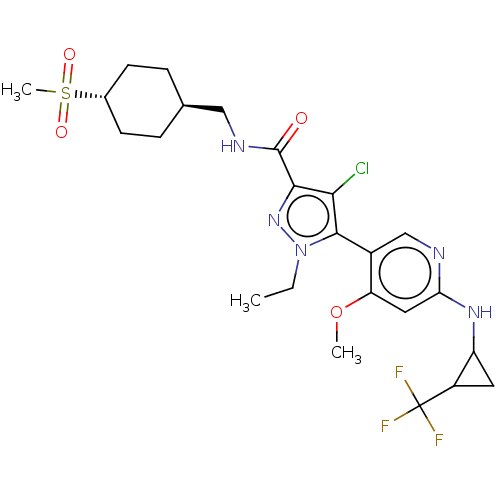 US10975057, Example 77 BDBM491789 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclopropyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction A
US10975057, Example 77 BDBM491789 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclopropyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction A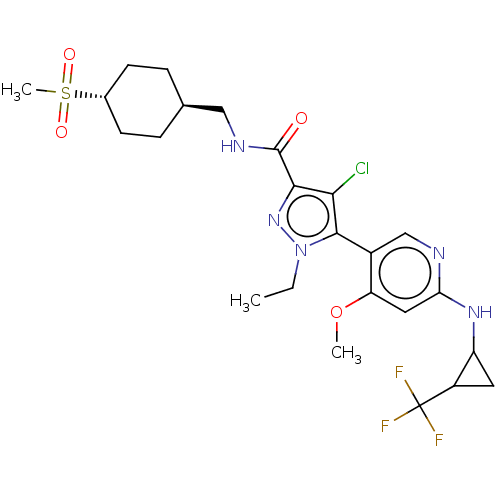 US10975057, Example 78 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclopropyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction B BDBM491790
US10975057, Example 78 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclopropyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction B BDBM491790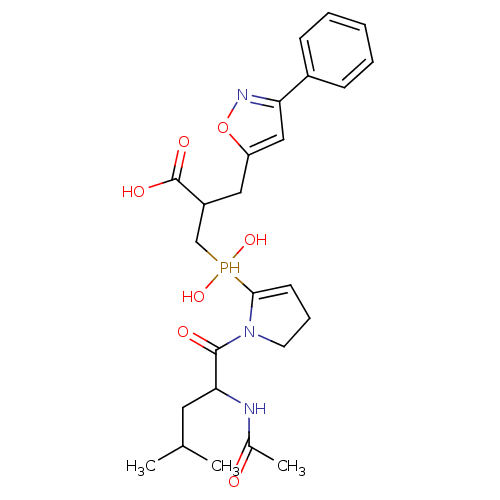 3-{[1-(2-acetamido-4-methylpentanoyl)pyrrolidin-2-yl](hydroxy)phosphoryl}-2-[(3-phenyl-1,2-oxazol-5-yl)methyl]propanoic acid The second fraction of the HPLC chromatogram. BDBM21473 Phosphinic Peptide Inhibitor, 40 FII
3-{[1-(2-acetamido-4-methylpentanoyl)pyrrolidin-2-yl](hydroxy)phosphoryl}-2-[(3-phenyl-1,2-oxazol-5-yl)methyl]propanoic acid The second fraction of the HPLC chromatogram. BDBM21473 Phosphinic Peptide Inhibitor, 40 FII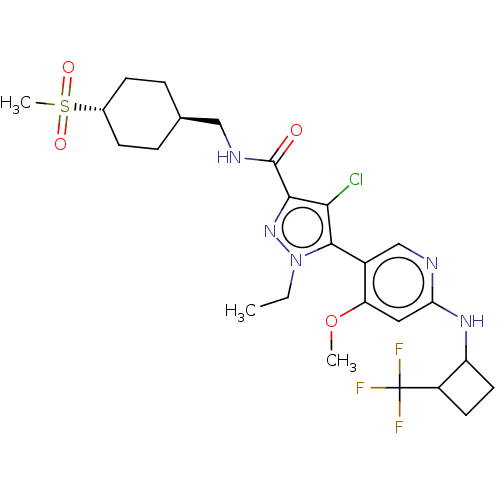 BDBM491785 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclobutyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction A US10975057, Example 72 US10975057, Example 71
BDBM491785 4-Chloro-1-ethyl-5-(4-methoxy-6-((2-(trifluoromethyl)cyclobutyl)amino)pyridin-3-yl)-N-(((1r,4r)-4-(methylsulfonyl)cyclohexyl)methyl)-1H-pyrazole-3-carboxamide, Fraction A US10975057, Example 72 US10975057, Example 71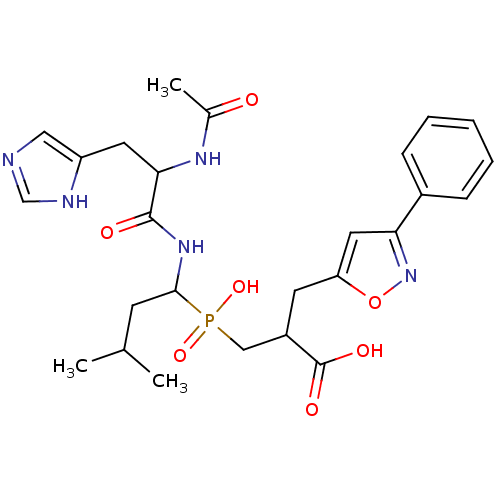 The second fraction of the HPLC chromatogram. Phosphinic Peptide Inhibitor, 44 FII BDBM21475 3-({1-[2-acetamido-3-(1H-imidazol-4-yl)propanamido]-3-methylbutyl}(hydroxy)phosphoryl)-2-[(3-phenyl-1,2-oxazol-5-yl)methyl]propanoic acid
The second fraction of the HPLC chromatogram. Phosphinic Peptide Inhibitor, 44 FII BDBM21475 3-({1-[2-acetamido-3-(1H-imidazol-4-yl)propanamido]-3-methylbutyl}(hydroxy)phosphoryl)-2-[(3-phenyl-1,2-oxazol-5-yl)methyl]propanoic acid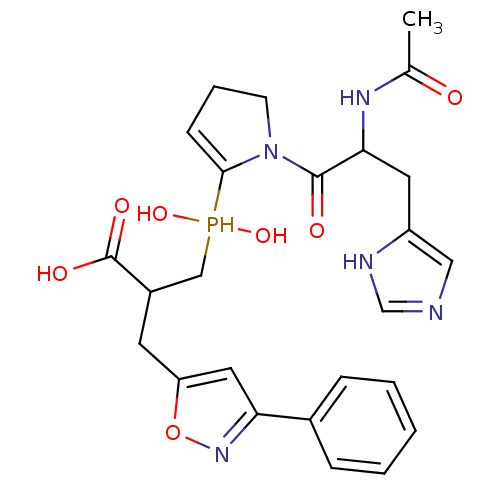 Phosphinic Peptide Inhibitor, 41 FII The second fraction of the HPLC chromatogram. 3-({1-[2-acetamido-3-(1H-imidazol-4-yl)propanoyl]pyrrolidin-2-yl}(hydroxy)phosphoryl)-2-[(3-phenyl-1,2-oxazol-5-yl)methyl]propanoic acid BDBM21474
Phosphinic Peptide Inhibitor, 41 FII The second fraction of the HPLC chromatogram. 3-({1-[2-acetamido-3-(1H-imidazol-4-yl)propanoyl]pyrrolidin-2-yl}(hydroxy)phosphoryl)-2-[(3-phenyl-1,2-oxazol-5-yl)methyl]propanoic acid BDBM21474