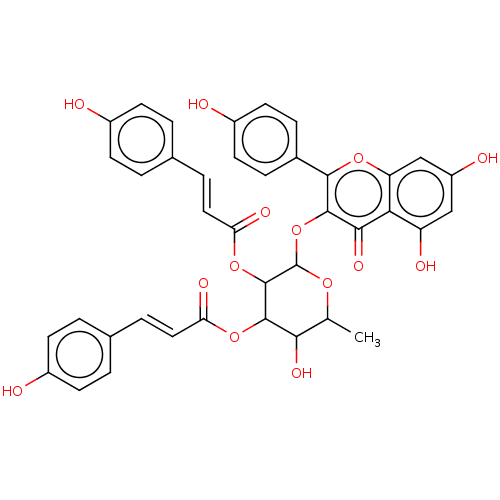
 BDBM241950 Neolitsea aciculata extract, 5
BDBM241950 Neolitsea aciculata extract, 5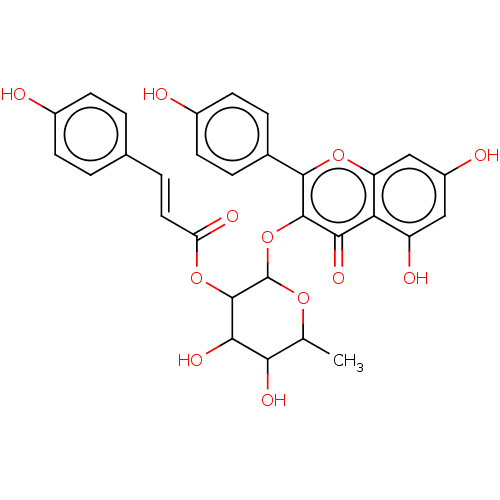 BDBM241951 Neolitsea aciculata extract, 6
BDBM241951 Neolitsea aciculata extract, 6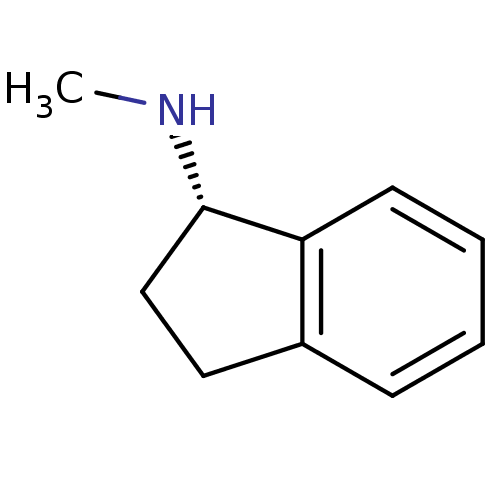 (1S)-N-methyl-2,3-dihydro-1H-inden-1-amine BDBM10996 rasagiline analog N-methyl-1(S)-aminoindan S-MAI
(1S)-N-methyl-2,3-dihydro-1H-inden-1-amine BDBM10996 rasagiline analog N-methyl-1(S)-aminoindan S-MAI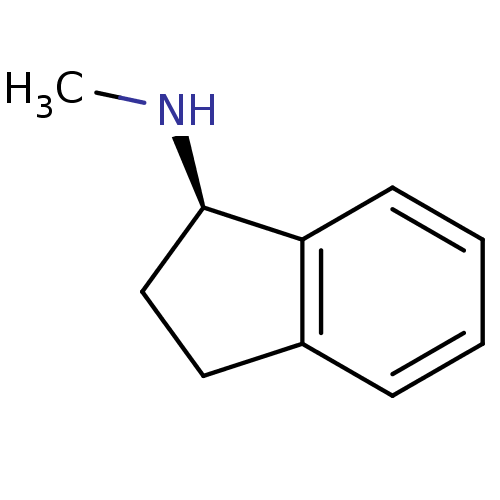 R-MAI rasagiline analog BDBM10995 N-methyl-1(R)-aminoindan (1R)-N-methyl-2,3-dihydro-1H-inden-1-amine
R-MAI rasagiline analog BDBM10995 N-methyl-1(R)-aminoindan (1R)-N-methyl-2,3-dihydro-1H-inden-1-amine
- Balestrieri, E; Pizzimenti, F; Ferlazzo, A; Giofrè, SV; Iannazzo, D; Piperno, A; Romeo, R; Chiacchio, MA; Mastino, A; Macchi, B Antiviral activity of seed extract from Citrus bergamia towards human retroviruses. Bioorg Med Chem 19: 2084-9 (2011)
- Rempel, V; Fuchs, A; Hinz, S; Karcz, T; Lehr, M; Koetter, U; Müller, CE Magnolia Extract, Magnolol, and Metabolites: Activation of Cannabinoid CB2 Receptors and Blockade of the Related GPR55. ACS Med Chem Lett 4: 41-5 (2013)
- Rainer, B; Revoltella, S; Mayr, F; Moesslacher, J; Scalfari, V; Kohl, R; Waltenberger, B; Pagitz, K; Siewert, B; Schwaiger, S; Stuppner, H From bench to counter: Discovery and validation of a peony extract as tyrosinase inhibiting cosmeceutical. Eur J Med Chem 184: (2019)
- Cuéllar, MJ; Giner, RM; Recio, MC; Just, MJ; Máñez, S; Cerdá, M; Hostettmann, K; Ríos, JL Zanhasaponins A and B, antiphospholipase A2 saponins from an antiinflammatory extract of Zanha africana root bark. J Nat Prod 60: 1158-60 (1998)
- Hüsch, J; Gerbeth, K; Fricker, G; Setzer, C; Zirkel, J; Rebmann, H; Schubert-Zsilavecz, M; Abdel-Tawab, M Effect of phospholipid-based formulations of Boswellia serrata extract on the solubility, permeability, and absorption of the individual boswellic acid constituents present. J Nat Prod 75: 1675-82 (2012)
- Park, MH; Kim, IS; Kim, SA; Na, CS; Hong, CY; Dong, MS; Yoo, HH Inhibitory effect of Rhus verniciflua Stokes extract on human aromatase activity; butin is its major bioactive component. Bioorg Med Chem Lett 24: 1730-3 (2014)
- Wu, T; Jiang, C; Wang, L; Morris-Natschke, SL; Miao, H; Gu, L; Xu, J; Lee, KH; Gu, Q 3,5-Diarylpyrazole Derivatives Obtained by Ammonolysis of the Total Flavonoids from Chrysanthemum indicum Extract Show Potential for the Treatment of Alzheimer's Disease. J Nat Prod 78: 1593-9 (2015)
- Kasangana, PB; Haddad, PS; Eid, HM; Nachar, A; Stevanovic, T Bioactive Pentacyclic Triterpenes from the Root Bark Extract of Myrianthus arboreus, a Species Used Traditionally to Treat Type-2 Diabetes. J Nat Prod 81: 2169-2176 (2018)
- Sharma, R; Gatchie, L; Williams, IS; Jain, SK; Vishwakarma, RA; Chaudhuri, B; Bharate, SB Glycyrrhiza glabra extract and quercetin reverses cisplatin resistance in triple-negative MDA-MB-468 breast cancer cells via inhibition of cytochrome P450 1B1 enzyme. Bioorg Med Chem Lett 27: 5400-5403 (2017)
- Rho, HS; Ahn, SM; Lee, BC; Kim, MK; Ghimeray, AK; Jin, CW; Cho, DH Changes in flavonoid content and tyrosinase inhibitory activity in kenaf leaf extract after far-infrared treatment. Bioorg Med Chem Lett 20: 7534-6 (2010)
- Wangtrakuldee, P; Byrd, MS; Campos, CG; Henderson, MW; Zhang, Z; Clare, M; Masoudi, A; Myler, PJ; Horn, JR; Cotter, PA; Hagen, TJ Discovery of Inhibitors of ACS Med Chem Lett 4: 699-703 (2013)
- Mathavan, I; Liu, LJ; Robinson, SW; El-Sakkary, N; Elatico, AJJ; Gomez, D; Nellas, R; Owens, RJ; Zuercher, W; Navratilova, I; Caffrey, CR; Beis, K Identification of Inhibitors of the ACS Med Chem Lett 13: 1715-1722 (2022)
- Harrison, LA; Atkinson, SJ; Bassil, A; Chung, CW; Grandi, P; Gray, JRJ; Levernier, E; Lewis, A; Lugo, D; Messenger, C; Michon, AM; Mitchell, DJ; Preston, A; Prinjha, RK; Rioja, I; Seal, JT; Taylor, S; Wall, ID; Watson, RJ; Woolven, JM; Demont, EH Identification of a Series of J Med Chem 64: 10742-10771 (2021)
- Bisson, WH; Cheltsov, AV; Bruey-Sedano, N; Lin, B; Chen, J; Goldberger, N; May, LT; Christopoulos, A; Dalton, JT; Sexton, PM; Zhang, XK; Abagyan, R Discovery of antiandrogen activity of nonsteroidal scaffolds of marketed drugs. Proc Natl Acad Sci U S A 104: 11927-32 (2007)
- Manickam, M; Pillaiyar, T; Boggu, P; Venkateswararao, E; Jalani, HB; Kim, ND; Lee, SK; Jeon, JS; Kim, SK; Jung, SH Discovery of enantioselectivity of urea inhibitors of soluble epoxide hydrolase. Eur J Med Chem 117: 113-24 (2016)
- Alnabulsi, S; Hussein, B; Santina, E; Alsalahat, I; Kadirvel, M; Magwaza, RN; Bryce, RA; Schwalbe, CH; Baldwin, AG; Russo, I; Stratford, IJ; Freeman, S Evaluation of analogues of furan-amidines as inhibitors of NQO2. Bioorg Med Chem Lett 28: 1292-1297 (2018)
- Suto, MJ; Gupta, V; Mathew, B; Zhang, W; Pallero, MA; Murphy-Ullrich, JE Identification of Inhibitors of Thrombospondin 1 Activation of TGF-β. ACS Med Chem Lett 11: 1130-1136 (2020)
- Liu, J; Fu, Z; Li, AR; Johnson, M; Zhu, L; Marcus, A; Danao, J; Sullivan, T; Tonn, G; Collins, T; Medina, J Optimization of a series of quinazolinone-derived antagonists of CXCR3. Bioorg Med Chem Lett 19: 5114-8 (2009)
- Madhav, H; Reddy, GS; Rizvi, Z; Jameel, E; Patel, TS; Rahman, A; Yadav, V; Fatima, S; Heyat, F; Pal, K; Minju-Op, A; Subbarao, N; Bhattacharjee, S; Dixit, BC; Sijwali, PS; Hoda, N Reinvestigation of diphenylmethylpiperazine analogues of pyrazine as new class of RSC Med Chem 15: 1022-1037 (2024)
- Bing, DH Nature of the active site of a subunit of the first component of human complement. Biochemistry 8: 4503-10 (1969)
- Bheemanaboina, RRY; de Souza, ML; Gonzalez, ML; Mahmood, SU; Eck, T; Kreiss, T; Aylor, SO; Roth, A; Lee, P; Pybus, BS; Colussi, DJ; Childers, WE; Gordon, J; Siekierka, JJ; Bhanot, P; Rotella, DP Discovery of Imidazole-Based Inhibitors of ACS Med Chem Lett 12: 1962-1967 (2021)
- Pajouhesh, H; Delwig, A; Beckley, JT; Klas, S; Monteleone, D; Zhou, X; Luu, G; Du Bois, J; Hunter, JC; Mulcahy, JV Discovery of Selective Inhibitors of Na ACS Med Chem Lett 13: 1763-1768 (2022)
- Blake, JF; Kallan, NC; Xiao, D; Xu, R; Bencsik, JR; Skelton, NJ; Spencer, KL; Mitchell, IS; Woessner, RD; Gloor, SL; Risom, T; Gross, SD; Martinson, M; Morales, TH; Vigers, GP; Brandhuber, BJ Discovery of pyrrolopyrimidine inhibitors of Akt. Bioorg Med Chem Lett 20: 5607-12 (2010)
- Graef, IA; Alhamadsheh, MM Identification of stabilizers of multimeric proteins US Patent US10278929 (2019)
- Castro, AC; Grogan, MJ; McGovern, KJ; Tremblay, MR Methods of use of cyclopamine analogs US Patent US10314827 (2019)
- Ferreira de Freitas, R; Liu, Y; Szewczyk, MM; Mehta, N; Li, F; McLeod, D; Zepeda-Velázquez, C; Dilworth, D; Hanley, RP; Gibson, E; Brown, PJ; Al-Awar, R; James, LI; Arrowsmith, CH; Barsyte-Lovejoy, D; Min, J; Vedadi, M; Schapira, M; Allali-Hassani, A Discovery of Small-Molecule Antagonists of the PWWP Domain of NSD2. J Med Chem 64: 1584-1592 (2021)
- Ontoria, JM; Paonessa, G; Ponzi, S; Ferrigno, F; Nizi, E; Biancofiore, I; Malancona, S; Graziani, R; Roberts, D; Willis, P; Bresciani, A; Gennari, N; Cecchetti, O; Monteagudo, E; Orsale, MV; Veneziano, M; Di Marco, A; Cellucci, A; Laufer, R; Altamura, S; Summa, V; Harper, S Discovery of a Selective Series of Inhibitors of Plasmodium falciparum HDACs. ACS Med Chem Lett 7: 454-9 (2016)
- Degorce, SL; Anjum, R; Dillman, KS; Drew, L; Groombridge, SD; Halsall, CT; Lenz, EM; Lindsay, NA; Mayo, MF; Pink, JH; Robb, GR; Scott, JS; Stokes, S; Xue, Y Optimization of permeability in a series of pyrrolotriazine inhibitors of IRAK4. Bioorg Med Chem 26: 913-924 (2018)
- Mayhoub, AS; Marler, L; Kondratyuk, TP; Park, EJ; Pezzuto, JM; Cushman, M Optimization of the aromatase inhibitory activities of pyridylthiazole analogues of resveratrol. Bioorg Med Chem 20: 2427-34 (2012)
- Kim, HS; Hammill, JT; Scott, DC; Chen, Y; Rice, AL; Pistel, W; Singh, B; Schulman, BA; Guy, RK Improvement of Oral Bioavailability of Pyrazolo-Pyridone Inhibitors of the Interaction of DCN1/2 and UBE2M. J Med Chem 64: 5850-5862 (2021)
- Bickel, D; Gohlke, H C-terminal modulators of heat shock protein of 90 kDa (HSP90): State of development and modes of action. Bioorg Med Chem 27: (2019)
- Aertgeerts, K; Skene, R; Yano, J; Sang, BC; Zou, H; Snell, G; Jennings, A; Iwamoto, K; Habuka, N; Hirokawa, A; Ishikawa, T; Tanaka, T; Miki, H; Ohta, Y; Sogabe, S Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem 286: 18756-65 (2011)
- Johnson, M; Li, AR; Liu, J; Fu, Z; Zhu, L; Miao, S; Wang, X; Xu, Q; Huang, A; Marcus, A; Xu, F; Ebsworth, K; Sablan, E; Danao, J; Kumer, J; Dairaghi, D; Lawrence, C; Sullivan, T; Tonn, G; Schall, T; Collins, T; Medina, J Discovery and optimization of a series of quinazolinone-derived antagonists of CXCR3. Bioorg Med Chem Lett 17: 3339-43 (2007)
- Casimiro-Garcia, A; Allais, C; Brennan, A; Choi, C; Dower, G; Farley, KA; Fleming, M; Flick, A; Frisbie, RK; Hall, J; Hepworth, D; Jones, H; Knafels, JD; Kortum, S; Lovering, FE; Mathias, JP; Mohan, S; Morgan, PM; Parng, C; Parris, K; Pullen, N; Schlerman, F; Stansfield, J; Strohbach, JW; Vajdos, FF; Vincent, F; Wang, H; Wang, X; Webster, R; Wright, SW Discovery of a Series of Pyrimidine Carboxamides as Inhibitors of Vanin-1. J Med Chem 65: 757-784 (2022)
- Medina, JR; Grant, SW; Axten, JM; Miller, WH; Donatelli, CA; Hardwicke, MA; Oleykowski, CA; Liao, Q; Plant, R; Xiang, H Discovery of a new series of Aurora inhibitors through truncation of GSK1070916. Bioorg Med Chem Lett 20: 2552-5 (2010)
- Gazzard, L; Appleton, B; Chapman, K; Chen, H; Clark, K; Drobnick, J; Goodacre, S; Halladay, J; Lyssikatos, J; Schmidt, S; Sideris, S; Wiesmann, C; Williams, K; Wu, P; Yen, I; Malek, S Discovery of the 1,7-diazacarbazole class of inhibitors of checkpoint kinase 1. Bioorg Med Chem Lett 24: 5704-9 (2014)
- Occhipinti, A; Berlicki, Ł; Giberti, S; Dziedzioła, G; Kafarski, P; Forlani, G Effectiveness and mode of action of phosphonate inhibitors of plant glutamine synthetase. Pest Manag Sci 66: 51-8 (2010)
- Bauman, DR; Whitehead, A; Contino, LC; Cui, J; Garcia-Calvo, M; Gu, X; Kevin, N; Ma, X; Pai, LY; Shah, K; Shen, X; Stribling, S; Zokian, HJ; Metzger, J; Shevell, DE; Waddell, ST Evaluation of selective inhibitors of 11ß-HSD1 for the treatment of hypertension. Bioorg Med Chem Lett 23: 3650-3 (2013)
- Smith, GF; Altman, MD; Andresen, B; Baker, J; Brubaker, JD; Chen, H; Chen, Y; Childers, M; Donofrio, A; Ferguson, H; Fischer, C; Fischmann, TO; Gibeau, C; Hicks, A; Jin, S; Kattar, S; Kleinschek, MA; Leccese, E; Lesburg, C; Li, C; Lim, J; Liu, D; Maclean, JKF; Mansoor, F; Moy, LY; Mulrooney, EF; Necheva, AS; Presland, J; Rakhilina, L; Yang, R; Torres, L; Zhang-Hoover, J; Northrup, A Identification of quinazoline based inhibitors of IRAK4 for the treatment of inflammation. Bioorg Med Chem Lett 27: 2721-2726 (2017)
- Angelastro, MR; Marquart, AL; Mehdi, S; Koehl, JR; Vaz, RJ; Bey, P; Peet, NP The synthesis of ketomethylene pseudopeptide analogues of dipeptide aldehyde inhibitors of calpain. Bioorg Med Chem Lett 9: 139-40 (1999)
- Kozlov, MV; Konduktorov, KA; Shcherbakova, AS; Kochetkov, SN Synthesis of N'-propylhydrazide analogs of hydroxamic inhibitors of histone deacetylases (HDACs) and evaluation of their impact on activities of HDACs and replication of hepatitis C virus (HCV). Bioorg Med Chem Lett 29: 2369-2374 (2019)
- Petrassi, HM; Lairson, LL; Chin, E; Schultz, PG; Yu, C; Yang, B; Grant, V; Li, Y; Pacheco, A; Chu, A; Johnson, K; Chatterjee, AK AGONISTS OF STIMULATOR OF INTERFERON GENES STING US Patent US20230357253 (2023)
- Foster, AB; Jarman, M; Leung, CS; Rowlands, MG; Taylor, GN; Plevey, RG; Sampson, P Analogues of aminoglutethimide: selective inhibition of aromatase. J Med Chem 28: 200-4 (1985)
- Phommart, S; Sutthivaiyakit, P; Chimnoi, N; Ruchirawat, S; Sutthivaiyakit, S Constituents of the leaves of Macaranga tanarius. J Nat Prod 68: 927-30 (2005)
- Wang, J; Zeng, W; Li, S; Shen, L; Gu, Z; Zhang, Y; Li, J; Chen, S; Jia, X Discovery and Assessment of Atropisomers of (±)-Lesinurad. ACS Med Chem Lett 8: 299-303 (2017)
- Lanman, BA; Allen, JR; Allen, JG; Amegadzie, AK; Ashton, KS; Booker, SK; Chen, JJ; Chen, N; Frohn, MJ; Goodman, G; Kopecky, DJ; Liu, L; Lopez, P; Low, JD; Ma, V; Minatti, AE; Nguyen, TT; Nishimura, N; Pickrell, AJ; Reed, AB; Shin, Y; Siegmund, AC; Tamayo, NA; Tegley, CM; Walton, MC; Wang, HL; Wurz, RP; Xue, M; Yang, KC; Achanta, P; Bartberger, MD; Canon, J; Hollis, LS; McCarter, JD; Mohr, C; Rex, K; Saiki, AY; San Miguel, T; Volak, LP; Wang, KH; Whittington, DA; Zech, SG; Lipford, JR; Cee, VJ Discovery of a Covalent Inhibitor of KRAS J Med Chem 63: 52-65 (2020)
- Gelin, CF; Stenne, B; Coate, H; Hiscox, A; Soyode-Johnson, A; Wall, JL; Lord, B; Schoellerman, J; Coe, KJ; Wang, K; Alcázar, J; Chrovian, CC; Dvorak, CA; Carruthers, NI; Koudriakova, T; Balana, B; Letavic, MA Discovery of a Series of Substituted 1 J Med Chem 66: 2877-2892 (2023)
- Zaware, N; Sharma, H; Yang, J; Devambatla, RK; Queener, SF; Anderson, KS; Gangjee, A Discovery of potent and selective inhibitors of ACS Med Chem Lett 4: 1148-1151 (2013)
- Lanter, JC; Chen, AY; Williamson, T; Koenig, G; Blain, JF; Burnett, DA Discovery of quinuclidine modulators of cellular progranulin. Bioorg Med Chem Lett 47: (2021)
- Rami, HK; Thompson, M; Wyman, P; Jerman, JC; Egerton, J; Brough, S; Stevens, AJ; Randall, AD; Smart, D; Gunthorpe, MJ; Davis, JB Discovery of small molecule antagonists of TRPV1. Bioorg Med Chem Lett 14: 3631-4 (2004)
- Germanas, JP; Wang, S; Miner, A; Hao, W; Ready, JM Discovery of small-molecule inhibitors of tyrosinase. Bioorg Med Chem Lett 17: 6871-5 (2007)
- Niu, Y; Li, Q; Tu, C; Li, N; Gao, L; Lin, H; Wang, Z; Zhou, Z; Li, L Hypouricemic Actions of the Pericarp of Mangosteen J Nat Prod 86: 24-33 (2023)
- Tomala, MD; Magiera-Mularz, K; Kubica, K; Krzanik, S; Zieba, B; Musielak, B; Pustula, M; Popowicz, GM; Sattler, M; Dubin, G; Skalniak, L; Holak, TA Identification of small-molecule inhibitors of USP2a. Eur J Med Chem 150: 261-267 (2018)
- Kelley, JL; Miller, CA; White, HL Inhibition of histidine decarboxylase. Derivatives of histidine. J Med Chem 20: 506-9 (1977)
- Li, L; Wu, T; Feng, J; Ren, P; Liu, Y Inhibitors of ERK and methods of use US Patent US10301317 (2019)
- Posakony, J; Hirao, M; Stevens, S; Simon, JA; Bedalov, A Inhibitors of Sir2: evaluation of splitomicin analogues. J Med Chem 47: 2635-44 (2004)
- Lin, H; He, B Methods of treatment using modulators of SIRT2 US Patent US9359293 (2016)
- Modulation of proteasome subunit selectivity of syringolins.
- Gajiwala, KS; Huh, CW; Jalaie, M; Patman, RL; Rui, EY; Sun, J; Wythes, MJ Modulators of STING (stimulator of interferon genes) US Patent US11964978 (2024)
- Miah, AH; Smith, IED; Rackham, M; Mares, A; Thawani, AR; Nagilla, R; Haile, PA; Votta, BJ; Gordon, LJ; Watt, G; Denyer, J; Fisher, DT; Dace, P; Giffen, P; Goncalves, A; Churcher, I; Scott-Stevens, P; Harling, JD Optimization of a Series of RIPK2 PROTACs. J Med Chem 64: 12978-13003 (2021)
- Kilburn, JP; Ascic, E; Marigo, M; David, L Prodrugs of modulators of the NMDA receptor US Patent US11358971 (2022)
- Kirby, IT; Person, A; Cohen, M Rational design of selective inhibitors of PARP4. RSC Med Chem 12: 1950-1957 (2021)
- Desjardins, M; Desgagnes, J; Lacoste, L; Yang, F; Morin, M; Lapointe, J; Chenevert, R Synthesis of inhibitors of glutamyl-tRNA synthetase Bioorg Med Chem Lett 7: 2363-2366 (1997)
- Dancer, JE; Ford, MJ; Hamilton, K; Kilkelly, M; Lindell, SD; O'Mahony, MJ; Saville-Stones, EA Synthesis of potent inhibitors of histidinol dehydrogenase Bioorg Med Chem Lett 6: 2131-2136 (1996)
- Barton, DH; Géro, SD; Lawrence, F; Robert-Gero, M; Quiclet-Sire, B; Samadi, M Total synthesis of uracil analogues of sinefungin. J Med Chem 35: 63-7 (1992)
- Ozawa, M; Morita, M; Hirai, G; Tamura, S; Kawai, M; Tsuchiya, A; Oonuma, K; Maruoka, K; Sodeoka, M Contribution of Cage-Shaped Structure of Physalins to Their Mode of Action in Inhibition of NF-kB Activation ACS Med Chem Lett 4: 730-5 (2013)
- Cardozo, MG; Iimura, Y; Sugimoto, H; Yamanishi, Y; Hopfinger, AJ QSAR analyses of the substituted indanone and benzylpiperidine rings of a series of indanone-benzylpiperidine inhibitors of acetylcholinesterase. J Med Chem 35: 584-9 (1992)
- Rich, DH; Sun, ET; Ulm, E Synthesis of analogues of the carboxyl protease inhibitor pepstatin. Effects of structure on inhibition of pepsin and renin. J Med Chem 23: 27-33 (1980)
- Cale, JM; Li, SH; Warnock, M; Su, EJ; North, PR; Sanders, KL; Puscau, MM; Emal, CD; Lawrence, DA Characterization of a novel class of polyphenolic inhibitors of plasminogen activator inhibitor-1. J Biol Chem 285: 7892-902 (2010)
- Sedrani, R; Jones, LH; Jutzi-Eme, AM; Schuler, W; Cottens, S Cleavage of the cyclohexyl-subunit of rapamycin results in loss of immunosuppressive activity. Bioorg Med Chem Lett 9: 459-62 (1999)
- Blass, BE Covalent Inhibitors of the TEC Family of Kinases and Their Methods of Use. ACS Med Chem Lett 9: 587-589 (2018)
- Ujjinamatada, RK; Bhan, A; Hosmane, RS Design of inhibitors against guanase: synthesis and biochemical evaluation of analogues of azepinomycin. Bioorg Med Chem Lett 16: 5551-4 (2006)
- Berlicki, L; Obojska, A; Forlani, G; Kafarski, P Design, synthesis, and activity of analogues of phosphinothricin as inhibitors of glutamine synthetase. J Med Chem 48: 6340-9 (2005)
- Di Chio, C; Previti, S; Amendola, G; Ravichandran, R; Wagner, A; Cosconati, S; Hellmich, UA; Schirmeister, T; Zappalà, M; Ettari, R Development of novel dipeptide nitriles as inhibitors of rhodesain of Trypanosoma brucei rhodesiense. Eur J Med Chem 236: (2022)
- Zhang, Y; Seigal, BA; Terrett, NK; Talbott, RL; Fargnoli, J; Naglich, JG; Chaudhry, C; Posy, SL; Vuppugalla, R; Cornelius, G; Lei, M; Wang, C; Zhang, Y; Schmidt, RJ; Wei, DD; Miller, MM; Allen, MP; Li, L; Carter, PH; Vite, GD; Borzilleri, RM Dimeric Macrocyclic Antagonists of Inhibitor of Apoptosis Proteins for the Treatment of Cancer. ACS Med Chem Lett 6: 770-5 (2015)
- Chowdhury, S; Chafeev, M; Liu, S; Sun, J; Raina, V; Chui, R; Young, W; Kwan, R; Fu, J; Cadieux, JA Discovery of XEN907, a spirooxindole blocker of NaV1.7 for the treatment of pain. Bioorg Med Chem Lett 21: 3676-81 (2011)
- Siu, T; Altman, MD; Baltus, GA; Childers, M; Ellis, JM; Gunaydin, H; Hatch, H; Ho, T; Jewell, J; Lacey, BM; Lesburg, CA; Pan, BS; Sauvagnat, B; Schroeder, GK; Xu, S Discovery of a Novel cGAMP Competitive Ligand of the Inactive Form of STING. ACS Med Chem Lett 10: 92-97 (2019)
- Porter, J; Lumb, S; Lecomte, F; Reuberson, J; Foley, A; Calmiano, M; le Riche, K; Edwards, H; Delgado, J; Franklin, RJ; Gascon-Simorte, JM; Maloney, A; Meier, C; Batchelor, M Discovery of a novel series of quinoxalines as inhibitors of c-Met kinase. Bioorg Med Chem Lett 19: 397-400 (2008)
- Ettari, R; Previti, S; Di Chio, C; Maiorana, S; Allegra, A; Schirmeister, T; Zappalà, M Drug Synergism: Studies of Combination of RK-52 and Curcumin against Rhodesain of ACS Med Chem Lett 11: 806-810 (2020)
- Kawatkar, SP; Barlaam, B; Kemmitt, P; Simpson, I; Watson, D; Wang, P; Lamont, S; Su, Q; Boiko, S; Ikeda, T; Patel, J; Pike, A; Pollard, H; Read, J; Sarkar, U; Wang, H; Wen, Q; Yan, Z; Dowling, JE; Dry, H; Edmondson, SD Identification of a novel series of azabenzimidazole-derived inhibitors of spleen tyrosine kinase. Bioorg Med Chem Lett 30: (2020)
- Hintermann, S; Chiesi, M; von Krosigk, U; Mathé, D; Felber, R; Hengerer, B Identification of a series of highly potent activators of the Nurr1 signaling pathway. Bioorg Med Chem Lett 17: 193-6 (2006)
- Szardenings, AK; Antonenko, V; Campbell, DA; DeFrancisco, N; Ida, S; Shi, L; Sharkov, N; Tien, D; Wang, Y; Navre, M Identification of highly selective inhibitors of collagenase-1 from combinatorial libraries of diketopiperazines. J Med Chem 42: 1348-57 (1999)
- Ouvry, G; Clary, L; Tomas, L; Aurelly, M; Bonnary, L; Borde, E; Bouix-Peter, C; Chantalat, L; Defoin-Platel, C; Deret, S; Forissier, M; Harris, CS; Isabet, T; Lamy, L; Luzy, AP; Pascau, J; Soulet, C; Taddei, A; Taquet, N; Thoreau, E; Varvier, E; Vial, E; Hennequin, LF Impact of Minor Structural Modifications on Properties of a Series of mTOR Inhibitors. ACS Med Chem Lett 10: 1561-1567 (2019)
- Chikhale, RV; Barmade, MA; Murumkar, PR; Yadav, MR Overview of the Development of DprE1 Inhibitors for Combating the Menace of Tuberculosis. J Med Chem 61: 8563-8593 (2018)
- Boivin, RP; Luu-The, V; Lachance, R; Labrie, F; Poirier, D Structure-activity relationships of 17alpha-derivatives of estradiol as inhibitors of steroid sulfatase. J Med Chem 43: 4465-78 (2000)
- Baldwin, RM; Fu, X; Kula, NS; Baldessarini, RJ; Amici, L; Innis, RB; Tamagnan, GD Synthesis and affinity of a possible byproduct of electrophilic radiolabeling of [123I]IBZM. Bioorg Med Chem Lett 13: 4015-7 (2003)
- Szkaradek, N; Rapacz, A; Pytka, K; Filipek, B; Siwek, A; Cegła, M; Marona, H Synthesis and preliminary evaluation of pharmacological properties of some piperazine derivatives of xanthone. Bioorg Med Chem 21: 514-22 (2013)
- Boyle, NA; Talesa, V; Giovannini, E; Rosi, G; Norton, SJ Synthesis and study of thiocarbonate derivatives of choline as potential inhibitors of acetylcholinesterase. J Med Chem 40: 3009-13 (1997)
- Scheeff, S; Rivière, S; Ruiz, J; Abdelrahman, A; Schulz-Fincke, AC; Köse, M; Tiburcy, F; Wieczorek, H; Gütschow, M; Müller, CE; Menche, D Synthesis of Novel Potent Archazolids: Pharmacology of an Emerging Class of Anticancer Drugs. J Med Chem 63: 1684-1698 (2020)
- Bänteli, R; Ernst, B Synthesis of sialyl Lewis(x) mimics. Modifications of the 6-position of galactose. Bioorg Med Chem Lett 11: 459-62 (2001)
- Ehlert, FJ; Griffin, MT; Glidden, PF The interaction of the enantiomers of aceclidine with subtypes of the muscarinic receptor. J Pharmacol Exp Ther 279: 1335-44 (1996)
- Millet Aguilar-Galindo, O; Laín Torre, A Use of inhibitors of porphobilinogen deaminase in the treatment of congenital erythropoietic porphyria US Patent US9138423 (2015)
- Gleeson, P; Bravi, G; Modi, S; Lowe, D ADMET rules of thumb II: A comparison of the effects of common substituents on a range of ADMET parameters. Bioorg Med Chem 17: 5906-19 (2009)
- Khan, MT; Khan, R; Wuxiuer, Y; Arfan, M; Ahmed, M; Sylte, I Identification of novel quinazolin-4(3H)-ones as inhibitors of thermolysin, the prototype of the M4 family of proteinases. Bioorg Med Chem 18: 4317-27 (2010)
- Rich, DH; Salituro, FG Synthesis of analogues of pepstatin. Effect of structure in subsites P1', P2', and P2 on inhibition of porcine pepsin. J Med Chem 26: 904-10 (1983)
- Rich, DH; Bernatowicz, MS Synthesis of analogues of the carboxyl protease inhibitor pepstatin. Effect of structure in subsite P3 on inhibition of pepsin. J Med Chem 25: 791-5 (1982)
- Rodriguez, M; Fulcrand, P; Laur, J; Aumelas, A; Bali, JP; Martinez, J Synthesis of gastrin antagonists, analogues of the C-terminal tetrapeptide of gastrin, by introduction of a beta-homo residue. J Med Chem 32: 522-8 (1989)
- León, F; Obeng, S; Mottinelli, M; Chen, Y; King, TI; Berthold, EC; Kamble, SH; Restrepo, LF; Patel, A; Gamez-Jimenez, LR; Lopera-Londoño, C; Hiranita, T; Sharma, A; Hampson, AJ; Canal, CE; McMahon, LR; McCurdy, CR Activity of J Med Chem 64: 13510-13523 (2021)
- Zhao, F; Atxabal, U; Mariottini, S; Yi, F; Lotti, JS; Rouzbeh, N; Liu, N; Bunch, L; Hansen, KB; Clausen, RP Derivatives of ( J Med Chem 65: 734-746 (2022)
- Yamada, A; Kazui, Y; Yoshioka, H; Tanatani, A; Mori, S; Kagechika, H; Fujii, S Development of ACS Med Chem Lett 7: 1028-1033 (2016)
- Stéen, EJL; Park, AY; Beaino, W; Gadhe, CG; Kooijman, E; Schuit, RC; Schreurs, M; Leferink, P; Hoozemans, JJM; Kim, JE; Lee, J; Windhorst, AD Development of J Med Chem 66: 12990-13006 (2023)
- Wong, SW; Vivash, L; Mudududdla, R; Nguyen, N; Hermans, SJ; Shackleford, DM; Field, J; Xue, L; Aprico, A; Hancock, NC; Haskali, M; Stashko, MA; Frye, SV; Wang, X; Binder, MD; Ackermann, U; Parker, MW; Kilpatrick, TJ; Baell, JB Development of [ Eur J Med Chem 226: (2021)
- Meng, W; Brigance, R; Mignone, J; Negash, L; Zhao, G; Ahmad, S; Wang, W; Moore, F; Ye, XY; Sun, JH; Mathur, A; Li, YX; Azzara, A; Ma, Z; Chu, CH; Cullen, MJ; Rooney, S; Harvey, S; Kopcho, L; Abell, L; O'Malley, K; Keim, W; Dierks, EA; Chang, S; Foster, KA; Harden, D; Dabros, M; Goti, V; De Oliveira, C; Krishna, G; Pelleymounter, MA; Whaley, J; Robl, JA; Cheng, D; Devasthale, P Discovery of J Med Chem 66: 13135-13147 (2023)
- Luo, G; Chen, L; Kostich, WA; Hamman, B; Allen, J; Easton, A; Bourin, C; Gulianello, M; Lippy, J; Nara, S; Maishal, TK; Thiyagarajan, K; Jalagam, P; Pattipati, SN; Dandapani, K; Dokania, M; Vattikundala, P; Sharma, V; Elavazhagan, S; Verma, MK; Das, ML; Wagh, S; Balakrishnan, A; Johnson, BM; Santone, KS; Thalody, G; Denton, R; Saminathan, H; Holenarsipur, VK; Kumar, A; Rao, A; Putlur, SP; Sarvasiddhi, SK; Shankar, G; Louis, JV; Ramarao, M; Conway, CM; Li, YW; Pieschl, R; Tian, Y; Hong, Y; Ditta, J; Mathur, A; Li, J; Smith, D; Pawluczyk, J; Sun, D; Yip, S; Wu, DR; Vetrichelvan, M; Gupta, A; Wilson, A; Gopinathan, S; Wason, S; Bristow, L; Albright, CF; Bronson, JJ; Macor, JE; Dzierba, CD Discovery of ( J Med Chem 65: 4457-4480 (2022)
- Perkins, JJ; McQuade, P; Bungard, CJ; Diamond, TL; Gantert, LT; Gotter, AL; Hanney, B; Hills, ID; Hurzy, DM; Joshi, A; Kern, JT; Schlegel, KS; Manikowski, JJ; Meng, Z; O'Brien, JA; Roecker, AJ; Smith, SM; Uslaner, JM; Hostetler, E; Meissner, RS Discovery of [ ACS Med Chem Lett 14: 986-992 (2023)
- Zhu, Y; Ma, Y; Zu, W; Song, J; Wang, H; Zhong, Y; Li, H; Zhang, Y; Gao, Q; Kong, B; Xu, J; Jiang, F; Wang, X; Li, S; Liu, C; Liu, H; Lu, T; Chen, Y Identification of J Med Chem 63: 6748-6773 (2020)
- Hu, XL; Lv, XY; Wang, R; Long, H; Feng, JH; Wang, BL; Shen, W; Liu, H; Xiong, F; Zhang, XQ; Ye, WC; Wang, H Optimization of J Med Chem 64: 7760-7777 (2021)
- Kudo, Y; Hanifin, CT; Kotaki, Y; Yotsu-Yamashita, M Structures of J Nat Prod 83: 2706-2717 (2020)
- Yoder, RJ; Zhuang, Q; Beck, JM; Franjesevic, A; Blanton, TG; Sillart, S; Secor, T; Guerra, L; Brown, JD; Reid, C; McElroy, CA; Doğan Ekici, Ö; Callam, CS; Hadad, CM Study of ACS Med Chem Lett 8: 622-627 (2017)
- Cheng, MC; Li, CY; Ko, HC; Ko, FN; Lin, YL; Wu, TS Antidepressant principles of the roots of Polygala tenuifolia. J Nat Prod 69: 1305-9 (2006)
- DeMartino, JK; Hwang, I; Connelly, S; Wilson, IA; Boger, DL Asymmetric synthesis of inhibitors of glycinamide ribonucleotide transformylase. J Med Chem 51: 5441-8 (2008)
- Garcia-Jimenez, A; Teruel-Puche, JA; Berna, J; Rodriguez-Lopez, JN; Tudela, J; Garcia-Ruiz, PA; Garcia-Canovas, F Characterization of the action of tyrosinase on resorcinols. Bioorg Med Chem 24: 4434-4443 (2016)
- Xu, W; Zhu, C; Cheng, W; Fan, X; Chen, X; Yang, S; Guo, Y; Ye, F; Shi, J Chemical Constituents of the Roots of Euphorbia micractina. J Nat Prod 72: 1620-6 (2009)
- Shao, L; Abolin, C; Hewitt, MC; Koch, P; Varney, M Derivatives of tramadol for increased duration of effect. Bioorg Med Chem Lett 16: 691-4 (2005)
- Zhang, X; Xu, F; Tong, L; Zhang, T; Xie, H; Lu, X; Ren, X; Ding, K Design and synthesis of selective degraders of EGFR Eur J Med Chem 192: (2020)
- Cody, WL; Doherty, AM; He, JX; DePue, PL; Rapundalo, ST; Hingorani, GA; Major, TC; Panek, RL; Dudley, DT; Haleen, SJ Design of a functional hexapeptide antagonist of endothelin. J Med Chem 35: 3301-3 (1992)
- Gabriel, B; Stubbs, MT; Bergner, A; Hauptmann, J; Bode, W; Stürzebecher, J; Moroder, L Design of benzamidine-type inhibitors of factor Xa. J Med Chem 41: 4240-50 (1998)
- Freeman-Cook, KD; Autry, C; Borzillo, G; Gordon, D; Barbacci-Tobin, E; Bernardo, V; Briere, D; Clark, T; Corbett, M; Jakubczak, J; Kakar, S; Knauth, E; Lippa, B; Luzzio, MJ; Mansour, M; Martinelli, G; Marx, M; Nelson, K; Pandit, J; Rajamohan, F; Robinson, S; Subramanyam, C; Wei, L; Wythes, M; Morris, J Design of selective, ATP-competitive inhibitors of Akt. J Med Chem 53: 4615-22 (2010)
- Development of a Series of Pyrrolopyridone MAT2A Inhibitors.
- Roth, AG; Redmer, S; Arenz, C Development of carbohydrate-derived inhibitors of acid sphingomyelinase. Bioorg Med Chem 18: 939-44 (2010)
- Chambers, RJ; Antognoli, GW; Cheng, JB; Marfat, A; Pillar, JS; Shirley, JT; Watson, JW Development of new chromanol antagonists of leukotriene D4. Bioorg Med Chem Lett 8: 1791-6 (1998)
- Ji, W; Wang, ES; Manz, TD; Jiang, J; Donovan, KA; Abulaiti, X; Fischer, ES; Cantley, LC; Zhang, T; Gray, NS Development of potent and selective degraders of PI5P4Kγ. Eur J Med Chem 247: (2023)
- Aldrich, LN; Burdette, JE; Carcache de Blanco, E; Coss, CC; Eustaquio, AS; Fuchs, JR; Kinghorn, AD; MacFarlane, A; Mize, BK; Oberlies, NH; Orjala, J; Pearce, CJ; Phelps, MA; Rakotondraibe, LH; Ren, Y; Soejarto, DD; Stockwell, BR; Yalowich, JC; Zhang, X Discovery of Anticancer Agents of Diverse Natural Origin. J Nat Prod 85: 702-719 (2022)
- Tanaka, Y; Kurasawa, O; Yokota, A; Klein, MG; Ono, K; Saito, B; Matsumoto, S; Okaniwa, M; Ambrus-Aikelin, G; Morishita, D; Kitazawa, S; Uchiyama, N; Ogawa, K; Kimura, H; Imamura, S Discovery of Novel Allosteric Inhibitors of Deoxyhypusine Synthase. J Med Chem 63: 3215-3226 (2020)
- Shinozuka, T; Ito, S; Kimura, T; Izumi, M; Wakabayashi, K Discovery of a Novel Class of ERRα Agonists. ACS Med Chem Lett 12: 817-821 (2021)
- Ferguson, FM; Ni, J; Zhang, T; Tesar, B; Sim, T; Kim, ND; Deng, X; Brown, JR; Zhao, JJ; Gray, NS Discovery of a Series of 5,11-Dihydro-6 ACS Med Chem Lett 7: 908-912 (2016)
- Wang, YH; Zhou, MZ; Ye, T; Wang, PP; Lu, R; Wang, YL; Liu, CX; Xiao, W; Li, JY; Meng, ZB; Xu, LL; Hu, QH; Jiang, C Discovery of a Series of 5-Amide-1 J Med Chem 65: 15967-15990 (2022)
- Pryde, DC; Marron, BE; West, CW; Reister, S; Amato, G; Yoger, K; Antonio, B; Padilla, K; Cox, PJ; Turner, J; Warmus, JS; Swain, NA; Omoto, K; Mahoney, JH; Gerlach, AC Discovery of a Series of Indazole TRPA1 Antagonists. ACS Med Chem Lett 8: 666-671 (2017)
- Crosignani, S; Missotten, M; Cleva, C; Dondi, R; Ratinaud, Y; Humbert, Y; Mandal, AB; Bombrun, A; Power, C; Chollet, A; Proudfoot, A Discovery of a novel series of CXCR3 antagonists. Bioorg Med Chem Lett 20: 3614-7 (2010)
- Hu, Y; Cole, D; Denny, RA; Anderson, DR; Ipek, M; Ni, Y; Wang, X; Thaisrivongs, S; Chamberlain, T; Hall, JP; Liu, J; Luong, M; Lin, LL; Telliez, JB; Gopalsamy, A Discovery of indazoles as inhibitors of Tpl2 kinase. Bioorg Med Chem Lett 21: 4758-61 (2011)
- Biswas, T; Green, KD; Garneau-Tsodikova, S; Tsodikov, OV Discovery of inhibitors of Bacillus anthracis primase DnaG. Biochemistry 52: 6905-10 (2013)
- Peng, X; Lanter, JC; Y-P Chen, A; Brand, MA; Wozniak, MK; Hoekman, S; Longin, O; Regeling, H; Zonneveld, W; P L Bell, R; Koenig, G; Hurst, RS; Blain, JF; Burnett, DA Discovery of oxazoline enhancers of cellular progranulin release. Bioorg Med Chem Lett 80: (2023)
- Shah, P; Cheasty, A; Foxton, C; Raynham, T; Farooq, M; Gutierrez, IF; Lejeune, A; Pritchard, M; Turnbull, A; Pang, L; Owen, P; Boyd, S; Stowell, A; Jordan, A; Hamilton, NM; Hitchin, JR; Stockley, M; MacDonald, E; Quesada, MJ; Trivier, E; Skeete, J; Ovaa, H; Moolenaar, WH; Ryder, H Discovery of potent inhibitors of the lysophospholipase autotaxin. Bioorg Med Chem Lett 26: 5403-5410 (2016)
- Butler, JR; Rescourio, G; Milgram, BC; Foti, RS; Kornecook, T; Ligutti, J; Moyer, BD; Taborn, K; Youngblood, BD; Yu, V; Shimanovich, R; Boezio, A; Weiss, M Discovery of pyridyl urea sulfonamide inhibitors of Na Bioorg Med Chem Lett 73: (2022)
- Emmitte, KA; Andrews, CW; Badiang, JG; Davis-Ward, RG; Dickson, HD; Drewry, DH; Emerson, HK; Epperly, AH; Hassler, DF; Knick, VB; Kuntz, KW; Lansing, TJ; Linn, JA; Mook, RA; Nailor, KE; Salovich, JM; Spehar, GM; Cheung, M Discovery of thiophene inhibitors of polo-like kinase. Bioorg Med Chem Lett 19: 1018-21 (2009)
- Walters, I; Austin, C; Austin, R; Bonnert, R; Cage, P; Christie, M; Ebden, M; Gardiner, S; Grahames, C; Hill, S; Hunt, F; Jewell, R; Lewis, S; Martin, I; David Nicholls, na; David Robinson, na Evaluation of a series of bicyclic CXCR2 antagonists. Bioorg Med Chem Lett 18: 798-803 (2008)
- Harner, MJ; Chauder, BA; Phan, J; Fesik, SW Fragment-based screening of the bromodomain of ATAD2. J Med Chem 57: 9687-92 (2014)
- Sampson, D; Bricker, B; Zhu, XY; Peprah, K; Lamango, NS; Setola, V; Roth, BL; Ablordeppey, SY Further evaluation of the tropane analogs of haloperidol. Bioorg Med Chem Lett 24: 4294-7 (2014)
- ELZEIN, E HETEROCYCLIC INHIBITORS OF CD73 FOR TREATMENT OF DISEASE US Patent US20240140979 (2024)
- De Savi, C; Waterson, D; Pape, A; Lamont, S; Hadley, E; Mills, M; Page, KM; Bowyer, J; Maciewicz, RA Hydantoin based inhibitors of MMP13--discovery of AZD6605. Bioorg Med Chem Lett 23: 4705-12 (2013)
- Jarman, M; Barrie, SE; Deadman, JJ; Houghton, J; McCague, R; Rowlands, MG Hydroxyperfluoroazobenzenes: novel inhibitors of enzymes of androgen biosynthesis. J Med Chem 33: 2452-5 (1990)
- CAO, J; COME, JH; DAKIN, LA; DENIS, F; DORSCH, WA; FORTIER, A; HAMEL, M; KRUEGER, EB; LEDFORD, B; NANTHAKUMAR, SS; NICOLAS, O; SAYEGH, C; SENTER, TJ; WANG, T; BRODNEY, M; HU, K; ROSE, P; GAGNON, K; SHI, Y; SHRESTHA, M; MEDEK, A; WITKOS, F INHIBITORS OF APOL1 AND METHODS OF USING SAME US Patent US20230271945 (2023)
- Haftchenary, S; Jouk, AO; Aubry, I; Lewis, AM; Landry, M; Ball, DP; Shouksmith, AE; Collins, CV; Tremblay, ML; Gunning, PT Identification of Bidentate Salicylic Acid Inhibitors of PTP1B. ACS Med Chem Lett 6: 982-6 (2015)
- Riboldi, GP; Zigweid, R; Myler, PJ; Mayclin, SJ; Couñago, RM; Staker, BL Identification of P218 as a potent inhibitor of RSC Med Chem 12: 103-109 (2021)
- Peace, S; Philp, J; Brooks, C; Piercy, V; Moores, K; Smethurst, C; Watson, S; Gaines, S; Zippoli, M; Mookherjee, C; Ife, R Identification of a sulfonamide series of CCR2 antagonists. Bioorg Med Chem Lett 20: 3961-4 (2010)
- Brown, A; Ellis, D; Wallace, O; Ralph, M Identification of amide bioisosteres of triazole oxytocin antagonists. Bioorg Med Chem Lett 20: 2224-8 (2010)
- Patouret, R; Doebelin, C; Garcia-Ordonez, RD; Chang, MR; Ruiz, C; Cameron, MD; Griffin, PR; Kamenecka, TM Identification of an aminothiazole series of RORβ modulators. Bioorg Med Chem Lett 28: 1178-1181 (2018)
- Xie, YF; Sircar, I; Lake, K; Komandla, M; Ligsay, K; Li, J; Xu, K; Parise, J; Schneider, L; Huang, D; Liu, J; Sakurai, N; Barbosa, M; Jack, R Identification of novel series of human CCR1 antagonists. Bioorg Med Chem Lett 18: 2215-21 (2008)
- Pitts, WJ; Vaccaro, W; Huynh, T; Leftheris, K; Roberge, JY; Barbosa, J; Guo, J; Brown, B; Watson, A; Donaldson, K; Starling, GC; Kiener, PA; Poss, MA; Dodd, JH; Barrish, JC Identification of purine inhibitors of phosphodiesterase 7 (PDE7). Bioorg Med Chem Lett 14: 2955-8 (2004)
- Mahasenan, KV; Ding, D; Gao, M; Nguyen, TT; Suckow, MA; Schroeder, VA; Wolter, WR; Chang, M; Mobashery, S In Search of Selectivity in Inhibition of ADAM10. ACS Med Chem Lett 9: 708-713 (2018)
- Abdel-Magid, AF Inhibitors of ATR Kinase for Treatment of Cancer. ACS Med Chem Lett 4: 688-9 (2013)
- Gray, NS; De Clercq, D; Jang, J; Janne, P; To, C; Eck, M; Park, E; Heppner, D Inhibitors of EGFR and methods of use thereof US Patent US11584746 (2023)
- Lazo, JS; Sharlow, ER; McQueeney, KE; Wipf, P; Salamoun, JM Inhibitors of PTP4A3 for the treatment of cancer US Patent US10308663 (2019)
- Han, H; Yoon, J; Janda, KD Investigations of azapeptides as mimetics of Leu-enkephalin. Bioorg Med Chem Lett 8: 117-20 (1999)
- Davis, DC; Bungard, JD; Chang, S; Rodriguez, AL; Blobaum, AL; Boutaud, O; Melancon, BJ; Niswender, CM; Jeffrey Conn, P; Lindsley, CW Lead optimization of the VU0486321 series of mGlu Bioorg Med Chem Lett 32: (2021)
- Borzilleri, RM; Zhang, Y; Miller, M; Fraley, A Macrocyclic compounds for inhibition of inhibitors of apoptosis US Patent US9605022 (2017)
- Ryu, K; Kim, M; Griesinger, C; Lee, D Method of increasing platelet counts of a subject US Patent US11744839 (2023)
- Hammock, BD; Hwang, SH; Hashimoto, K; Ren, Q Methods of inhibiting formation of alpha synuclein aggregates US Patent US12251379 (2025)
- Vacca, J Modulators of HSD17B13 and methods of use thereof US Patent US11957687 (2024)
- Kumaravel, G; Boettcher, BR; Shapiro, MJ; Petter, C Peptide mimics of glycylproline as inhibitors of prolidase Bioorg Med Chem Lett 5: 2825-2828 (1995)
- Lee, MJ; Nagasawa, HT; Elberling, JA; DeMaster, EG Prodrugs of nitroxyl as inhibitors of aldehyde dehydrogenase. J Med Chem 35: 3648-52 (1992)
- Morgan, RK; Kirby, IT; Vermehren-Schmaedick, A; Rodriguez, K; Cohen, MS Rational Design of Cell-Active Inhibitors of PARP10. ACS Med Chem Lett 10: 74-79 (2019)
- Riefolo, F; Sortino, R; Matera, C; Claro, E; Preda, B; Vitiello, S; Traserra, S; Jiménez, M; Gorostiza, P Rational Design of Photochromic Analogues of Tricyclic Drugs. J Med Chem 64: 9259-9270 (2021)
- Narwal, M; Venkannagari, H; Lehtiö, L Structural basis of selective inhibition of human tankyrases. J Med Chem 55: 1360-7 (2012)
- Kalbfleisch, JJ; Reed, CW; Park, C; Spearing, PK; Quitalig, MC; Jenkins, MT; Rodriguez, AL; Blobaum, AL; Conn, PJ; Niswender, CM; Lindsley, CW Synthesis and SAR of a series of mGlu Bioorg Med Chem Lett 30: (2020)
- Igarashi, Y; Ichikawa, M; Ichikawa, Y Synthesis of a new inhibitor of α-fucosidase Bioorg Med Chem Lett 6: 553-558 (1996)
- Lee, S; Jung, KY; Park, J; Cho, JH; Kim, YC; Chang, S Synthesis of potent chemical inhibitors of dynamin GTPase. Bioorg Med Chem Lett 20: 4858-64 (2010)
- Tinto, F; Villano, R; Kostrzewa, M; Ligresti, A; Straker, H; Manzo, E Synthesis of the Major Mammalian Metabolites of THCV. J Nat Prod 83: 2060-2065 (2020)
- Bhat, L; Bhat, SR Synthesis, methods of using, and compositions of cycloalkylmethylamines US Patent US9302981 (2016)
- La, DS; Peterson, EA; Bode, C; Boezio, AA; Bregman, H; Chu-Moyer, MY; Coats, J; DiMauro, EF; Dineen, TA; Du, B; Gao, H; Graceffa, R; Gunaydin, H; Guzman-Perez, A; Fremeau, R; Huang, X; Ilch, C; Kornecook, TJ; Kreiman, C; Ligutti, J; Jasmine Lin, MH; McDermott, JS; Marx, I; Matson, DJ; McDonough, SI; Moyer, BD; Nho Nguyen, H; Taborn, K; Yu, V; Weiss, MM The discovery of benzoxazine sulfonamide inhibitors of Na Bioorg Med Chem Lett 27: 3477-3485 (2017)
- Anthony Romero, F; Hastings, NB; Moningka, R; Guo, Z; Wang, M; Di Salvo, J; Lei, Y; Trusca, D; Deng, Q; Tong, V; Terebetski, JL; Ball, RG; Ujjainwalla, F The discovery of potent antagonists of NPBWR1 (GPR7). Bioorg Med Chem Lett 22: 1014-8 (2012)
- Chiosis, G; Greengard, P; Dou, F; Luo, W; He, H; Zatorska, D Treatment of neurodegenerative diseases through inhibition of HSP90 US Patent US10336757 (2019)
- Unzue, A; Jessen-Trefzer, C; Spiliotopoulos, D; Gaudio, E; Tarantelli, C; Dong, J; Zhao, H; Pachmayr, J; Zahler, S; Bernasconi, E; Sartori, G; Cascione, L; Bertoni, F; Śledź, P; Caflisch, A; Nevado, C Understanding the mechanism of action of pyrrolo[3,2- RSC Med Chem 11: 665-675 (2020)
- Cioffi, G; D'Auria, M; Braca, A; Mendez, J; Castillo, A; Morelli, I; De Simone, F; De Tommasi, N Antioxidant and free-radical scavenging activity of constituents of the leaves of Tachigalia paniculata. J Nat Prod 65: 1526-9 (2002)
- Pauli-Magnus, C; von Richter, O; Burk, O; Ziegler, A; Mettang, T; Eichelbaum, M; Fromm, MF Characterization of the major metabolites of verapamil as substrates and inhibitors of P-glycoprotein. J Pharmacol Exp Ther 293: 376-82 (2000)
- Queiroz, MM; Queiroz, EF; Zeraik, ML; Ebrahimi, SN; Marcourt, L; Cuendet, M; Castro-Gamboa, I; Hamburger, M; da Silva Bolzani, V; Wolfender, JL Chemical composition of the bark of Tetrapterys mucronata and identification of acetylcholinesterase inhibitory constituents. J Nat Prod 77: 650-6 (2014)
- Rogers, JL; Bayeh, L; Scheuermann, TH; Longgood, J; Key, J; Naidoo, J; Melito, L; Shokri, C; Frantz, DE; Bruick, RK; Gardner, KH; MacMillan, JB; Tambar, UK Development of inhibitors of the PAS-B domain of the HIF-2a transcription factor. J Med Chem 56: 1739-47 (2013)
- Lu, T; Connolly, PJ; Philippar, U; Sun, W; Cummings, MD; Barbay, K; Gys, L; Van Nuffel, L; Austin, N; Bekkers, M; Shen, F; Cai, A; Attar, R; Meerpoel, L; Edwards, J Discovery and optimization of a series of small-molecule allosteric inhibitors of MALT1 protease. Bioorg Med Chem Lett 29: (2019)
- Crosignani, S; Jorand-Lebrun, C; Campbell, G; Prêtre, A; Grippi-Vallotton, T; Quattropani, A; Bouscary-Desforges, G; Bombrun, A; Missotten, M; Humbert, Y; Frémaux, C; Pâquet, M; El Harkani, K; Bradshaw, CG; Cleva, C; Abla, N; Daff, H; Schott, O; Pittet, PA; Arrighi, JF; Gaudet, M; Johnson, Z Discovery of a Novel Series of CRTH2 (DP2) Receptor Antagonists Devoid of Carboxylic Acids. ACS Med Chem Lett 2: 938-942 (2011)
- Oost, TK; Sun, C; Armstrong, RC; Al-Assaad, AS; Betz, SF; Deckwerth, TL; Ding, H; Elmore, SW; Meadows, RP; Olejniczak, ET; Oleksijew, A; Oltersdorf, T; Rosenberg, SH; Shoemaker, AR; Tomaselli, KJ; Zou, H; Fesik, SW Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem 47: 4417-26 (2004)
- PubChem, PC Dose response confirmation of the uHTS fluorescent assay for identification of inhibitors of ATG4B. PubChem Bioassay (2011)
- Jackson, CM; Blass, B; Coburn, K; Djandjighian, L; Fadayel, G; Fluxe, AJ; Hodson, SJ; Janusz, JM; Murawsky, M; Ridgeway, JM; White, RE; Wu, S Evolution of thiazolidine-based blockers of human Kv1.5 for the treatment of atrial arrhythmias. Bioorg Med Chem Lett 17: 282-4 (2006)
- Kundu, B; Bauser, M; Betschinger, J; Kraas, W; Jung, G Identification of a potent analogue of Nazumamide A through iteration of combinatorial tetrapeptide libraries. Bioorg Med Chem Lett 8: 1669-72 (1999)
- Dios, A; Mitchell, RA; Aljabari, B; Lubetsky, J; O'Connor, K; Liao, H; Senter, PD; Manogue, KR; Lolis, E; Metz, C; Bucala, R; Callaway, DJ; Al-Abed, Y Inhibition of MIF bioactivity by rational design of pharmacological inhibitors of MIF tautomerase activity. J Med Chem 45: 2410-6 (2002)
- McCague, R; Rowlands, MG; Barrie, SE; Houghton, J Inhibition of enzymes of estrogen and androgen biosynthesis by esters of 4-pyridylacetic acid. J Med Chem 33: 3050-5 (1990)
- Alvarado, M; Decara, J; Luque, MJ; Hernandez-Folgado, L; Gómez-Cañas, M; Gómez-Ruiz, M; Fernández-Ruiz, J; Elguero, J; Jagerovic, N; Serrano, A; Goya, P; de Fonseca, FR Novel antiobesity agents: synthesis and pharmacological evaluation of analogues of Rimonabant and of LH21. Bioorg Med Chem 21: 1708-16 (2013)
- Skerlj, RT; Bastos, CM; Booker, ML; Kramer, ML; Barker, RH; Celatka, CA; Munoz, B; Sidhu, AB; Cortese, JF; Wittlin, S; Papastogiannidis, P; Angulo-Barturen, I; Jimenez-Diaz, MB; Sybertz, E Optimization of Potent Inhibitors of P. falciparum Dihydroorotate Dehydrogenase for the Treatment of Malaria. ACS Med Chem Lett 2: 708-713 (2011)
- Ran, F; Liu, Y; Wang, C; Xu, Z; Zhang, Y; Liu, Y; Zhao, G; Ling, Y Review of the development of BTK inhibitors in overcoming the clinical limitations of ibrutinib. Eur J Med Chem 229: (2022)
- PubChem, PC SAR analysis of Antagonists of XIAP-Bir3 domain of IAP-family anti-apoptotic proteins PubChem Bioassay (2009)
- Jamieson, C; Maclean, JK; Brown, CI; Campbell, RA; Gillen, KJ; Gillespie, J; Kazemier, B; Kiczun, M; Lamont, Y; Lyons, AJ; Moir, EM; Morrow, JA; Pantling, J; Rankovic, Z; Smith, L Structure based evolution of a novel series of positive modulators of the AMPA receptor. Bioorg Med Chem Lett 21: 805-11 (2011)
- Le Marec, O; Neveu, C; Lefranc, B; Dubessy, C; Boutin, JA; Do-Régo, JC; Costentin, J; Tonon, MC; Tena-Sempere, M; Vaudry, H; Leprince, J Structure-activity relationships of a series of analogues of the RFamide-related peptide 26RFa. J Med Chem 54: 4806-14 (2011)
- Wustrow, DJ; Wise, LD; Cody, DM; MacKenzie, RG; Georgic, LM; Pugsley, TA; Heffner, TG Studies of the active conformation of a novel series of benzamide dopamine D2 agonists. J Med Chem 37: 4251-7 (1995)
- Noël, S; Hoegy, F; Rivault, F; Rognan, D; Schalk, IJ; Mislin, GL Synthesis and biological properties of thiazole-analogues of pyochelin, a siderophore of Pseudomonas aeruginosa. Bioorg Med Chem Lett 24: 132-5 (2013)
- Ocain, TD; Rich, DH Synthesis of sulfur-containing analogues of bestatin. Inhibition of aminopeptidases by alpha-thiolbestatin analogues. J Med Chem 31: 2193-9 (1988)
- Epps, DE; Cheney, J; Schostarez, H; Sawyer, TK; Prairie, M; Krueger, WC; Mandel, F Thermodynamics of the interaction of inhibitors with the binding site of recombinant human renin. J Med Chem 33: 2080-6 (1990)
- Samir, M; Ramadan, M; Abdelrahman, MH; Elbastawesy, MAI; Halby, HM; Abdel-Aziz, M; Abuo-Rahma, GEA Bioorg Med Chem 73: (2022)
- MAI, W; LIU, X; SHI, W; DENG, Z; ZHU, W; LI, Z; ZOU, H US Patent US20250042896 (2025)
- Rotili, D; Tarantino, D; Carafa, V; Paolini, C; Schemies, J; Jung, M; Botta, G; Di Maro, S; Novellino, E; Steinkühler, C; De Maria, R; Gallinari, P; Altucci, L; Mai, A J Med Chem 55: 8193-7 (2012)
- Vyskocil, S; Cardin, D; Ciavarri, J; Conlon, J; Cullis, C; England, D; Gershman, R; Gigstad, K; Gipson, K; Gould, A; Greenspan, P; Griffin, R; Gulavita, N; Harrison, S; Hu, Z; Hu, Y; Hata, A; Huang, J; Huang, SC; Janowick, D; Jones, M; Kolev, V; Langston, SP; Lee, HM; Li, G; Lok, D; Ma, L; Mai, D; Malley, J; Matsuda, A; Mizutani, H; Mizutani, M; Molchanova, N; Nunes, E; Pusalkar, S; Renou, C; Rowland, S; Sato, Y; Shaw, M; Shen, L; Shi, Z; Skene, R; Soucy, F; Stroud, S; Xu, H; Xu, T; Abu-Yousif, AO; Zhang, J J Med Chem 64: 6902-6923 (2021)
- Gong, XW; Mai, JH; Xu, YH Bioorg Med Chem Lett 22: 2388-92 (2012)
- Farand, J; Mai, N; Chandrasekhar, J; Newby, ZE; Van Veldhuizen, J; Loyer-Drew, J; Venkataramani, C; Guerrero, J; Kwok, A; Li, N; Zherebina, Y; Wilbert, S; Zablocki, J; Phillips, G; Watkins, WJ; Mourey, R; Notte, GT Bioorg Med Chem Lett 26: 5926-5930 (2016)
- Thanh, ND; Hai, DS; Ngoc Bich, VT; Thu Hien, PT; Ky Duyen, NT; Mai, NT; Dung, TT; Toan, VN; Kim Van, HT; Dang, LH; Toan, DN; Thanh Van, TT Eur J Med Chem 167: 454-471 (2019)
- Li, L; Zhao, H; Peng, X; Liu, J; Mai, R; Chen, J; Lin, L; Chen, T; Yan, J; Shi, J; Chen, J Bioorg Med Chem 71: (2022)
- Pizzonero, M; Akkari, R; Bock, X; Gosmini, R; De Lemos, E; Duthion, B; Newsome, G; Mai, TT; Roques, V; Jary, H; Lefrancois, JM; Cherel, L; Quenehen, V; Babel, M; Merayo, N; Bienvenu, N; Mammoliti, O; Coti, G; Palisse, A; Cowart, M; Shrestha, A; Greszler, S; Van Der Plas, S; Jansen, K; Claes, P; Jans, M; Gees, M; Borgonovi, M; De Wilde, G; Conrath, K J Med Chem 67: 5216-5232 (2024)
- Labéguère, F; Dupont, S; Alvey, L; Soulas, F; Newsome, G; Tirera, A; Quenehen, V; Mai, TTT; Deprez, P; Blanqué, R; Oste, L; Le Tallec, S; De Vos, S; Hagers, A; Vandevelde, A; Nelles, L; Vandervoort, N; Conrath, K; Christophe, T; van der Aar, E; Wakselman, E; Merciris, D; Cottereaux, C; da Costa, C; Saniere, L; Clement-Lacroix, P; Jenkins, L; Milligan, G; Fletcher, S; Brys, R; Gosmini, R J Med Chem 63: 13526-13545 (2020)
- Tu, G; Li, S; Huang, H; Li, G; Xiong, F; Mai, X; Zhu, H; Kuang, B; Xu, WF Bioorg Med Chem 16: 6663-8 (2008)
- Wang, B; Mai, YC; Li, Y; Hou, JQ; Huang, SL; Ou, TM; Tan, JH; An, LK; Li, D; Gu, LQ; Huang, ZS Eur J Med Chem 45: 1415-23 (2010)
- Li, PH; Zeng, P; Chen, SB; Yao, PF; Mai, YW; Tan, JH; Ou, TM; Huang, SL; Li, D; Gu, LQ; Huang, ZS J Med Chem 59: 238-52 (2016)
- Mai, ZP; Zhou, K; Ge, GB; Wang, C; Huo, XK; Dong, PP; Deng, S; Zhang, BJ; Zhang, HL; Huang, SS; Ma, XC J Nat Prod 78: 2372-80 (2015)
- XIONG, B; LI, J; LIU, T; ZHOU, Y; LI, C; LI, N; KAN, W; SU, M; SHENG, L; HU, X US Patent US20250042862 (2025)
- Cheng, J; Li, Y; Wang, X; Dong, G; Sheng, C J Med Chem 63: 7892-7905 (2020)
- Zhang, XJ; Liu, MH; Luo, YS; Han, GY; Ma, ZQ; Huang, F; Wang, Y; Miao, ZY; Zhang, WN; Sheng, CQ; Yao, JZ Eur J Med Chem 217: (2021)
- Feng, L; Chen, X; Sheng, G; Li, Y; Li, Y; Zhang, Y; Yao, K; Wu, Z; Zhang, R; Kiboku, T; Kawasaki, A; Horimoto, K; Tang, Y; Sun, M; Han, F; Chen, D J Med Chem 66: 14609-14622 (2023)
- Sun, J; Lv, PC; Guo, FJ; Wang, XY; Xiao-Han, na; Zhang, Y; Sheng, GH; Qian, SS; Zhu, HL Eur J Med Chem 81: 420-6 (2014)
- Zhang, P; Hao, J; Liu, J; Lu, Q; Sheng, H; Zhang, L; Sun, H J Nat Prod 72: 1414-8 (2009)
- Li, X; Sheng, J; Huang, G; Ma, R; Yin, F; Song, D; Zhao, C; Ma, S Eur J Med Chem 97: 32-41 (2015)
- Sheng, K; Song, Y; Lei, F; Zhao, W; Fan, L; Wu, L; Liu, Y; Wu, S; Zhang, Y Eur J Med Chem 227: (2022)
- Sheng L
- Ni, WW; Liu, Q; Ren, SZ; Li, WY; Yi, LL; Jing, H; Sheng, LX; Wan, Q; Zhong, PF; Fang, HL; Ouyang, H; Xiao, ZP; Zhu, HL Bioorg Med Chem 26: 4145-4152 (2018)
- Leng, J; Zhao, Y; Sheng, P; Xia, Y; Chen, T; Zhao, S; Xie, S; Yan, X; Wang, X; Yin, Y; Kong, L J Med Chem 65: 16774-16800 (2022)
- Sheng, R; Lin, X; Zhang, J; Chol, KS; Huang, W; Yang, B; He, Q; Hu, Y Bioorg Med Chem 17: 6692-8 (2009)
- Minutolo, F; Antonello, M; Bertini, S; Ortore, G; Placanica, G; Rapposelli, S; Sheng, S; Carlson, KE; Katzenellenbogen, BS; Katzenellenbogen, JA; Macchia, M J Med Chem 46: 4032-42 (2003)
- Plouvier, B; Beatch, GN; Jung, GL; Zolotoy, A; Sheng, T; Clohs, L; Barrett, TD; Fedida, D; Wang, WQ; Zhu, JJ; Liu, Y; Abraham, S; Lynn, L; Dong, Y; Wall, RA; Walker, MJ J Med Chem 50: 2818-41 (2007)
- Sheng, W; Yang, L; Pan, Z US Patent US10106508 (2018)
- Fu, RG; Sun, Y; Sheng, WB; Liao, DF Eur J Med Chem 136: 195-211 (2017)
- Zhang, X; Sheng, X; Shen, J; Zhang, S; Sun, W; Shen, C; Li, Y; Wang, J; Lv, H; Cui, M; Zhu, Y; Huang, L; Hao, D; Qi, Z; Sun, G; Mao, W; Pan, Y; Shen, L; Li, X; Hu, G; Gong, Z; Han, S; Li, J; Chen, S; Tu, R; Wang, X; Wu, C ACS Med Chem Lett 11: 1863-1868 (2020)
- Sheng, XC; Appleby, T; Butler, T; Cai, R; Chen, X; Cho, A; Clarke, MO; Cottell, J; Delaney, WE; Doerffler, E; Link, J; Ji, M; Pakdaman, R; Pyun, HJ; Wu, Q; Xu, J; Kim, CU Bioorg Med Chem Lett 22: 2629-34 (2012)
- Zhou, Z; Li, Y; Ma, X; Cao, B; Peng, T; Sheng, Y; Peng, H; Li, R; Cao, Y; Xi, R; Li, F; Wang, M; Sun, H; Zhang, G; Zhang, H; Hu, K; Xiao, W; Wang, F J Med Chem 64: 7404-7421 (2021)
- Yang, KS; Ma, XR; Ma, Y; Alugubelli, YR; Scott, DA; Vatansever, EC; Drelich, AK; Sankaran, B; Geng, ZZ; Blankenship, LR; Ward, HE; Sheng, YJ; Hsu, JC; Kratch, KC; Zhao, B; Hayatshahi, HS; Liu, J; Li, P; Fierke, CA; Tseng, CK; Xu, S; Liu, WR ChemMedChem (2020)
- Xu, B; Feng, Y; Cheng, H; Song, Y; Lv, B; Wu, Y; Wang, C; Li, S; Xu, M; Du, J; Peng, K; Dong, J; Zhang, W; Zhang, T; Zhu, L; Ding, H; Sheng, Z; Welihinda, A; Roberge, JY; Seed, B; Chen, Y Bioorg Med Chem Lett 21: 4465-70 (2011)
- Apel, C; Gény, C; Dumontet, V; Birlirakis, N; Roussi, F; Pham, VC; Doan Thi Mai, H; Nguyen, VH; Chau, VM; Litaudon, M J Nat Prod 77: 1430-7 (2014)
- Bedoya, LM; Beltrán, M; Sancho, R; Olmedo, DA; Sánchez-Palomino, S; del Olmo, E; López-Pérez, JL; Muñoz, E; San Feliciano, A; Alcamí, J Bioorg Med Chem Lett 15: 4447-50 (2005)
- Jackson, ER; San Jose, G; Brothers, RC; Edelstein, EK; Sheldon, Z; Haymond, A; Johny, C; Boshoff, HI; Couch, RD; Dowd, CS Bioorg Med Chem Lett 24: 649-53 (2014)
- Bacalhau, P; San Juan, AA; Marques, CS; Peixoto, D; Goth, A; Guarda, C; Silva, M; Arantes, S; Caldeira, AT; Martins, R; Burke, AJ Bioorg Chem 67: 1-8 (2016)
- Barlaam, B; Casella, R; Cidado, J; Cook, C; De Savi, C; Dishington, A; Donald, CS; Drew, L; Ferguson, AD; Ferguson, D; Glossop, S; Grebe, T; Gu, C; Hande, S; Hawkins, J; Hird, AW; Holmes, J; Horstick, J; Jiang, Y; Lamb, ML; McGuire, TM; Moore, JE; O'Connell, N; Pike, A; Pike, KG; Proia, T; Roberts, B; San Martin, M; Sarkar, U; Shao, W; Stead, D; Sumner, N; Thakur, K; Vasbinder, MM; Varnes, JG; Wang, J; Wang, L; Wu, D; Wu, L; Yang, B; Yao, T J Med Chem 63: 15564-15590 (2020)
- Lanman, BA; Reed, AB; Cee, VJ; Hong, FT; Pettus, LH; Wurz, RP; Andrews, KL; Jiang, J; McCarter, JD; Mullady, EL; San Miguel, T; Subramanian, R; Wang, L; Whittington, DA; Wu, T; Zalameda, L; Zhang, N; Tasker, AS; Hughes, PE; Norman, MH Bioorg Med Chem Lett 24: 5630-4 (2014)
- Goldstein, DM; Alfredson, T; Bertrand, J; Browner, MF; Clifford, K; Dalrymple, SA; Dunn, J; Freire-Moar, J; Harris, S; Labadie, SS; La Fargue, J; Lapierre, JM; Larrabee, S; Li, F; Papp, E; McWeeney, D; Ramesha, C; Roberts, R; Rotstein, D; San Pablo, B; Sjogren, EB; So, OY; Talamas, FX; Tao, W; Trejo, A; Villasenor, A; Welch, M; Welch, T; Weller, P; Whiteley, PE; Young, K; Zipfel, S J Med Chem 49: 1562-75 (2006)
- Gargantilla, M; Francés, C; Adhav, A; Forcada-Nadal, A; Martínez-Gualda, B; Martí-Marí, O; López-Redondo, ML; Melero, R; Marco-Marín, C; Gougeard, N; Espinosa, C; Rubio-Del-Campo, A; Ruiz-Partida, R; Hernández-Sierra, MDP; Villamayor-Belinchón, L; Bravo, J; Llacer, JL; Marina, A; Rubio, V; San-Félix, A; Geller, R; Pérez-Pérez, MJ J Med Chem 66: 10432-10457 (2023)
- Kaiho, T; San-Nohe, K; Kajiya, S; Suzuki, T; Otsuka, K; Ito, T; Kamiya, J; Maruyama, M J Med Chem 32: 351-7 (1989)
- Rabal, O; San José-Enériz, E; Agirre, X; Sánchez-Arias, JA; de Miguel, I; Ordoñez, R; Garate, L; Miranda, E; Sáez, E; Vilas-Zornoza, A; Pineda-Lucena, A; Estella, A; Zhang, F; Wu, W; Xu, M; Prosper, F; Oyarzabal, J J Med Chem 64: 3392-3426 (2021)
- Lamotte, Y; Faucher, N; Sançon, J; Pineau, O; Sautet, S; Fouchet, MH; Beneton, V; Tousaint, JJ; Saintillan, Y; Ancellin, N; Nicodeme, E; Grillot, D; Martres, P Bioorg Med Chem Lett 24: 1098-103 (2014)
- Sluis-Cremer, N; Hamamouch, N; San Félix, A; Velazquez, S; Balzarini, J; Camarasa, MJ J Med Chem 49: 4834-41 (2006)
- San Sebastián, E; Zimmerman, T; Zubia, A; Vara, Y; Martin, E; Sirockin, F; Dejaegere, A; Stote, RH; Lopez, X; Pantoja-Uceda, D; Valcárcel, M; Mendoza, L; Vidal-Vanaclocha, F; Cossío, FP; Blanco, FJ J Med Chem 56: 735-47 (2013)
- García-Aparicio, C; Bonache, MC; De Meester, I; San-Félix, A; Balzarini, J; Camarasa, MJ; Velazquez, S J Med Chem 49: 5339-51 (2006)
- Cossio Mora, FP; Arias Echeverría, LL; Vara Zalazar, YI; Aldaba Arévalo, E; San Sebastián Larzabal, E; Zubia Olascoaga, A US Patent US8835659 (2014)
- Cossío Mora, FP; Zubia Olascoaga, A; Vara Salazar, Y; San Sebastián Larzabal, EI; Otaegui Ansa, D; Masdeu Margalef, M; Aldaba Arévalo, E US Patent US8685992 (2014)
- Aguirre Ena, X; Oyarzabal Santamarina, J; Prósper Cardoso, F; Rabal Gracia, MO; San José Enériz, E; Sánchez Arias, JA US Patent US10407423 (2019)
- Ilyinsky, NS; Shchyolkina, AK; Borisova, OF; Mamaeva, OK; Zvereva, MI; Azhibek, DM; Livshits, MA; Mitkevich, VA; Balzarini, J; Sinkevich, YB; Luzikov, YN; Dezhenkova, LG; Kolotova, ES; Shtil, AA; Shchekotikhin, AE; Kaluzhny, DN Eur J Med Chem 85: 605-14 (2014)
- Funk, OF; Kettmann, V; Drimal, J; Langer, T J Med Chem 47: 2750-60 (2004)
- Xu, Q; Kulkarni, AA; Sajith, AM; Hussein, D; Brown, D; Güner, OF; Reddy, MD; Watkins, EB; Lassègue, B; Griendling, KK; Bowen, JP Bioorg Med Chem 26: 989-998 (2018)
- Aktaş, DA; Akinalp, G; Sanli, F; Yucel, MA; Gambacorta, N; Nicolotti, O; Karatas, OF; Algul, O; Burmaoglu, S Bioorg Med Chem Lett 30: (2020)
- ChEMBL_2346015 Inhibition of HDAC in human HeLa nuclear extract
- ChEMBL_2346044 Inhibition of HDAC1 in human HeLa nuclear extract
- ChEMBL_2346045 Inhibition of HDAC2 in human HeLa nuclear extract
- ChEMBL_472921 (CHEMBL921164) Inhibition of HDAC1 in rat liver extract
- ChEBML_28421 Inhibition of AICAR formyltransferase from extract of Manca human lymphoma cells
- ChEMBL_1552208 (CHEMBL3761210) Inhibition of HDAC in human HeLa nuclear extract
- ChEMBL_1875110 (CHEMBL4376399) Inhibition of HDAC in human K562 nuclear extract
- ChEMBL_1919932 (CHEMBL4422777) Inhibition of HDAC in human HeLa nuclear extract
- ChEMBL_2439282 Inhibition of HDAC in human HeLa cells nuclear extract
- ChEMBL_69929 (CHEMBL678809) Inhibition of GAR formyltransferase from extract of Manca human lymphoma cells
- ChEMBL_1919890 (CHEMBL4422735) Inhibition of HDAC1/HDAC2 in human HeLa nuclear extract
- ChEMBL_852177 (CHEMBL2157598) Inhibition of aCDase expressed in human HL60 cell extract
- ChEMBL_432005 (CHEMBL918789) Inhibition of telomerase in JR8 cell extract by TRAP assay
- ChEMBL_472923 (CHEMBL921166) Inhibition of HDAC1 in rat liver extract by trypsin assay
- ChEMBL_873193 (CHEMBL2183424) Inhibition of DNA-PK isolated from human HeLa cell extract
- ChEBML_54534 Inhibition of DNA-dependent protein kinase (DNA-PK) of HeLa cell nuclear cell extract
- ChEMBL_209485 (CHEMBL810077) Inhibition of pure human thymidylate synthase from extract of Manca human lymphoma cells
- ChEMBL_87718 (CHEMBL697246) Inhibition of histone deacetylase (HDAC) activity in HeLa cell nuclear extract
- ChEMBL_28279 (CHEMBL645820) Inhibition of AGT activity to 50% of control rate in HT-29 cell extract
- ChEMBL_756042 (CHEMBL1804152) Inhibition of NFkappa p65 isolated from nuclear extract of human HeLa cells by ELISA
- ChEMBL_2105981 (CHEMBL4814656) Inhibition of telomerase in human SGC-7901 cell extract by TRAP assay
- ChEMBL_2201302 (CHEMBL5114010) Inhibition of telomerase derived from human A2780 cell extract by TRAP assay
- ChEMBL_2275541 Inhibition of human HDAC in human HeLa cell nuclear extract by fluorescence assay
- ChEMBL_457624 (CHEMBL923825) Inhibition of HDAC activity in HeLa cell nuclear extract by fluorescent assay
- ChEMBL_583224 (CHEMBL1055023) Inhibition of ALR2 from Sprague-Dawley albino rat lens extract by spectrophotometrically
- ChEMBL_610879 (CHEMBL1065034) Inhibition of HDAC in human HeLa cell nuclear extract by fluorescence assay
- ChEMBL_956022 (CHEMBL2380117) Inhibition of telomerase in human HL60 cell extract by TRAP-LIG assay
- ChEMBL_34783 (CHEMBL646224) In vitro inhibition of Angiotensin I converting enzyme isolated from rabbit lung extract.
- ChEMBL_653705 (CHEMBL1227029) Inhibition of HDAC in human HeLa cell extract by fluorescence plate reader assay
- ChEMBL_1434455 (CHEMBL3388272) Inhibition of HDAC in human HeLa cell extract after 15 mins by fluorescence assay
- ChEMBL_1580211 (CHEMBL3811519) Inhibition of HDAC1 in human Jurkat cells extract after 30 mins by immunoprecipitation assay
- ChEMBL_1580212 (CHEMBL3811520) Inhibition of HDAC3 in human Jurkat cells extract after 30 mins by immunoprecipitation assay
- ChEMBL_1990603 (CHEMBL4624338) Inhibition of NF-kappaB p65 in human HeLa cells nuclear extract by chemiluminescent assay
- ChEMBL_557374 (CHEMBL955704) Inhibition of PSMA in human LNCaP cell extract using [3H]NAAG by radiometric assay
- ChEMBL_2201829 (CHEMBL5114537) Inhibition of recombinant human Top1 derived from human MCF7 cell extract incubated for 30 mins
- ChEMBL_2213599 (CHEMBL5126731) Inhibition of HDAC1 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate
- ChEMBL_2213600 (CHEMBL5126732) Inhibition of HDAC2 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate
- ChEMBL_2213601 (CHEMBL5126733) Inhibition of HDAC8 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate
- ChEMBL_2272806 Inhibition of HDAC1 in human HeLa nuclear extract incubated for 30 mins by fluorescence based analysis
- ChEMBL_2339423 Inhibition of HDAC in human HeLa nuclear extract measured after 30 mins by fluorescence based assay
- ChEMBL_2488043 Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate
- ChEMBL_422105 (CHEMBL907102) In vitro inhibition of histone deacetylase activity using HeLa cell nuclear extract as enzyme source
- ChEMBL_557375 (CHEMBL955705) Inhibition of PSMA in human LNCaP cell extract using [3H]NAAG by fluorescence-based assay
- ChEMBL_776071 (CHEMBL1912767) Inhibition of HDAC in human HeLa cell extract assessed as fluorophore release by fluorescence spectrophotometry
- ChEMBL_1700237 (CHEMBL4051219) Inhibition of HDAC 10 in human HeLa nuclear extract measured after 60 mins by fluorometric analysis
- ChEMBL_2201312 (CHEMBL5114020) Inhibition of telomerase derived from human K562 cell extract incubated for 30 mins by TRAP assay
- ChEMBL_223549 (CHEMBL845855) concentration required to reduce AGT activity to 50% of control rate in HT-29 cell extract.
- ChEMBL_2236190 (CHEMBL5150086) Inhibition of HDAC in human HeLa nuclear extract measured after 30 mins by fluorescence based assay
- ChEMBL_2239302 (CHEMBL5153198) Inhibition of HDAC in human NALM-6 nuclear extract incubated for 48 hrs by fluorometric analysis
- ChEMBL_2274123 Inhibition of human HDAC using human HeLa cell nuclear extract measured after 60 mins by fluorescence assay
- ChEMBL_2373527 Inhibition of HDAC in human HeLa cells nuclear extract incubated for 30 mins by multiplate reader analysis
- ChEMBL_2373528 Inhibition of HDAC1 in human HeLa cells nuclear extract incubated for 30 mins by multiplate reader analysis
- ChEMBL_2460780 Inhibition of HDAC in human HeLa cells nuclear extract incubated for 30 mins by multiplate reader analysis
- ChEMBL_2525403 Inhibition of human HeLa cell nuclear extract purified DNA-PK using p53 peptide as substrate by ELISA
- ChEMBL_28280 (CHEMBL645821) concentration required to reduce AGT activity to 50% of control rate in HT-29 cell extract.
- ChEMBL_614728 (CHEMBL1114231) Inhibition of human HDAC in human HeLa cell nuclear extract after 15 mins by colorimetric assay
- ChEMBL_753553 (CHEMBL1799171) Inhibition of HDAC6 in human HeLa cell nuclear extract after 30 mins by fluorescence microplate reader
- ChEMBL_832314 (CHEMBL2066852) Inhibition of DNMT1 in human HeLa cell nuclear extract assessed as methylated substrate level by ELISA
- ChEMBL_966564 (CHEMBL2399492) Inhibition of HDAC isolated from human HeLa cell nuclear extract after 30 mins by fluorescence assay
- ChEMBL_977181 (CHEMBL2416774) Inhibition of HDAC in human HeLa cell extract using Fluor deLys as substrate by fluorimetric assay
- ChEMBL_1527400 (CHEMBL3636775) Inhibition of human HDAC in HeLa cell nuclear extract by fluorometric assay using Fluor de Lys substrate
- ChEMBL_1728624 (CHEMBL4143902) Inhibition of HDAC in human HeLa nuclear extract using Fluor de Lys as substrate by fluorimetric method
- ChEMBL_1839042 (CHEMBL4339257) Inhibition of HDAC (unknown origin) in human HeLa cell nuclear extract using Color de Lys as substrate
- ChEMBL_2150392 (CHEMBL5034854) Inhibition of human HDAC using human HeLa cell nuclear extract measured after 60 mins by fluorescence assay
- ChEMBL_2201002 (CHEMBL5113710) Inhibition of HDAC1 derived from human HeLa nuclear extract using COLOR DE LYS substrate by colorimetric assay
- ChEMBL_2488159 Inhibition of human HDAC extracted from human HeLa cell nuclear extract incubated for 30 mins by fluorometric analysis
- ChEMBL_1772528 (CHEMBL4224640) Inhibition of chymotrypsin-like activity of 20S proteasome in human PC3 cell extract using Suc-LLVYaminoluciferin as substrate after 2 hrs
- ChEMBL_1772529 (CHEMBL4224641) Inhibition of chymotrypsin-like activity of 20S proteasome in human LNCAP cell extract using Suc-LLVYaminoluciferin as substrate after 2 hrs
- ChEMBL_873187 (CHEMBL2183418) Inhibition of DNA-PK isolated from human HeLa cell extract assessed as inhibition of p53 peptide fragment phosphorylation after 10 mins
- ChEMBL_629937 (CHEMBL1109181) Inhibition of NFkappa p50 isolated from nuclear extract of human HeLa cells assessed as blockade of binding to biotinylated consesus sequence by chemiluminescence assay
- ChEMBL_629938 (CHEMBL1109182) Inhibition of NFkappa p65 isolated from nuclear extract of human HeLa cells assessed as blockade of binding to biotinylated consesus sequence by chemiluminescence assay
- ChEMBL_749381 (CHEMBL1785171) Inhibition of NFkappa p65 in nuclear extract of human HeLa cells assessed as blockade of NFkappa p65 binding to biotinylated-consesus sequence by ELISA
- ChEMBL_1825529 (CHEMBL4325293) Inhibition of PI3Kdelta in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825530 (CHEMBL4325294) Inhibition of VPS34 in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1986966 (CHEMBL4620513) Inhibition of HDAC in human HeLa cell nuclear extract using fluor-de-lys as substrate by spectrofluorometric analysis
- ChEMBL_2047319 (CHEMBL4702018) Inhibition of HDAC in human HeLa nuclear extract using fluoroscence-labeled acetylated peptide as substrate by fluorometric assay
- ChEMBL_2206578 (CHEMBL5119286) Inhibition of HDAC in human HeLa nuclear extract incubated for 30 mins by fluorescence-based Glo-luminescence assay
- ChEMBL_2488178 Inhibition of HDAC3 in human HeLa cell nuclear extract incubated for 30 mins by fluorescence based microplate reader analysis
- ChEMBL_873195 (CHEMBL2183783) Inhibition of ATM isolated from human HeLa cell extract using glutathione S-transferase-p53N66 as substrate by ELISA
- ChEMBL_714888 (CHEMBL1663834) Inhibition of NF-kappaB p65 isolated from nuclear extract of human HeLa cells assessed as blockade of binding to biotinylated consesus sequence by chemiluminescence assay
- ChEMBL_2355383 Inhibition of HDAC in human HeLa cell nuclear extract using Kac fluorogenic peptide as substrate containing residues 379-382 of p53 by fluorescence assay
- ChEMBL_306888 (CHEMBL828694) In vitro inhibitory concentration against histone deacetylase of DU-145 prostate cell nuclear extract as deacetylation of biotinylated [3H]-acetyl histone H4 peptide
- ChEBML_1684531 Inhibition of AChE1 in Anopheles gambiae body extract using acetylthiocholine iodide as substrate measured over 60 secs by Ellman's method
- ChEMBL_143356 (CHEMBL751280) Inhibitory activity evaluated from soluble cell extract of Neuronal nitric oxide synthase and partially purified by DEAE-sepharose chromatography
- ChEMBL_2157455 (CHEMBL5042115) Inhibition of HDAC in human HeLa nuclear extract using fluorogenic substrate incubated for 30 mins by fluorescence based assay
- ChEMBL_2274135 Inhibition of human HDAC1 using human HeLa cell nuclear extract at 1 uM measured after 60 mins by fluorescence assay
- ChEMBL_2274136 Inhibition of human HDAC2 using human HeLa cell nuclear extract at 1 uM measured after 60 mins by fluorescence assay
- ChEMBL_2274137 Inhibition of human HDAC3 using human HeLa cell nuclear extract at 1 uM measured after 60 mins by fluorescence assay
- ChEMBL_87390 (CHEMBL691505) Tested for Histone deacetylase enzyme inhibition assay using Eimeria tenella extract
- ChEMBL_2310516 Inhibition of recombinant Top1 in Leishmania donovani Ag83 whole cell extract assessed as relaxation of supercoiled pBluescript SK(+) DNA measured by agarose gel electrophoresis analysis
- ChEMBL_1351241 (CHEMBL3271696) Inhibition of HDAC in human HeLa cell nuclear extract using acetylated lysine as substrate after 30 mins by spectrophotometric analysis
- ChEMBL_143357 (CHEMBL751281) Inhibitory activity evaluated from soluble cell extract of human Neuronal nitric oxide synthase and partially purified by DEAE-sepharose chromatography
- ChEMBL_143358 (CHEMBL751652) Inhibitory activity evaluated from soluble cell extract of human nNeuronal nitric oxide synthase and partially purified by DEAE-sepharose chromatography
- ChEMBL_1589349 (CHEMBL3830593) Inhibition of full length BRPF1 in human HUT78 cell nuclear/chromatin extract after 45 mins by chemoproteomic competition binding assay
- ChEMBL_164142 (CHEMBL771466) Compound concentration which displaces 50% of [125I]-labeled 2-5A probe bound to RNase L from mouse L cell extract
- ChEMBL_1684530 (CHEMBL4035009) Inhibition of AChE1 in Anopheles gambiae head extract using acetylthiocholine iodide as substrate measured over 60 secs by Ellman's method
- ChEMBL_1684531 (CHEMBL4035010) Inhibition of AChE1 in Anopheles gambiae body extract using acetylthiocholine iodide as substrate measured over 60 secs by Ellman's method
- ChEMBL_1700230 (CHEMBL4051212) Inhibition of HDAC 1 in human HeLa nuclear extract using HDAC substrate-3 measured after 60 mins by fluorometric analysis
- ChEMBL_1700231 (CHEMBL4051213) Inhibition of HDAC 2 in human HeLa nuclear extract using HDAC substrate-3 measured after 60 mins by fluorometric analysis
- ChEMBL_1700232 (CHEMBL4051214) Inhibition of HDAC 3 in human HeLa nuclear extract using HDAC substrate-3 measured after 60 mins by fluorometric analysis
- ChEMBL_2030958 (CHEMBL4685116) Inhibition of HDAC in human HeLa cell nuclear extract using fluorescence substrate incubated for 30 mins by fluorescence based assay
- ChEMBL_2032352 (CHEMBL4686510) Inhibition of tissue extract derived mEH (unknown origin) using [3H]trans-stilbene oxide as substrate by liquid scintillation counting method
- ChEMBL_2119132 (CHEMBL4828198) Inhibition of PI3Kdelta in human HL-60 cell extract measured after 2 hrs by kinobeads based chemoproteomic competition binding assay
- ChEMBL_2119133 (CHEMBL4828199) Inhibition of VPS34 in human HL-60 cell extract measured after 2 hrs by kinobeads based chemoproteomic competition binding assay
- ChEMBL_809835 (CHEMBL2014772) Inhibition of HDAC6 in human HeLa cells nuclear extract using Fluor-de-Lys as substrate after 30 mins by spectrophotometry
- ChEMBL_812542 (CHEMBL2014417) Inhibition of HDAC1 in human HeLa cells nuclear extract using Fluor-de-Lys as substrate after 30 mins by spectrophotometry
- ChEMBL_89193 (CHEMBL701145) Inhibitory activity evaluated for soluble cell extract of human Inducible nitric oxide synthase and partially purified by DEAE-sepharose chromatography
- ChEMBL_974420 (CHEMBL2412317) Inhibition of HDAC in human HeLa cell extract using fluor de Lys as substrate after 15 mins by fluorometric analysis
- ChEMBL_54532 (CHEMBL664849) Affinity for DNA-dependent protein kinase(DNA-PK) from HeLa cell extract
- ChEMBL_769098 (CHEMBL1832655) Inhibition of STAT3 in mouse NIH3T3/vSrc nuclear extract assessed as disruption of the Stat3-DNA complex pre-incubated for 30 mins by EMSA analysis
- ChEBML_1572320 Inhibition of HDAC in human HeLa nuclear extract using BOC-Ac-Lys-AMC as substrate incubated for 90 mins by fluorescence assay
- ChEMBL_1282340 (CHEMBL3100391) Inhibition of HDAC in human HeLa cell nuclear extract using fluor de Lys as substrate after 15 mins by fluorimetric analysis
- ChEMBL_1551034 (CHEMBL3761048) Inhibition of HDAC in human HeLa cell nuclear extract using BML-KI104 Fluor de Lys as substrate by fluorescence-based assay
- ChEMBL_1551259 (CHEMBL3761968) Inhibition of HDAC in human HeLa cells nuclear extract using Fluor de lys as substrate after 15 mins by fluorometric analysis
- ChEMBL_1570662 (CHEMBL3795030) Inhibition of HDAC in human HeLa nuclear extract using fluor de lys as substrate after 10 to 15 mins by spectrofluorometry
- ChEMBL_1919885 (CHEMBL4422730) Inhibition of HDAC in human HeLa nuclear extract using Fluor-de-lys as substrate measured after 60 mins by fluorescence assay
- ChEMBL_1919886 (CHEMBL4422731) Inhibition of HDAC in human HeLa cytosolic extract using Fluor-de-lys as substrate measured after 60 mins by fluorescence assay
- ChEMBL_2131671 (CHEMBL4841186) Inhibition of HDAC in human HeLa nuclear extract using Fluor de Lys as substrate incubated for 20 mins by fluorometric assay
- ChEMBL_2224455 (CHEMBL5137968) Inhibition of HDAC6 derived from human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2224456 (CHEMBL5137969) Inhibition of HDAC1 derived from human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2337887 Inhibition of HDAC in human nuclear extract using Ac-Arg-Gly-Lys(Ac)-AMC as substrate incubated overnight by fluorescence based assay
- ChEMBL_2492889 Inhibition of HDAC1 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2492890 Inhibition of HDAC2 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2492891 Inhibition of HDAC3 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2492892 Inhibition of HDAC6 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_2492893 Inhibition of HDAC7 derived from human HeLa cell extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate by fluorescence based assay
- ChEMBL_809834 (CHEMBL2014771) Inhibition of HDAC3-NCoR2 in human HeLa cells nuclear extract using Fluor-de-Lys as substrate after 30 mins by spectrophotometry
- ChEMBL_816565 (CHEMBL2025279) Inhibition of HDAC1 in human HeLa cell nuclear extract using Fluor de Lys as substrate after 15 mins by fluorometric analysis
- ChEMBL_816566 (CHEMBL2025280) Inhibition of HDAC6 in human HeLa cell nuclear extract using Fluor de Lys as substrate after 15 mins by fluorometric analysis
- ChEMBL_816567 (CHEMBL2025281) Inhibition of HDAC8 in human HeLa cell nuclear extract using Fluor de Lys as substrate after 15 mins by fluorometric analysis
- ChEMBL_2310567 Inhibition of recombinant Top1 in Antimony resistant Leishmania donovani BHU575 whole cell extract assessed as relaxation of supercoiled pBluescript SK(+) DNA measured by agarose gel electrophoresis analysis
- ChEMBL_87532 (CHEMBL694903) In vitro inhibitory activity against histone deacetylase (HDAC) isolated from HeLa nuclear extract
- ChEMBL_87542 (CHEMBL695140) In vitro inhibitory activity against human histone deacetylase (HDAC) using HeLa nuclear extract
- ChEMBL_1524328 (CHEMBL3632020) Inhibition of Stat3 dimer DNA binding activity in human U251MG cells nuclear extract after 1.5 hrs by EMSA using radiolabeled probe hSIE
- ChEMBL_1524329 (CHEMBL3632021) Inhibition of Stat3 dimer DNA binding activity in human U373MG cells nuclear extract after 1.5 hrs by EMSA using radiolabeled probe hSIE
- ChEMBL_1545338 (CHEMBL3751357) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (Ac)-AMC as substrate after 60 mins by fluorometric analysis
- ChEMBL_1545348 (CHEMBL3751367) Inhibition of HDAC1 in human HeLa cell nuclear extract using Boc-Lys (Ac)-AMC as substrate after 60 mins by fluorometric analysis
- ChEMBL_1545349 (CHEMBL3751368) Inhibition of HDAC6 in human HeLa cell nuclear extract using Boc-Lys (Ac)-AMC as substrate after 60 mins by fluorometric analysis
- ChEMBL_1545404 (CHEMBL3751629) Inhibition of HDAC8 in human HeLa cell nuclear extract using Boc-Lys (Ac)-AMC as substrate after 60 mins by fluorometric analysis
- ChEMBL_1572320 (CHEMBL3795851) Inhibition of HDAC in human HeLa nuclear extract using BOC-Ac-Lys-AMC as substrate incubated for 90 mins by fluorescence assay
- ChEMBL_1700233 (CHEMBL4051215) Inhibition of HDAC 8 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1700234 (CHEMBL4051216) Inhibition of HDAC 4 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1700235 (CHEMBL4051217) Inhibition of HDAC 5 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1700236 (CHEMBL4051218) Inhibition of HDAC 9 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1700238 (CHEMBL4051220) Inhibition of HDAC 11 in human HeLa nuclear extract using fluorogenic HDAC class 2A substrate measured after 60 mins by fluorometric analysis
- ChEMBL_1774366 (CHEMBL4231358) Inhibition of HDAC in human HeLa nuclear extract using Ac-Lys(Ac)-pNA as substrate measured after 30 mins by fluorometric analysis
- ChEMBL_1875119 (CHEMBL4376408) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate measured after 30 mins by fluorescence assay
- ChEMBL_1919962 (CHEMBL4422807) Inhibition of HDAC2 in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate measured after 30 mins by fluorescence assay
- ChEMBL_1919963 (CHEMBL4422808) Inhibition of HDAC6 in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate measured after 30 mins by fluorescence assay
- ChEMBL_1919964 (CHEMBL4422809) Inhibition of HDAC8 in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate measured after 30 mins by fluorescence assay
- ChEMBL_2197604 (CHEMBL5110120) Inhibition of HDAC1/HDAC2 in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate and measured by fluorometric method
- ChEMBL_2224821 (CHEMBL5138334) Displacement of [3H]-acetylated histones HDAC6 derived from human K562 cell nuclear extract incubated for 10 mins by liquid scintillation counting method
- ChEMBL_2273494 Inhibition of human recombinant STAT3 assessed as reduction in DNA binding activity with HepG2 nuclear extract incubated for 1 hr by ELISA assay
- ChEMBL_881914 (CHEMBL2212517) Inhibition of topoisomerase-1 in human U251 cells assessed as inhibition of hypoxia-induced HIF-1alpha accumulation in nuclear extract after 6 to 24 hrs by immunoblot analysis
- ChEMBL_1769411 (CHEMBL4221523) Inhibition of HDAC1/CoREST3 in HEK293 whole cell extract using fluorescent acetylated histone peptide as substrate after 60 mins by fluorescence based assay
- ChEMBL_2052178 (CHEMBL4707179) Inhibition of HDAC1 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate measured after 2 hrs by fluorescence based assay
- ChEMBL_2224822 (CHEMBL5138335) Displacement of [3H]-acetylated histones from HDAC1 derived from human K562 cell nuclear extract incubated for 10 mins by liquid scintillation counting method
- ChEMBL_2224823 (CHEMBL5138336) Displacement of [3H]-acetylated histones from HDAC3 derived from human K562 cell nuclear extract incubated for 10 mins by liquid scintillation counting method
- ChEMBL_2452840 Inhibition of HDAC2 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_2452841 Inhibition of HDAC4 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_2452842 Inhibition of HDAC7 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_2452843 Inhibition of HDAC9 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_2452845 Inhibition of HDAC11 in human HeLa cells nuclear extract using BocLys(acetyl)-AMC as substrate incubated for 1 hr by fluorescence plate reader analysis
- ChEMBL_65296 (CHEMBL676785) Inhibitory activity evaluated from soluble cell extract of human endothelia constitutive enzyme (Endothelial nitric oxide synthase) and partially purified by DEAE-sepharose chromatography
- ChEMBL_1618726 (CHEMBL3860895) Inhibition of HDAC1/HDAC2 in human HeLa cell nuclear extract preincubated for 20 mins followed by addition of HDAC green as substrate measured after 60 mins by fluorescence analysis
- ChEMBL_2114766 (CHEMBL4823707) Inhibition of DNA-PK isolated from human HeLa nuclear extract using full length His-tagged p53 as substrate measured after 75 mins in presence of ATP by HTRF assay
- ChEMBL_1590578 (CHEMBL3829047) Inhibition of HDAC in human HeLa cell nuclear extract using Ac-Leu-Gly-Lys (Ac)-AMC as substrate after 30 mins by fluorescence assay
- ChEMBL_809836 (CHEMBL2014773) Inhibition of HDAC8 in human HeLa cells nuclear extract using Arg-His-Lys(Ac)-Lys(Ac)-AMC as substrate after 30 mins by spectrophotometry
- ChEMBL_1663210 (CHEMBL4012891) Binding affinity to TNKS1 in human HeLa cell extract after 2 hrs by HTS assay
- ChEMBL_2067854 (CHEMBL4723107) Inhibition of tyrosinase in human HBL cell extract using L-DOPA as substrate measured every 10 mins for 1 hr by MBTH based spectrophotometric method
- ChEMBL_2171587 (CHEMBL5056721) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as fluorogenic substrate measured after 1 hr by flourescence plate reader method
- ChEMBL_653215 (CHEMBL1226418) Inhibition of eIF4A-mediated cap-dependent protein synthesis in FF-HCV-Ren mRNA transfected Swiss mouse Krebs2 cell extract by [35S]methionine metabolic labeling study
- ChEMBL_1663203 (CHEMBL4012884) Binding affinity to PARP1 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663204 (CHEMBL4012885) Binding affinity to PARP2 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663205 (CHEMBL4012886) Binding affinity to PARP4 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663206 (CHEMBL4012887) Binding affinity to PARP11 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663208 (CHEMBL4012889) Binding affinity to PARP14 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663209 (CHEMBL4012890) Binding affinity to PARP16 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_1663213 (CHEMBL4012894) Binding affinity to PARP10 in human Jurkat cell extract after 45 mins by mass spectrometric analysis
- ChEMBL_2232525 (CHEMBL5146297) Inhibition of CDK2 (unknown origin) in baculovirus infected Sf9 insect cell extract using histone H1 as substrate incubated for 10 mins in presence of [gamma-32P]ATP by radiometric scintillation assay
- ChEMBL_1437098 (CHEMBL3381112) Inhibition of HDAC1/HDAC2 in human HeLa cell extract incubated for 5 mins prior to substrate addition measured after 30 mins by microtitre plate reader analysis
- ChEMBL_1634410 (CHEMBL3877202) Inhibition of immobilized N-LY294002 bead binding to BRD2 (unknown origin) expressed in HEK293T cell nuclear extract incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1634411 (CHEMBL3877203) Inhibition of immobilized N-LY294002 bead binding to BRD3 (unknown origin) expressed in HEK293T cell nuclear extract incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1763006 (CHEMBL4198253) Inhibition of HDAC1/2 in human K562 nuclear extract using (QSY-7)-RGGRGLGK(Ac)-GGARRHRK(TAMRA)NH2 as substrate incubated for 30 mins by fluorescence assay
- ChEMBL_984068 (CHEMBL2434426) Inhibition of GST-tagged full length XIAP (unknown origin) assessed as caspase 3/7 reactivation in S-100 cell extract after 4 hrs by fluorescence assay
- ChEBML_90523 Compound was tested for its ability to penetrate the cell wall and inhibit macrophage ACAT in cell culture using murine IC-21 macrophages (MAI)
- KINOMEscan Enzyme Binding Assays Kinase enzyme binding affinities of compounds disclosed herein were determined using the KINOMEscan technology performed by DiscoveRx Corporation, San Diego, Calif., USA (www.kinomescan.com).
- ChEMBL_1477045 (CHEMBL3428001) Inhibition of HDAC in human HeLa cell nuclear extract using Fluor de Lys as substrate incubated with compound for 30 mins by microtiter-plate reading flourimeter analysis
- ChEMBL_1763011 (CHEMBL4198258) Inhibition of HDAC in human HeLa nuclear extract preincubated for 10 mins followed by Boc-Lys(acetyl)-AMC substrate addition measured after 30 mins by fluorescence assay
- Enzyme Activity Assay α-Amylase activity was assayed with the chromogenic substrate RBB-starch. An enzyme aliquot was incubated (20 min, 26°C) with 0.3% RBB-starch in 0.1 M Britton-Robinson buffer at the pH optimum of the enzyme (6.5 for Aca s 4 and A. siro extract, 7.0 for D. farinae extract, 6.9 for PPA) or at pH 4.5-9.0 (pH profiling). The reaction was stopped with 0.2 M NaOH, the mixture was centrifuged (10000 g, 10 min), and the absorbance at 620 nm of the supernatant was measured against a control sample (incubated in the absence of enzyme/extract).
- ChEMBL_1555945 (CHEMBL3767300) Inhibition of HDAC1/HDAC2 in human HeLa cells nuclear extract preincubated for 5 mins before Boc-Lys (acetyl)-AMC substrate addition for 30 mins by microplate reader analysis
- ChEMBL_1835547 (CHEMBL4335680) Inhibition of TNF alpha stimulated NF-KappaB p65 in human HeLa nuclear extract assessed as decrease in NF-KappaB translocation to nucleus measured after 5 hrs by ELISA
- ChEMBL_1913618 (CHEMBL4416201) Binding affinity to CDPK4 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr in presence of ATP-competitive kinase inhibitor by kinobeads based pull down assay
- ChEMBL_1913620 (CHEMBL4416203) Binding affinity to CDPK1 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr in presence of ATP-competitive kinase inhibitor by kinobeads based pull down assay
- ChEMBL_2260085 (CHEMBL5215096) Inhibition of HDAC1 in human HeLa cell nuclear extract using Bos-Lys(acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition incubated for 30 mins
- ChEBML_1769460 Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys-(Ac)-AMC as substrate preincubated for 15 mins followed by substrate addition measured after 60 mins by fluorescence assay
- ChEMBL_1432943 (CHEMBL3383918) Inhibition of 5-LO in human PMNL S100 extract using arachidonic acid as substrate incubated for 15 mins prior to substrate addition measured after 10 mins by HPLC analysis
- ChEMBL_2057508 (CHEMBL4712509) Inhibition of oligonucleotide [32P]-labelled 5'-AGCITCATTTCCCGTAAATCCCTA probe binding to STAT1 homodimer in mouse NIH3T3 nuclear extract preincubated for 30 mins followed by hSIE probe addition by EMSA analysis
- ChEMBL_2093845 (CHEMBL4775108) Inhibition of human HeLa nuclear extract derived ATM using glutathioneS-transferase-p53N66 as substrate preincubated for 10 mins followed by ATP addition and measured after 1 hr by ELISA
- ChEMBL_2277623 Inhibition of human HDAC4 in HeLa cells extract incubated for 5 mins followed by substrate addition and measured after 15 mins using Fluor-de-Lys as substrate by spectrofluorometric analysis
- ChEMBL_2277624 Inhibition of human HDAC2 in HeLa cells extract incubated for 5 mins followed by substrate addition and measured after 15 mins using Fluor-de-Lys as substrate by spectrofluorometric analysis
- ChEMBL_2277625 Inhibition of human HDAC7 in HeLa cells extract incubated for 5 mins followed by substrate addition and measured after 15 mins using Fluor-de-Lys as substrate by spectrofluorometric analysis
- ChEMBL_2277626 Inhibition of human HDAC8 in HeLa cells extract incubated for 5 mins followed by substrate addition and measured after 15 mins using Fluor-de-Lys as substrate by spectrofluorometric analysis
- ChEMBL_1825526 (CHEMBL4325290) Binding affinity to PI3Kdelta in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825527 (CHEMBL4325291) Binding affinity to VPS34 in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825557 (CHEMBL4325321) Binding affinity to PI3Kalpha in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825558 (CHEMBL4325322) Binding affinity to PI3Kbeta in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1825559 (CHEMBL4325323) Binding affinity to PI3Kgamma in human HL60 cell extract measured after 2 hrs by kinobeads based pull down assay
- ChEMBL_1444367 (CHEMBL3372358) Inhibition of HDAC in human HeLa cell extract using Boc-Lys (acetyl)-AMC as substrate incubated for 10 mins prior to substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_1634379 (CHEMBL3877171) Inhibition of immobilized N-LY294002 bead binding to C-terminal Flag-tagged BRD4 (unknown origin) expressed in HEK293T cell nuclear extract incubated for 1 hr by LC-MS/MS analysis
- ChEMBL_1769460 (CHEMBL4221572) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys-(Ac)-AMC as substrate preincubated for 15 mins followed by substrate addition measured after 60 mins by fluorescence assay
- ChEMBL_1784393 (CHEMBL4255910) Inhibition of STAT3 DNA binding activity in mouse NIH/3T3 nuclear extract preincubated for 30 mins followed by [32P]hSIE addition measured after 30 mins by electrophoretic mobility shift assay
- ChEMBL_1797560 (CHEMBL4269677) Inhibition of ATR in human HeLa cell nuclear extract using GST-fused p53N66 as substrate preincubated for 10 mins followed by ATP addition and measured after 1 hr by ELISA
- ChEMBL_1867771 (CHEMBL4368746) Inhibition of HDAC4 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_1867772 (CHEMBL4368747) Inhibition of HDAC5 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_1867773 (CHEMBL4368748) Inhibition of HDAC7 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_1867774 (CHEMBL4368749) Inhibition of HDAC9 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_1867775 (CHEMBL4368750) Inhibition of HDAC6 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorometric assay
- ChEMBL_2057507 (CHEMBL4712508) Inhibition of oligonucleotide [32P]-labelled 5'-AGCITCATTTCCCGTAAATCCCTA probe binding to STAT1/STAT3 heterodimer in mouse NIH3T3 nuclear extract preincubated for 30 mins followed by hSIE probe addition by EMSA analysis
- ChEMBL_2197605 (CHEMBL5110121) Inhibition of HDAC1 in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(triflouroacetyl)-AMC as substrate incubated for 30 mins and measured by fluorescence assay
- ChEMBL_2197606 (CHEMBL5110122) Inhibition of HDAC6 in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(triflouroacetyl)-AMC as substrate incubated for 30 mins and measured by fluorescence assay
- ChEMBL_2197607 (CHEMBL5110123) Inhibition of HDAC8 in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(triflouroacetyl)-AMC as substrate incubated for 30 mins and measured by fluorescence assay
- ChEMBL_1895949 (CHEMBL4397984) Inhibition of HDAC in human HeLa cell nuclear extract assessed as decrease in deacetylation of FLUOR DE LYS Green substrate preincubated for 10 mins followed by substrate addition and measured after 30 mins by fluorescence based assay
- ChEMBL_1763428 (CHEMBL4198675) Inhibition of HDAC1/2 in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_1769470 (CHEMBL4221582) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 10 mins followed by substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_1769471 (CHEMBL4221583) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_1781393 (CHEMBL4252910) Inhibition of HDAC in human HeLa cell extract using Fluor de Lys-Green as substrate preincubated for 5 mins followed by substrate addition and measured after 1 hr by fluorescence assay
- ChEMBL_1847057 (CHEMBL4347598) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_1936352 (CHEMBL4482111) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC as substrate preincubated for 10 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_1936353 (CHEMBL4482112) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_830578 (CHEMBL2061433) Inhibition of human Tdp1 in Tdp1-deficient chicken DT40 whole cell extract using 5'-[32P]-labeled single-stranded DNA oligonucleotide containing 3'-phosphotyrosine as substrate after 15 mins by PAGE analysis
- ChEMBL_1913617 (CHEMBL4416200) Binding affinity to CDPK4 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr by kinobeads based pull down assay
- ChEMBL_1913619 (CHEMBL4416202) Binding affinity to CDPK1 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr by kinobeads based pull down assay
- ChEMBL_1913623 (CHEMBL4416206) Binding affinity to CK1 in Plasmodium falciparum 3D7 blood stage extract incubated for 1 hr by kinobeads based pull down assay
- ChEMBL_1551388 (CHEMBL3762509) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 15 mins followed by substrate addition measured after 60 mins by fluorescence-based assay
- ChEMBL_1683516 (CHEMBL4033995) Inhibition of HDAC1/2 in human HeLa cell nuclear extract using COLOR DE LYS as substrate pretreated for 5 mins followed by substrate addition measured after 30 mins by UV-absorption method
- ChEMBL_1893047 (CHEMBL4394968) Inhibition of HDAC in human HeLa cell nuclear extract using fluor-de-lys-green as substrate preincubated for 10 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2057493 (CHEMBL4712494) Inhibition of oligonucleotide [32P]-labelled 5'-AGCITCATTTCCCGTAAATCCCTA probe binding to STAT3 SH2 domain in mouse NIH3T3/v-Src nuclear extract preincubated for 30 mins followed by hSIE probe addition by EMSA analysis
- ChEMBL_2213936 (CHEMBL5127068) Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by microplate reader analysis
- ChEMBL_2268733 Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence based analysis
- ChEMBL_2268738 Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys (acetyl)-AMC as substrate preincubated for 15 mins followed by substrate addition and measured after 60 mins by fluorescence based analysis
- ChEMBL_2273479 Inhibition of recombinant STAT3 (unknown origin) expressed in baculovirus in Sf9 insect cells assessed as reduction in DNA binding activity with NIH3T3 nuclear extract incubated for 30 mins by electrophoretic mobility shift assay
- ChEMBL_2488040 Inhibition of HDAC in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 15 mins followed by substrate addition and measured after 60 mins by fluorescence based analysis
- ChEMBL_1553268 (CHEMBL3767500) Inhibition of HDAC1/2 in human HeLa cell nuclear extract using color de Lys as substrate preincubated for 5 mins followed by substrate addition measured after 30 min by microtiter plate reader analysis
- ChEMBL_1750439 (CHEMBL4185199) Inhibition of HDAC1 in Plasmodium falciparum 3D7 nuclear extract using Ac-RGK(Ac)-AMC fluorogenic peptide as substrate preincubated for 1 hr followed by substrate addition measured after 10 min by fluorescence assay
- ChEMBL_1760362 (CHEMBL4195370) Inhibition of HDAC in human HeLa cell nuclear extract using fluorogenic Boc-Lys(acetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by fluorescence-based assay
- ChEMBL_2050950 (CHEMBL4705649) Inhibition of FAM-Bim peptide binding to human full-length N-terminal His6-tagged Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract measured after 30 mins by fluorescence polarization assay
- ChEMBL_2031685 (CHEMBL4685843) Inhibition of class 1 HDAC in human HeLa cell nuclear extract using Boc-Lys(Ac)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2273504 Inhibition of recombinant STAT3 (unknown origin) expressed in Escherichia coli BL21 (DE3) cells assessed as reduction in DNA binding activity with SK-MEL-5 nuclear extract incubated for 30 mins by electrophoretic mobility shift assay
- ChEMBL_2475467 Inhibition of C-terminal 6His-tagged recombinant human STAT3 (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 10 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475468 Inhibition of C-terminal 6His-tagged recombinant human STAT3 (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475469 Inhibition of C-terminal 6His-tagged recombinant human STAT3 (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 60 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2298866 Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2496980 Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2496998 Inhibition of HDAC1 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2496999 Inhibition of HDAC2 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2497000 Inhibition of HDAC3 in human HeLa nuclear extract using Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_887090 (CHEMBL2214315) Inhibition of Apaf-1-caspase 9-cytochrome c-caspase 3 complex in human HEK293 cytosolic extract using afc-DEVD as substrate preincubated for 30 mins before substrate addition measured after 30 mins by fluorescence assay
- ChEMBL_2018354 (CHEMBL4671932) Inhibition of HDAC in human HeLa nuclear extract using Boc-Lys(acetyl)-AMC or Boc-Lys (triflouroacetyl)-AMC as substrate preincubated for 5 mins followed by substrate addition and measured after 30 mins by fluorescence assay
- ChEMBL_2260034 (CHEMBL5215045) Inhibition of HDAC-1 (unknown origin) expressed in Escherichia coli BL21 (DE3) in HeLa nuclear extract using KI177 as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by Bradford reagent method
- ChEMBL_2050946 (CHEMBL4705645) Binding affinity to human full-length N-terminal His6-tagged Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract by isothermal titration calorimetry
- ChEMBL_34793 (CHEMBL643770) In vitro inhibitory activity against Angiotensin I converting enzyme (ACE) isolated from rabbit lung extract using hippuryl-L-histidyl-L-leucine (HHL) as the substrate
- ChEMBL_1738015 (CHEMBL4153765) Inhibition of ATM derived from human HeLa cell nuclear extract using glutathione S-transferase p53N66 as substrate preincubated for 10 mins followed by ATP addition and subsequent incubation for 1 hr measured after 1.5 hrs by ELISA
- Reporter Assay For TLR8 and TLR7 activity testing, HEK-Blue human TLR8 or TLR7 cells, respectively, (Invivogen, San Diego, Calif., USA) transfected with a SEAP reporter (secreted embryonic alkaline phosphatase) construct were used, in which the reporter expression is regulated by the NF-κB promoter upon stimulation for 24 hr. The reporter activity was determined using Quanti Blue kit (Invivogen, San Diego, Calif., USA) at a wavelength of 640 nm.
- ChEMBL_2475470 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2260035 (CHEMBL5215046) Inhibition of HDAC-6 (unknown origin) expressed in Escherichia coli BL21 (DE3) in HeLa nuclear extract using Boc-Lys(TFA)-AMC as substrate preincubated for 5 mins followed by substrate addition measured after 30 mins by Bradford reagent method
- ChEMBL_2050947 (CHEMBL4705646) Binding affinity to human full-length N-terminal His6-tagged prephosphorylated Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract by isothermal titration calorimetry
- ChEMBL_2475471 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 C328A mutant (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475472 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 C426A mutant (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475473 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 C468A mutant (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- ChEMBL_2475474 Inhibition of recombinant wild type tyrosine phosphorylated C-terminal 6His tagged human STAT3 C542S mutant (127 to 711 residues) DNA-binding activity transfected in v-Src transformed mouse NIH3T3 cell nuclear extract preincubated for 30 mins followed by addition of radiolabeled hSIE and measured after 30 mins by EMSA analysis
- HDAC1 Assay Human recombinant full length HDAC1 expressed in a baculovirus expression system was purchased from BPS BioSciences (San Diego, Calif., U.S.A.). The substrate used in the HDAC1 assay was 5 μM of acetyl-Gly-Ala-Lys(acetyl)-AMC.
- HDAC6 Assay Human recombinant full length HDAC6 expressed in a baculovirus expression system was purchased from BPS BioSciences (San Diego, Calif., U.S.A.). The substrate used in the HDAC1 assay was 5 μM of acetyl-Gly-Ala-Lys (acetyl)-AMC.
- Enzyme Assay ATR for use in the in vitro enzyme assay was obtained from HeLa nuclear extract (CIL Biotech, Mons, Belgium) by immunoprecipitation with rabbit polycolonal antiserum raised to amino acids 400-480 of ATR (Tibbetts R S et, al, 1999, GenesDev.13:152-157).
- Biological Assay For mTOR enzyme activity assays, mTOR protein was isolated from HeLa cell cytoplasmic extract by immunoprecipitation, and activity determined essentially as described previously using recombinant PHAS-1as a substrate (ref 21).
- Dose Response confirmation of uHTS for the identification of inhibitors of NALP1 in yeast using a luminescent assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1 U01 AI078048 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego, CA NLR family proteins are an important component of the innate immune system of vertebrates. These proteins possess a nucleotide-binding oligomerization domain, called NACHT, in combination with variable numbers of Leucine-Rich Repeat (LRR) domains that bind molecules produced by pathogens and probably also products of tissue injury. Among the effector mechanisms of NLR family proteins is activation of Caspase-1, which cleaves and activates pro-inflammatory cytokines. We present here a unique primary assay, in which we have reconstituted the mammalian Caspase mediated IL-1 activation pathway consisting of NLRP1 (NALP1), ASC, and Caspase-1 in Sacchar
- Dose Response confirmation of uHTS for the identification of inhibitors of NALP3 in yeast using a luminescent assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1 U01 AI078048 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA NLR family proteins are an important component of the innate immune system of vertebrates. These proteins possess a nucleotide-binding oligomerization domain, called NACHT, in combination with variable numbers of Leucine-Rich Repeat (LRR) domains that bind molecules produced by pathogens and probably also products of tissue injury. Among the effector mechanisms of NLR family proteins is activation of Caspase-1, which cleaves and activates pro-inflammatory cytokines. We present here a unique primary assay, in which we have reconstituted the mammalian Caspase mediated IL-1 activation pathway consisting of NLRP3 (NALP3), ASC, and Caspase-1 in Saccharo
- Dose Response confirmation of uHTS small molecule inhibitors of Plasmodium falciparum Glucose-6-phosphate dehydrogenase via a fluorescence intensity assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R21AI082434-01 Assay Provider: Lars Bode, Ph.D., University of California San Diego, San Diego, CA Tropical malaria caused by the protozoan parasite Plasmodium falciparum is responsible for up to three million deaths annually. Due to increasing regional distribution and resistances against the clinically used antimalarials, novel antimalarial drugs - which have new mechanisms of action and are suitable for combination therapies - are urgently required. Plasmodium falciparum Glucose-6-phosphate dehydrogenase (PfGluPho) is a potential novel target for antimalarial drug design. Glucose-6-Phosphate Dehydrogenase (G6PD) reaction is the first and rate-limiting step in the pentose phosphate pathway (PPP), catalyzed by a bifunctional enzyme Plasmodium fal
- Dose Response orthogonal assay utilizing the direct end-point detection of NADPH for uHTS small molecule inhibitors of Plasmodium falciparum Glucose-6-phosphate dehydrogenase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R21AI082434-01 Assay Provider: Lars Bode, Ph.D., University of California San Diego, San Diego, CA Tropical malaria caused by the protozoan parasite Plasmodium falciparum is responsible for up to three million deaths annually. Due to increasing regional distribution and resistances against the clinically used antimalarials, novel antimalarial drugs - which have new mechanisms of action and are suitable for combination therapies - are urgently required. Plasmodium falciparum Glucose-6-phosphate dehydrogenase (PfGluPho) is a potential novel target for antimalarial drug design. Glucose-6-Phosphate Dehydrogenase (G6PD) reaction is the first and rate-limiting step in the pentose phosphate pathway (PPP), catalyzed by a bifunctional enzyme Plasmodium fal
- Dose Response orthogonal kinetic assay utilizing the direct detection of NADPH for uHTS small molecule inhibitors of Plasmodium falciparum Glucose-6-phosphate dehydrogenase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R21AI082434-01 Assay Provider: Lars Bode, Ph.D., University of California San Diego, San Diego, CA Tropical malaria caused by the protozoan parasite Plasmodium falciparum is responsible for up to three million deaths annually. Due to increasing regional distribution and resistances against the clinically used antimalarials, novel antimalarial drugs - which have new mechanisms of action and are suitable for combination therapies - are urgently required. Plasmodium falciparum Glucose-6-phosphate dehydrogenase (PfGluPho) is a potential novel target for antimalarial drug design. Glucose-6-Phosphate Dehydrogenase (G6PD) reaction is the first and rate-limiting step in the pentose phosphate pathway (PPP), catalyzed by a bifunctional enzyme Plasmodium fal
- Pharmacological Assay For TLR8 and TLR7 activity testing, HEK-Blue human TLR8 or TLR7 cells (Invivogen, San Diego, Calif., USA) are used, respectively. These cells are designed for studying the stimulation of human TLR8 or TLR7 by monitoring the activation of NF-κB. A SEAP (secreted embryonic alkaline phosphatase) reporter gene is placed under the control of the IFN-b minimal promoter fused to five NF-κB and AP-1-binding sites. Therefore the reporter expression is regulated by the NF-κB promoter upon stimulation of human TLR8 or TLR7 for 20 hours. The cell culture supernatant SEAP reporter activity was determined using Quanti Blue kit (Invivogen, San Diego, Ca, USA) at a wavelength of 640 nm, a detection medium that turns purple/blue in the presence of alkaline phosphatase.
- Pharmacological Assay For TLR8 and TLR7 activity testing, HEK-Blue human TLR8 or TLR7 cells (Invivogen, San Diego, Calif., USA) are used, respectively. These cells are designed for studying the stimulation of human TLR8 or TLR7 by monitoring the activation of NF-κB. A SEAP (secreted embryonic alkaline phosphatase) reporter gene is placed under the control of the IFN-b minimal promoter fused to five NF-κB and AP-1-binding sites. Therefore the reporter expression is regulated by the NF-κB promoter upon stimulation of human TLR8 or TLR7 for 20 hours. The cell culture supernatant SEAP reporter activity was determined using Quanti Blue kit (Invivogen, San Diego, Ca, USA) at a wavelength of 640 nm, a detection medium that turns purple/blue in the presence of alkaline phosphatase.
- Pharmacological Assay For TLR8 and TLR7 activity testing, HEK-Blue human TLR8 or TLR7 cells (Invivogen, San Diego, Calif., USA) are used, respectively. These cells are designed for studying the stimulation of human TLR8 or TLR7 by monitoring the activation of NF-κB. A SEAP (secreted embryonic alkaline phosphatase) reporter gene is placed under the control of the IFN-b minimal promoter fused to five NF-κB and AP-1-binding sites. Therefore the reporter expression is regulated by the NF-κB promoter upon stimulation of human TLR8 or TLR7 for 20 hours. The cell culture supernatant SEAP reporter activity was determined using Quanti Blue kit (Invivogen, San Diego, Calif., USA) at a wavelength of 640 nm, a detection medium that turns purple/blue in the presence of alkaline phosphatase.
- ChEMBL_2050942 (CHEMBL4705641) Binding affinity to human full-length N-terminal His6-tagged Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract after 30 mins using FAM-labelled compound by fluorescence polarization assay
- Fluor de Lys Assay Fluor de Lys is a fluorescence based HDAC activity assay comprising a combination of fluorogenic Histone deAcetylase Lysyl substrate and a developer. The kit is a highly sensitive and convenient alternative to radiolabeled, acetylated histones or peptide/HPLC methods for the assay of histone deacetylases. This assay is based on the ability of HeLa nuclear extract, which is enriched in HDAC activity, to mediate the deacetylation of the acetylated lysine side chain of the Fluor de Lys substrate. The assay procedure requires two steps. First, incubation of the HeLa nuclear extract with the Fluor de Lys substrate results in substrate deacetylation and thus sensitizes it to the second step. In the second step, treatment of the deacetylated substrate with the Fluor de Lys developer produces a fluorophore. The substrate-developer reaction, under normal circumstances goes to completion in less than 1 min at 25 C.
- HDAC enzyme inhibitionAssay HDAC enzyme inhibition assays were performed using purified HDACs 1-10 essentially as described in Beckers et al., 2007, Int. J. Cancer., 121:1138-48 and Perez-Balado et al., 2007, J. Med. Chem., 50:2497-2505. Inhibition assays using nuclear extract were performed essentially as described in Herman et al., 2006, Nat. Chem. Biol., 2:551-558. Briefly, the purified HDACs or nuclear extract were incubated with an acetylated substrate in the absence of the compound to be assayed and with increasing concentrations of the compound. The rate of substrate deacetylation was measured under each condition, and half-maximal inhibitory concentration with regard to each HDAC was determined by standard means.
- Dose Response confirmation of inhibitors of NALP1 in yeast using a Caspase-1-ASC counter screen. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1 U01 AI078048 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego, CA NLR family proteins are an important component of the innate immune system of vertebrates. These proteins possess a nucleotide-binding oligomerization domain, called NACHT, in combination with variable numbers of Leucine-Rich Repeat (LRR) domains that bind molecules produced by pathogens and probably also products of tissue injury. Among the effector mechanisms of NLR family proteins is activation of Caspase-1, which cleaves and activates pro-inflammatory cytokines. We present here a unique primary assay, in which we have reconstituted the mammalian Caspase mediated IL-1 activation pathway consisting of NLRP1 (NALP1), ASC, and Caspase-1 in Sacchar
- Dose Response confirmation of inhibitors of NALP3 in yeast using a Caspase-1-ASC counter screen Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1 U01 AI078048 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA NLR family proteins are an important component of the innate immune system of vertebrates. These proteins possess a nucleotide-binding oligomerization domain, called NACHT, in combination with variable numbers of Leucine-Rich Repeat (LRR) domains that bind molecules produced by pathogens and probably also products of tissue injury. Among the effector mechanisms of NLR family proteins is activation of Caspase-1, which cleaves and activates pro-inflammatory cytokines. We present here a unique primary assay, in which we have reconstituted the mammalian Caspase mediated IL-1 activation pathway consisting of NLRP3 (NALP3), ASC, and Caspase-1 in Saccharo
- SAR analysis of NF-kB dependent luciferase using DAP as an inducer Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory diso
- SAR analysis of NF-kappaB dependent luciferase using DAP as an inducer - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory diso
- SAR analysis of NF-kappaB dependent luciferase using Doxorucibin as an inducer - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory disorders,
- SAR analysis of compounds that inhibit NOD1 - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory diso
- SAR analysis of compounds that inhibit NOD1 - Set 3 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory diso
- SAR analysis of compounds that inhibit NOD1 revised Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory diso
- SAR analysis of compounds that inhibit NOD2 - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory disor
- SAR analysis of compounds that inhibit NOD2 - Set 3 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory disor
- SAR analysis of compounds that inhibit NOD2 revised Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory disor
- uHTS luminescence assay for the identification of compounds that inhibit NOD1 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory disor
- uHTS luminescence assay for the identification of compounds that inhibit NOD2 in MDP treated cells Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego, CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. Mutations in the NOD1 and NOD2 genes are associated with a number of human inflammatory dis
- ChEMBL_2050943 (CHEMBL4705642) Binding affinity to human full-length N-terminal His6-tagged prephosphorylated Bcl2 (2 to 206 residues) expressed in Escherichia coli S12 extract after 30 mins using FAM-labelled compound by fluorescence polarization assay
- Dose response confirmation of the uHTS fluorescent assay for identification of inhibitors of ATG4B Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH090871-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Autophagy is an evolutionarily conserved process whereby cells catabolize damaged proteins and organelles for purposes of generating substrates for sustaining ATP production during times of nutrient deprivation. The autophagic process involves membrane vesicles engulfing cytosol and organelles, delivering their contents to lysosomes for digestion. The genes responsible for autophagy have been identified, largely through genetic analysis of yeast, Saccharomyces cerevisiae, and are conserved in mammals, plants, and essentially all eukaryotes. While autophagy is critical for cell survival in the context of nutrient deprivation, circumstances
- Dose response confirmation of the uHTS fluorescent assay for identification of inhibitors of ATG4B. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH090871-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Autophagy is an evolutionarily conserved process whereby cells catabolize damaged proteins and organelles for purposes of generating substrates for sustaining ATP production during times of nutrient deprivation. The autophagic process involves membrane vesicles engulfing cytosol and organelles, delivering their contents to lysosomes for digestion. The genes responsible for autophagy have been identified, largely through genetic analysis of yeast, Saccharomyces cerevisiae, and are conserved in mammals, plants, and essentially all eukaryotes. While autophagy is critical for cell survival in the context of nutrient deprivation, circumstances
- SAR analysis of an In Vitro TNAP Dose Response Luminescent Assay Sanford-Burnham Center for Chemical Genomics (SBCCG) Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Number: MH077602-01 Assay Provider: Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute, San Diego, CA This TNAP dose response assay is developed and performed for the purpose of SAR study on analogs of hits originally identified in the TNAP luminescent HTS assay (AID 518) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing phosphate and alcohol. APs are dimeric enzymes found in the most organism. In human, four isozymes of APs have been identified. Three isozymes are tissue-specific and the fourth one is tissue-nonsepecifc, named TNAP. TNAP deficiency is associated with defective bone mineralization in the form of rickets and osteomalacia. Therefore, there are therapeutic potentials of inhibiting TNAP activity
- PDE3A Enzyme Inhibition Assay For the determination of the in vitro effect of example compounds on the PDE3A reactions 2 μl of the respective example compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3A cell extract from Sf9 cells overexpressing human full length PDE3A (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3A cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:5000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H]adenosine 3′, 5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac).
- Human Glucose-6-Phosphate Dehydrogenase Dose Response Selectivity Assay for Inhibitors of Plasmodium falciparum Glucose-6-Phosphate Dehydrogenase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R21AI082434-01 Assay Provider: Lars Bode, Ph.D., University of California San Diego, San Diego, CA Tropical malaria caused by the protozoan parasite Plasmodium falciparum is responsible for up to three million deaths annually. Due to increasing regional distribution and resistances against the clinically used antimalarials, novel antimalarial drugs - which have new mechanisms of action and are suitable for combination therapies - are urgently required. Plasmodium falciparum Glucose-6-phosphate dehydrogenase (PfGluPho) is a potential novel target for antimalarial drug design. Glucose-6-Phosphate Dehydrogenase (G6PD) reaction is the first and rate-limiting step in the pentose phosphate pathway (PPP), catalyzed by a bifunctional enzyme Plasmodium fal
- Dose Response confirmation of Inhibitors of Mdm2/MdmX interaction in luminescent format Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: R03 MH089489-01 Assay Provider: Dr. Geoffrey M. Wahl, Salk Institute for Biological Studies, San Diego, CA A wild type but attenuated p53 is retained in approximately 50% of human tumors, and reactivation of p53 in such tumors is an attractive chemotherapeutic strategy. p53 activity is restricted in vivo by mdm2 and mdmx, and knockout of either of these proteins is embryonic lethal in a p53-dependent manner (1, 2). Both proteins bind to p53 via a hydrophobic N-terminal pocket and block p53-dependent transcription of genes required for tumor suppression. Efforts to reactivate p53 with small molecules have focused on inhibition of the mdm2/p53 interaction, which leads to increased p53 levels and activity. However, recent reports indicate that targetin
- SAR analysis of NF-kappaB dependent luciferase using PMA/Ionomycin as an inducer - 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. NF-kB pathway activated by antigen receptors is critical for acquired (as opposed to innate)
- SAR assay for compounds that inhibit PHOSPHO1 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084086-01 Assay Provider: Dr. Jose Luis Milan, Sanford-Burnham Medical Research Institute, San Diego CA Mineralization of cartilage and bone occurs by a series of physicochemical and biochemical processes that together facilitate the deposition of hydroxyapatite (HA) in specific areas of the extracellular matrix (ECM). Experimental evidence has pointed to the presence of HA crystals along collagen fibrils in the ECM and also within the lumen of chondroblast- and osteoblast-derived matrix vesicles (MVs). Dr. Milan's working model is that bone mineralization is first initiated within the lumen of MVs. In a second step, HA crystals grow beyond the confines of the MVs and become exposed to the extracellular milieu where they continue to
- uHTS identification of small molecule antagonists of the binding of Siah-1 and a peptide ligand via a fluorescence polarization assay. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1 R03 MH086475-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Proteasomal degradation typically requires post-translational modification of target proteins with K48-linked polyubiquitin chains. This process of protein proteolysis plays a key role in normal cellular function. The E3 ubiquitin ligase, Siah-1, facilitates the transfer of ubiquitin to its substrate proteins destined for degradation by way of its RING domain. Siah-1 is a member of a family of highly conserved RING domain proteins, which regulate a variety of cellular functions, including cell cycle arrest, tumor suppression, and apoptosis through the beta-catenin degradation pathway. Siah-1 has also been identified as a p53-inducible gene, functi
- Her-2 Degradation Assay EC50 was defined as the concentration of the compound at which there was 50% degradation of the Her-2/neu protein in MCF7 breast carcinoma cells. Samples were analyzed using a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA) equipped with an argon-ion laser that emits 488 nm light for excitation of the phycoerythrin fluorochrome. A fluorescence histogram was generated, and the mean fluorescence intensity (MFI) of each sample was determined using Cellquest software.
- Kinase Screen Assay KINOMEscan (Ambit Biosciences, San Diego, Calif.), a high-throughput method for screening small molecular agents against a large panel of human kinases, was utilized for compound 2. The technology is a competition binding assay that profiled the selectivity of compound 2 against 350 kinases, each fused to a proprietary tag. The quantity of each kinase bound to an immobilized, active site-directed ligand was measured in the presence and absence of compound 2.
- Enzyme inhibition assay The nucleus extract was extracted from HDAC8 enzyme was expressed in Escherichia coli. Boc-Lys (acetyl)-AMC was used as the substrate of HDAC. SAHA which is the HDAC inhibitor on market was used as a positive control. The compounds were diluted to six concentrations (25, 5, 1, 0.2, 0.04 and 0.008 uM/L) to investigate their ability of inhibiting HDAC activity.
- Counter Screen using XIAP-Bir3 of the Chemical Antagonists of IAP-family anti-apoptotic proteins confirmation assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Proposal Number: MH081277-01 Assay Provider: John C. Reed, Sanford-Burnham Medical Research Institute, San Diego, CA This dose response assay is developed and performed as a counter screen to compounds in the Chemical Antagonists of IAP-family anti-apoptotic proteins confirmation (AID 1449) and to study the structure-activity relationship on analogs of the confirmed hits. Compounds are either acquired from commercial sources or synthesized internally. Apoptosis plays an essential role in many aspects of normal development and physiology, becoming dysregulated in myriad diseases characterized by insufficient or excessive cell death. Caspases are intracellular proteases that are suppressed by Inhibitor of Apoptosis Proteins (IAPs), a family of evolutionarily conse
- Dose Response concentration confirmation of uHTS hits from a small molecule activators of human intestinal alkaline phosphatase via a luminescent assay - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response concentration confirmation of uHTS hits from a small molecule activators of human intestinal alkaline phosphatase via a luminescent assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response confirmation of inhibitors of Sentrin-specific proteases (SENPs) using a Caspase-3 Selectivity assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network(MLPCN) Grant Proposal Number: 1R21 NS061758-01 fast track Assay Provider: Dr. Guy Salvesen, Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Modification of proteins by SUMO is a dynamic and reversible process. SUMOylation/deSUMOylation cycle regulates SUMOs function. Sentrin-specific proteases (SENPs) are involved in both the maturation of SUMO precursors (endopeptidase cleavage) and deconjugation of the targets (isopeptidase cleavage) [1-3]. There are seven SENPs (1, 2, 3, 5, 6, 7, 8) in humans, and several of these have been characterized as SUMO (or Nedd8) specific enzymes. SENP8 is not a SUMO protease, instead it functions on a small ubiquitin related protein Nedd8. The objective of this project is to generate small molecule inhibitors specific for
- Dose Response confirmation of inhibitors of Sentrin-specific proteases (SENPs) using a Luminescent Interference Counterscreen assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network(MLPCN)Grant Proposal Number: 1R21 NS061758-01 fast track Assay Provider: Dr. Guy Salvesen, Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Modification of proteins by SUMO is a dynamic and reversible process. SUMOylation/deSUMOylation cycle regulates SUMOs function. Sentrin-specific proteases (SENPs) are involved in both the maturation of SUMO precursors (endopeptidase cleavage) and deconjugation of the targets (isopeptidase cleavage) [1-3]. There are seven SENPs (1, 2, 3, 5, 6, 7, 8) in humans, and several of these have been characterized as SUMO (or Nedd8) specific enzymes. SENP8 is not a SUMO protease, instead it functions on a small ubiquitin related protein Nedd8. The objective of this project is to generate small molecule inhibitors specific for SENP8
- Dose Response confirmation of uHTS activators of Human Intestinal Alkaline Phosphatase using Placental Alkaline Phosphatase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response confirmation of uHTS activators of Human Intestinal Alkaline Phosphatase using Tissue Nonspecific Alkaline Phosphatase. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response confirmation of uHTS activators of Mouse Intestinal Alkaline Phosphatase using Human Intestinal Alkaline Phosphatase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response confirmation of uHTS for inhibitors of Sentrin-specific protease 6 (SENP6) using a Luminescent assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network(MLPCN) Grant Proposal Number: 1R21 NS061758-01 fast track Assay Provider: Dr. Guy Salvesen, Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Modification of proteins by SUMO is a dynamic and reversible process. SUMOylation/deSUMOylation cycle regulates SUMOs function. Sentrin-specific proteases (SENPs) are involved in both the maturation of SUMO precursors (endopeptidase cleavage) and deconjugation of the targets (isopeptidase cleavage) [1-3]. There are seven SENPs (1, 2, 3, 5, 6, 7, 8) in humans, and several of these have been characterized as SUMO (or Nedd8) specific enzymes. The objective of this project is to generate small molecule inhibitors specific for SENP6 (the deSUMOylating enzyme). 1536-well chemiluminescent screening assay utilizes RLRGG-
- Dose Response confirmation of uHTS for inhibitors of Sentrin-specific protease 8 (SENP8) using a Luminescent assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network(MLPCN)Grant Proposal Number: 1R21 NS061758-01 fast track Assay Provider: Dr. Guy Salvesen, Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Modification of proteins by SUMO is a dynamic and reversible process. SUMOylation/deSUMOylation cycle regulates SUMOs function. Sentrin-specific proteases (SENPs) are involved in both the maturation of SUMO precursors (endopeptidase cleavage) and deconjugation of the targets (isopeptidase cleavage) [1-3]. There are seven SENPs (1, 2, 3, 5, 6, 7, 8) in humans, and several of these have been characterized as SUMO (or Nedd8) specific enzymes. SENP8 is not a SUMO protease, instead it functions on a small ubiquitin related protein Nedd8. The objective of this project is to generate small molecule inhibitors specific for SENP8
- Dose Response confirmation of uHTS hits from a small molecule inhibitors of human intestinal alkaline phosphatase via a luminescent assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response confirmation of uHTS hits from a small molecule inhibitors of mouse intestinal alkaline phosphatase via a luminescent assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response confirmation of uHTS inhibitors of Human Intestinal Alkaline Phosphatase using Mouse Intestinal Alkaline Phosphatase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response confirmation of uHTS inhibitors of Human Intestinal Alkaline Phosphatase using Placental Alkaline Phosphatase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of I
- Dose Response confirmation of uHTS inhibitors of Mouse Intestinal Alkaline Phosphatase using Placental Alkaline Phosphatase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response confirmation of uHTS inhibitors of Mouse Intestinal Alkaline Phosphatase using Tissue Nonspecific Alkaline Phosphatase. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of
- Dose Response selectivity of inhibitors of STriatal-Enriched Phosphatase (STEP) in the Lymphoid Phosphatase (PTPN22) Inhibition Assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R03MH095532-01 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Disturbance of the dynamic balance between protein tyrosine phosphorylation and dephosphorylation is crucial for the development of many serious conditions, including cancer, diabetes, and autoimmune disorders. This is the first time that tyrosine phosphatase inhibitors are being proposed to improve cognitive function in Alzheimer's disease (AD). STriatal-Enriched Phosphatase (STEP) is a brain-specific protein tyrosine phosphatase that is highly expressed in regions where consolidation of memory occurs and regulates the internalization of NMDARs. Our recent work demonstrates that STEP is elevated in the prefrontal cortex of human AD patients
- Dose Response selectivity of inhibitors of STriatal-Enriched Phosphatase (STEP) in the dual-specificity protein-tyrosine phosphatase VHR Inhibition Assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R03MH095532-01 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Disturbance of the dynamic balance between protein tyrosine phosphorylation and dephosphorylation is crucial for the development of many serious conditions, including cancer, diabetes, and autoimmune disorders. This is the first time that tyrosine phosphatase inhibitors are being proposed to improve cognitive function in Alzheimer's disease (AD). STriatal-Enriched Phosphatase (STEP) is a brain-specific protein tyrosine phosphatase that is highly expressed in regions where consolidation of memory occurs and regulates the internalization of NMDARs. Our recent work demonstrates that STEP is elevated in the prefrontal cortex of human AD patients
- Dose Response selectivity of inhibitors of Striatal-Enriched Phosphatase (STEP) in a SHP2 (PTPN11) Inhibition Assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R03MH095532-01 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Disturbance of the dynamic balance between protein tyrosine phosphorylation and dephosphorylation is crucial for the development of many serious conditions, including cancer, diabetes, and autoimmune disorders. This is the first time that tyrosine phosphatase inhibitors are being proposed to improve cognitive function in Alzheimer's disease (AD). STriatal-Enriched Phosphatase (STEP) is a brain-specific protein tyrosine phosphatase that is highly expressed in regions where consolidation of memory occurs and regulates the internalization of NMDARs. Our recent work demonstrates that STEP is elevated in the prefrontal cortex of human AD patients
- Dose response confirmation of uHTS small molecule inhibitors of Striatal-Enriched Phosphatase via a fluorescence intensity assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R03MH095532-01 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Disturbance of the dynamic balance between protein tyrosine phosphorylation and dephosphorylation is crucial for the development of many serious conditions, including cancer, diabetes, and autoimmune disorders. This is the first time that tyrosine phosphatase inhibitors are being proposed to improve cognitive function in Alzheimer's disease (AD). STriatal-Enriched Phosphatase (STEP) is a brain-specific protein tyrosine phosphatase that is highly expressed in regions where consolidation of memory occurs and regulates the internalization of NMDARs. Our recent work demonstrates that STEP is elevated in the prefrontal cortex of human AD patients
- Dose response orthogonal assay of uHTS small molecule inhibitors of Striatal-Enriched Phosphatase via a colorimetric intensity assay. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R03MH095532-01 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Disturbance of the dynamic balance between protein tyrosine phosphorylation and dephosphorylation is crucial for the development of many serious conditions, including cancer, diabetes, and autoimmune disorders. This is the first time that tyrosine phosphatase inhibitors are being proposed to improve cognitive function in Alzheimer's disease (AD). STriatal-Enriched Phosphatase (STEP) is a brain-specific protein tyrosine phosphatase that is highly expressed in regions where consolidation of memory occurs and regulates the internalization of NMDARs. Our recent work demonstrates that STEP is elevated in the prefrontal cortex of human AD patients
- HTS TR-FRET-based dose response confirmatory assay for Siah-1 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH086475-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Proteasomal degradation typically requires post-translational modification of target proteins with K48-linked polyubiquitin chains. This process of protein proteolysis plays a key role in normal cellular function. The E3 ubiquitin ligase, Siah-1, facilitates the transfer of ubiquitin to its substrate proteins destined for degradation by way of its RING domain. Siah-1 is a member of a family of highly conserved RING domain proteins, which regulate a variety of cellular functions, including cell cycle arrest, tumor suppression, and apoptosis through the beta-catenin degradation pathway. Siah-1 has also been identified as a p53-inducible gene,
- SAR Analysis of small molecule inhibitors of Sentrin-specific protease 8 (SENP8) using a Luminescent assay - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network(MLPCN)Grant Proposal Number: 1R21 NS061758-01 fast track Assay Provider: Dr. Guy Salvesen, Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Modification of proteins by SUMO is a dynamic and reversible process. SUMOylation/deSUMOylation cycle regulates SUMOs function. Sentrin-specific proteases (SENPs) are involved in both the maturation of SUMO precursors (endopeptidase cleavage) and deconjugation of the targets (isopeptidase cleavage) [1-3]. There are seven SENPs (1, 2, 3, 5, 6, 7, 8) in humans, and several of these have been characterized as SUMO (or Nedd8) specific enzymes. SENP8 is not a SUMO protease, instead it functions on a small ubiquitin related protein Nedd8. The objective of this project is to generate small molecule inhibitors specific for SENP8
- Counterscreen for activators of the Protein Kinase A-R1A (PKA-R1A) complex Source (MLPCN Center Name): The Scripps Research Institute Molecular Screening Center (SRIMSC) Affiliation: The Scripps Research Institute, TSRI Assay Provider: Susan Taylor, University of California, San Diego (UCSD) Network: Molecular Library Probe Production Centers Network (MLPCN) Grant Proposal Number: R01 GM34921 Grant Proposal PI: Susan Taylor, University of California, San Diego (UCSD) External Assay ID: PKAR2B_ACT_FP_1536_3XEC50 DCSRUN PKAR1A Name: Counterscreen for activators of the Protein Kinase A-R1A (PKA-R1A) complex: fluorescence polarization-based biochemical high throughput dose response assay to identify activators of the Protein Kinase A-R2B (PKA-R2B) complex. Description: Protein phosphorylation is one of the most important mechanisms for regulation in mammalian cells, and cAMP is an ancient second messenger signaling molecule. Protein kinase A (PKA) is a ubiquitous cAMP-dependent serine/threonine protein kinase that phosphorylates intracellular protein substrat
- Counterscreen for activators of the Protein Kinase A-R2B (PKA-R2B) complex Source (MLPCN Center Name): The Scripps Research Institute Molecular Screening Center (SRIMSC) Affiliation: The Scripps Research Institute, TSRI Assay Provider: Susan Taylor, University of California, San Diego (UCSD) Network: Molecular Library Probe Production Centers Network (MLPCN) Grant Proposal Number: R01 GM34921 Grant Proposal PI: Susan Taylor, University of California, San Diego (UCSD) External Assay ID: PKAR1A_ACT_FP_1536_3XEC50 DCSRUN PKAR2B Name: Counterscreen for activators of the Protein Kinase A-R2B (PKA-R2B) complex: fluorescence polarization-based biochemical high throughput dose response assay to identify activators of the Protein Kinase A-R1A (PKA-R1A) complex. Description: Protein phosphorylation is one of the most important mechanisms for regulation in mammalian cells, and cAMP is an ancient second messenger signaling molecule. Protein kinase A (PKA) is a ubiquitous cAMP-dependent serine/threonine protein kinase that phosphorylates intracellular protein substrat
- Pharmacological Test For TLR8 and TLR7 activity testing, HEK-Blue human TLR8 or TLR7 cells (Invivogen, San Diego, Calif., USA) are used, respectively. These cells are designed for studying the stimulation of human TLR8 or TLR7 by monitoring the activation of NF-κB. A SEAP (secreted embryonic alkaline phosphatase) reporter gene is placed under the control of the IFN-b minimal promoter fused to five NF-κB and AP-1-binding sites. Therefore the reporter expression is regulated by the NF-κB promoter upon stimulation of human TLR8 or TLR7 for 20 hours. The cell culture supernatant SEAP reporter activity was determined using Quanti Blue kit (Invivogen, San Diego, Ca, USA) at a wavelength of 640 nm, a detection medium that turns purple/blue in the presence of alkaline phosphatase. EC50 values were determined using Activity Base analysis (ID Business Solution, Limited).
- SAR analysis of compounds that potentiate TRAIL-induced apoptosis in PPC-1 cells. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: X01 MH083230-01 Assay Provider: Dr. Dmitri Rozanov, Sanford-Burnham Medical Research Institute, San Diego CA This dose response assay is developed and performed to confirm hits originally identified in "uHTS for the identification of compounds that potentiate TRAIL-induced apoptosis of cancer cells" (AID 1443) and to study the structure-activity relationship on analogs of the confirmed hits. Compounds are either acquired from commercial sources or synthesized internally. Cytotoxic chemotherapy induces apoptosis via a pathway involving mitochondria, sometimes referred to as the "intrinsic pathway." An acquired resistance to anticancer drugs commonly results from the accumulation of defects in components of the mitochondrial pathway for apoptosis. Discov
- Electrophoretic Mobility Shift Assay (EMSA) His10-Stat3 was expressed in Sf9 cells from a baculovirus encoding the recombinant protein. A nuclear extract of the Sf9 cells was incubated with 32P-labeled high affinity c-fos sis inducible element (hSIE) either alone or in the presence of inhibitor. After 20 min of incubation, samples were electrophoresed on polyacrylamide gels. The gels were dried, exposed to a phosphorimager screen and scanned. IC50 values were derived from plots of spot intensity versus phosphopeptide concentration. The affinity of peptides to the SH2 domain is measured by the intensity of the radioactivity of the Stat3-DNA complex band in the electrophoresis gel.
- Fluorescence Polarization Assay FPAs were performed using various concentrations of GST-Bcl-2 family proteins, incubated with FITC-conjugated peptide substrates in the presence of inhibitor compound. After 30 min of incubation at room temperature, the polarization values in millipolarization units were measured at excitation/emission wavelengths of 480/535 nm with a multilabel plate reader (PerkinElmer). IC50 was determined by fitting the experimental data to a sigmoidal dose-response nonlinear regression model (SigmaPlot 10.0.1, Systat Software, Inc., San Jose, CA). Data reported are the mean of three independent experiments.
- Dose Response confirmation of Inhibitors of Mdm2/MdmX interaction using a Brca1/Bard1 BiLC Counterscreen assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: R03 MH089489-01 Assay Provider: Dr. Geoffrey M. Wahl, Salk Institute for Biological Studies, San Diego, CA A wild type but attenuated p53 is retained in approximately 50% of human tumors, and reactivation of p53 in such tumors is an attractive chemotherapeutic strategy. p53 activity is restricted in vivo by mdm2 and mdmx, and knockout of either of these proteins is embryonic lethal in a p53-dependent manner (1, 2). Both proteins bind to p53 via a hydrophobic N-terminal pocket and block p53-dependent transcription of genes required for tumor suppression. Efforts to reactivate p53 with small molecules have focused on inhibition of the mdm2/p53 interaction, which leads to increased p53 levels and activity. However, recent reports indicate that targetin
- HTS dose response assay for identification of inhibitors of TNFa-specific NF-kB induction Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Multiple cellular stimuli acting through various pathways lead to NF-kB induction. The assay described below uses tumor necrosis factor alpha (TNF-a), a canonical NF-kB inducer, and is designed for identification of hits specific to TNF-a-modulated pathways. We utilized this assay to assess selectivity of hits emerging from the primary screening of the library in NOD1- and NOD2-specific assays (AIDs 1578 and 1566). The HEK-293-T NF-kB-Luc cell line designed for luminescent detection of NF-kB induction was utilized in this assay. This assay is the dose response follow-up to "HTS assay for identification of inhibitors of TNF-a-specific NF-kB induc
- Dose Response confirmation of uHTS for the identification of UBC13 Polyubiquitin Inhibitors via a TR-FRET Assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R03 MH085677-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Tumor Necrosis Factor Receptor-Associated Factors (TRAFs) are a family of adapter proteins that bind an unusual ubiquitin-conjugating enzyme, Ubc13, which produces polyubiquitin chains linked at lysine 63 of ubiquitin. These lysine 63-linked ubiquitin polymers trigger changes in protein activity. Ubiquitination by Ubc13 of TRAFs and the various protein kinases to which TRAFs bind is recognized as a critical step in signaling by TNFRs, TLRs, NLRs, and T-cell and B-cell antigen receptors (TCR/BCR) during innate and acquired immune responses. Since aberrant signaling by these receptor systems is linked to a wide variety of autoimmune, inflammato
- Dose Response confirmation of uHTS for the identification of UBC13 Polyubiquitin Inhibitors via a TR-FRET Assay reconfirm Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R03 MH085677-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Tumor Necrosis Factor Receptor-Associated Factors (TRAFs) are a family of adapter proteins that bind an unusual ubiquitin-conjugating enzyme, Ubc13, which produces polyubiquitin chains linked at lysine 63 of ubiquitin. These lysine 63-linked ubiquitin polymers trigger changes in protein activity. Ubiquitination by Ubc13 of TRAFs and the various protein kinases to which TRAFs bind is recognized as a critical step in signaling by TNFRs, TLRs, NLRs, and T-cell and B-cell antigen receptors (TCR/BCR) during innate and acquired immune responses. Since aberrant signaling by these receptor systems is linked to a wide variety of autoimmune, inflammato
- Dose Response confirmation of uHTS hits from a small molecule inhibitors of human intestinal alkaline phosphatase via a luminescent assay - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological functio
- Dose Response confirmation of uHTS inhibitors of Human Intestinal Alkaline Phosphatase using Tissue Nonspecific Alkaline Phosphatase. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological functio
- Dose Response confirmation of uHTS inhibitors of Mouse Intestinal Alkaline Phosphatase using Human Intestinal Alkaline Phosphatase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Assay Provider Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute(SBMRI, San Diego, CA) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological functio
- Dose response counterscreen of uHTS hits for ATG4B inhibitors in a Phospholipase A2 assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH090871-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA This assay is a counterscreen for the primary screen (AID504462) which looked for inhibitors of Autophagin 1 (ATG4B). In screening for compounds that inhibit ATG4B, the High Throughput Screening (HTS) assay utilized a cleavable form of Phospholipase A2 (PLA2), which is expressed as a fusion protein with the Autophagin substrate LC3/ATG8 appended to its N-terminus, as the substrate for the primary enzymatic reaction. The addition of sequences to the N-terminus of PLA2 inhibits the activity of this enzyme. Cleavage by proteases removing the N-terminal extension then restores enzyme activity, constituting the basis for a protease assay. The sub
- Dose response counterscreen of uHTS hits for ATG4B inhibitors in a Phospholipase A2 assay Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH090871-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA This assay is a counterscreen for the primary screen (AID504462) which looked for inhibitors of Autophagin 1 (ATG4B). In screening for compounds that inhibit ATG4B, the High Throughput Screening (HTS) assay utilized a cleavable form of Phospholipase A2 (PLA2), which is expressed as a fusion protein with the Autophagin substrate LC3/ATG8 appended to its N-terminus, as the substrate for the primary enzymatic reaction. The addition of sequences to the N-terminus of PLA2 inhibits the activity of this enzyme. Cleavage by proteases removing the N-terminal extension then restores enzyme activity, constituting the basis for a protease assay. The sub
- SAR analysis of Antagonists of XIAP-Bir3 domain of IAP-family anti-apoptotic proteins - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network(MLPCN)Grant Proposal Number: MH081277-01 Assay Provider: John C. Reed, Sanford-Burnham Medical Research Institute San Diego, CA This dose response assay is developed and performed as a counter screen to compounds in the Chemical Antagonists of IAP-family anti-apoptotic proteins confirmation (AID 1449) and to study the structure-activity relationship on analogs of the confirmed hits. Compounds are either acquired from commercial sources or synthesized internally. This assay was performed in the assay providers' laboratory. Apoptosis plays an essential role in many aspects of normal development and physiology, becoming dysregulated in myriad diseases characterized by insufficient or excessive cell death. Caspases are intracellular proteases that are suppressed by Inhibit
- SAR analysis of Antagonists of XIAP-Bir3 domain of IAP-family anti-apoptotic proteins Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Proposal Number: MH081277-01 Assay Provider: John C. Reed, Sanford-Burnham Medical Research Institute, San Diego, CA This dose response assay is developed and performed as a counter screen to compounds in the Chemical Antagonists of IAP-family anti-apoptotic proteins confirmation (AID 1449) and to study the structure-activity relationship on analogs of the confirmed hits. Compounds are either acquired from commercial sources or synthesized internally. This assay was performed in the assay providers' laboratory. Apoptosis plays an essential role in many aspects of normal development and physiology, becoming dysregulated in myriad diseases characterized by insufficient or excessive cell death. Caspases are intracellular proteases that are suppressed by Inhibi
- SAR analysis of inhibitors of TNFa specific NF-kB induction - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Multiple cellular stimuli acting through various pathways lead to NF-kB induction. The assay described below uses tumor necrosis factor alpha (TNFa), a canonical NF-kB inducer, and is designed for identification of hits specific to TNFa-modulated pathways. We utilized this assay to assess selectivity of hits emerging from the primary screening of the library in NOD1- and NOD2-specific assays (AIDs 1578 and 1566). The HEK-293-T NF-kB-Luc cell line designed for luminescent detection of NF-kB induction was utilized in this assay. This dose response assay is developed and performed to confirm hits originally identified in "uHTS luminescence assay f
- SAR analysis of inhibitors of TNFa specific NF-kB induction - Set 3 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Multiple cellular stimuli acting through various pathways lead to NF-kB induction. The assay described below uses tumor necrosis factor alpha (TNFa), a canonical NF-kB inducer, and is designed for identification of hits specific to TNFa-modulated pathways. We utilized this assay to assess selectivity of hits emerging from the primary screening of the library in NOD1- and NOD2-specific assays (AIDs 1578 and 1566). The HEK-293-T NF-kB-Luc cell line designed for luminescent detection of NF-kB induction was utilized in this assay. This dose response assay is developed and performed to confirm hits originally identified in "uHTS luminescence a
- SAR analysis of inhibitors of TNFa specific NF-kB induction revised Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Multiple cellular stimuli acting through various pathways lead to NF-kB induction. The assay described below uses tumor necrosis factor alpha (TNFa), a canonical NF-kB inducer, and is designed for identification of hits specific to TNFa-modulated pathways. We utilized this assay to assess selectivity of hits emerging from the primary screening of the library in NOD1- and NOD2-specific assays (AIDs 1578 and 1566). The HEK-293-T NF-kB-Luc cell line designed for luminescent detection of NF-kB induction was utilized in this assay. This dose response assay is developed and performed to confirm hits originally identified in "uHTS luminescence assay f
- SAR assay for compounds inhibiting TNAP in the absence of phosphate acceptor performed in a luminescent assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Number: MH077602-01 Assay Provider: Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute, San Diego, CA This TNAP dose response assay is developed and performed for the purpose of SAR study on analogs of hits originally identified in the TNAP luminescent HTS assay (AID 518) Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing phosphate and alcohol. APs are dimeric enzymes found in the most organism. In human, four isozymes of APs have been identified. Three isozymes are tissue-specific and the fourth one is tissue-nonsepecifc, named TNAP. TNAP overexpression is associated with excessive calcification observed in different tissues. Therefore, there are therapeutic potentials of in
- uHTS identification of small molecule inhibitors of LYP via a fluorescence intensity assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1R21NS056945-01 Assay Provider: Dr. Nunzio Bottini, San Diego Institute for Allergy and Immunology CA LYP, is a lymphocyte specific protein tyrosine phosphatase that plays a critical regulatory role in T cell receptor signaling. The PTPN22 gene encodes this phosphatase. A single-nucleotide polymorphism in PTPN22 is associated with a number of autoimmune disorders, including type 1 diabetes, rheumatoid arthritis, juvenile rheumatoid arthritis, systemic lupus erythematosus and Grave's disease. The autoimmunity-predisposing allele is a gain-of-function mutant suggesting that a specific small-molecule inhibitor could eliminate its effect. Finding specific inhibitors of protein phosphatases has proven extremely difficult. The goal of this project is to f
- Chemical Antagonists IAP-family anti-apoptotic proteins Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Proposal Number: MH081277-01 Assay Provider: John C. Reed, Sanford-Burnham Medical Research Institute, San Diego, CA Apoptosis plays an essential role in many aspects of normal development and physiology, becoming dysregulated in myriad diseases characterized by insufficient or excessive cell death. Caspases are intracellular proteases that are suppressed by Inhibitor of Apoptosis Proteins (IAPs), a family of evolutionarily conserved anti-apoptotic proteins. Proteins released from mitochondria (SMAC and HtrA2) can competitively displace IAPs from the Caspases, thus helping to drive apoptosis. It has been shown that only a few residues at the N-terminus of activated SMAC protein (4-mer) are sufficient to affect the release of IAPs from Caspases. Thus, it is plausib
- Chemical Antagonists of IAP-family anti-apoptotic proteins confirmation Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Proposal Number: MH081277-01 Assay Provider: John C. Reed, Sanford-Burnham Medical Research Institute, San Diego, CA This XIAP dose response assay is developed and performed to confirm hits originally identified in the XIAP HTS binding assay (AID 1018) and to study the structure-activity relationship on analogs of the confirmed hits. Compounds are either acquired from commercial sources or synthesized internally. Apoptosis plays an essential role in many aspects of normal development and physiology, becoming dysregulated in myriad diseases characterized by insufficient or excessive cell death. Caspases are intracellular proteases that are suppressed by Inhibitor of Apoptosis Proteins (IAPs), a family of evolutionarily conserved anti-apoptotic proteins. Proteins
- SAR analysis of Antagonists of IAP-family anti-apoptotic proteins Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Proposal Number: MH081277-01 Assay Provider: John C. Reed, Sanford-Burnham Medical Research Institute, San Diego, CA This XIAP dose response assay is developed and performed to confirm hits originally identified in the XIAP HTS binding assay (AID 1018) and to study the structure-activity relationship on analogs of the confirmed hits. Compounds are either acquired from commercial sources or synthesized internally. The assay was performed in the assay providers' laboratory. Apoptosis plays an essential role in many aspects of normal development and physiology, becoming dysregulated in myriad diseases characterized by insufficient or excessive cell death. Caspases are intracellular proteases that are suppressed by Inhibitor of Apoptosis Proteins (IAPs), a fami
- Biological Activity Assay The KDM1A demethylase enzyme activity can obtained from mammalian cells or tissues expressing KDM1A from an endogenous or recombinant gene and purified or assayed from a whole cell extract. These methods can be used to determine the concentration of the disclosed compounds can inhibit fifty percent of the enzyme activity (IC50). In one aspect, the disclosed compounds exhibit inhibition fifty percent of the KDM1A enzyme activity at a concentration of less than 500 nM, less than 100 nM, less than 50 nM or less than 10 nM.
- In vitro LSD1 Inhibition Assay Briefly, the compounds in DMSO were added into the LSD1 in the reaction buffer consisting of 50 mM Tris HCl, pH 7.5, and 1% DMSO, using Acoustic Technology (Echo 550) in nanoliter range and incubated for 15 min at room temperature. The reactions were initiated by adding peptide substrate [10 µM Histone H3 (1 21) K4me2 peptide (AnaSpec, San Jose, CA, USA)] to the reaction mixtures and incubated for 60 min at room temperature. The reactions were terminated by the addition of the detection mixture consisting final concentration of 0.1 Unit/mL horseradish peroxidase (Sigma, San Jose, CA, USA) and 10 µM Amplex UltraRed (Life Technologies, Grand Island, NY, USA) and fluorescence at Ex/Em = 535/590 nm was measured with kinetic mode of 5 min interval in envision for 30 min.
- Enzymatic Assay The aim of this in vitro assay was to measure the inhibition of HCV NS3/4A protease complexes by the compounds of the present invention. This assay provides an indication of how effective compounds of the present invention would be in inhibiting HCV NS3/4A proteolytic activity. The inhibition of full-length hepatitis C NS3 protease enzyme was measured essentially as described in Poliakov, 2002 Prot Expression & Purification 25 363 371. Briefly, the hydrolysis of a depsipeptide substrate, Ac-DED(Edans)EEAbu [COO]ASK(Dabcyl)-NH2 (AnaSpec, San Jose, USA), was measured spectrofluorometrically in the presence of a peptide cofactor, KKGSVVIVGRIVLSGK ( ke Engstrom, Department of Medical Biochemistry and Microbiology, Uppsala University, Sweden). [Landro, 1997 #Biochem 36 9340-9348].
- Electrophoretic Mobility Shift Assay (EMSA) STAT proteins were expressed in Sf9 cells from a baculovirus encoding the recombinant protein. A nuclear extract of the Sf9 cells was incubated with 32P-labeled high affinity c-fos sis inducible element (hSIE) either alone or in the presence of inhibitor. After incubation, samples were electrophoresed on polyacrylamide gels. The gels were dried, exposed to a phosphorimager screen and scanned. IC50 values were derived from plots of spot intensity versus phosphopeptide concentration. The affinity of peptides to the SH2 domain is measured by the intensity of the radioactivity of the Stat-DNA complex band in the electrophoresis gel.
- PDE3B Enzyme Inhibition Assay The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of example compounds on the PDE3B reactions 2 μl of the respective example compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3B cell extract from Sf9 cells overexpressing human full length PDE3B (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3B cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:6000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H]adenosine 3′, 5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3B activity vs log compound concentration.
- Confirmation of compounds inhibiting phosphomannose isomerase (PMI) via a fluorescence intensity assay using a high concentration of mannose 6-phosphate. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Number: R03 MH082386-01 Assay Provider: Dr. Hudson H. Freeze, Sanford-Burnham Medical Research Institute, San Diego, CA Congenital Disorders of Glycosylation (CDG) are autosomal recessive defects in the synthesis of N-linked oligosaccharide chains. CDG group I (CDG-I) defects are defined as those caused by mutations in genes encoding enzymes used for the synthesis and transfer of lipid linked oligosaccharide (LLO) to newly synthesized proteins in the lumen of the ER. The steps in this pathway and the genes encoding them are very similar from yeast to human. It requires 30-40 single gene products, each dependent on the previous step in the linear sequence to produce and transfer the LLO to protein. Therefore, mutations in any step may cause a type of CDG. There is
- HTS fluorescence polarization-based dose response confirmatory screen for the Siah-1 primary assay utilizing an alternative fluorophore, fluorescein-labeled plectin Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH086475-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Proteasomal degradation typically requires post-translational modification of target proteins with K48-linked polyubiquitin chains. This process of protein proteolysis plays a key role in normal cellular function. The E3 ubiquitin ligase, Siah-1, facilitates the transfer of ubiquitin to its substrate proteins destined for degradation by way of its RING domain. Siah-1 is a member of a family of highly conserved RING domain proteins, which regulate a variety of cellular functions, including cell cycle arrest, tumor suppression, and apoptosis through the beta-catenin degradation pathway. Siah-1 has also been identified as a p53-inducible gene,
- In Vitro Inhibition Assay This work was performed at the MDS Pharma Services, Pharmacology Laboratories, Taiwan. The assay was an in vitro evaluation of the ability of an extract or a pure compound to inhibit the steroid 5alpha -reductase enzyme from metabolizing testosterone into dihydrotestosterone. This is an enzyme-immunoassay (EIA) for quantitative determination of testosterone in human serum or plasma. The significance of this type of inhibition is that it can lead to eradication of benign prostatic hyperplasia (BPH). Two distinct isozymes are found in mice, rats, monkeys and humans: type 1 and II. Each of these isozymes is differentially expressed in tissues and developmental stages. In human, type 1 steroid 5alpha -reductase is predominant in the sebaceous glands of most regions of skin, including scalp and liver and is responsible for approximately one third of circulating DHT. Inhibitors of steroid 5alpha -reductase may be of benefit in the treatment of androgenetic alopecia.
- Radioligand Labeled Binding Assay Log IC50 values for each test compound were determined from nonlinear regression analysis of data collected from two independent experiments performed in duplicates (40 independent experimental values) using GraphPad Prizm 4 software (GraphPad, San Diego, California). The inhibition constant (Ki) was calculated from the antilogarithmic IC50 value by the Cheng and Prusoff equation.
- Determination of kinetic parameters and the Ki of effective compounds using the FRET peptide Kinetic parameters were obtained using various concentrations of FRET peptide in the fluorescent assay. The maximal velocity (Vmax) and Michaelis Menten constant (Km) were calculated from the Eadie Hofstee plot. If the type of inhibitionwas found to be competitive using a Lineweaver Burk double reciprocal plot, then the inhibitory constant (Ki) for 3CLpro was estimated using the equation:Ki = IC50/(1+[substrate]/Km).Plots were performed, and kinetic parameters were calculated, using Prism software (Graphpad Software, San Diego, CA).
- HBSAg ELISA assay HepG2-Clone42 cells were seeded in into black clear-bottom 96-well plates at a concentration of 6.0×104 cells/well. 24 hours post-seeding, the cells were treated with 200 μl/well of media containing five-fold serial dilutions of compounds in DMSO. DMSO alone was used as the no drug control. The final DMSO concentration in all wells was 0.5%. The HBsAg ELISA kit (Alpha Diagnostic International, San Antonio, Tex., USE, Catalog #4110) was used to determine the level (semi-quantitative) of secreted HBV sAg.
- Counter screen SAR assay for PMM2 inhibitors via a fluorescence intensity assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Probe Production Centers Network (MLPCN) Grant Number: R03 MH082386-01 Assay Provider: Dr. Hudson H. Freeze, Sanford-Burnham Medical Research Institute, San Diego, CA Congenital Disorders of Glycosylation (CDGs) are rare genetic disorders in the synthesis of N-linked glycan chains. Mutations in PMM2, encoding phosphomannomutase 2 (PMM2, Man-6-P-> Man-1-P) cause the most common form, CDG-Ia. Patients have a host of problems including hypotonia, variable psychomotor retardation, seizures, peripheral neuropathy, cardiomyopathy, and protein losing enteropathy. There is no therapy for this disorder. A current approach to ameliorate the physiological conditions associated with CDG-Ia is to provide high influx of mannose for patience. We previously developed a HTS assay through the MLSCN to identify inhibitors of phos
- Dose Response confirmation of UBC13 Polyubiquitin Inhibitors using a Bfl-1 counterscreen Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1R03 MH085677-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute, San Diego CA Tumor Necrosis Factor Receptor-Associated Factors (TRAFs) are a family of adapter proteins that bind an unusual ubiquitin-conjugating enzyme, Ubc13, which produces polyubiquitin chains linked at lysine 63 of ubiquitin. These lysine 63-linked ubiquitin polymers trigger changes in protein activity. Ubiquitination by Ubc13 of TRAFs and the various protein kinases to which TRAFs bind is recognized as a critical step in signaling by TNFRs, TLRs, NLRs, and T-cell and B-cell antigen receptors (TCR/BCR) during innate and acquired immune responses. Since aberrant signaling by these receptor systems is linked to a wide variety of autoimmune, inflammato
- Fluorescence polarization-based biochemical high throughput dose response assay for activators of the Protein Kinase A-R1A (PKA-R1A) complex Source (MLPCN Center Name): The Scripps Research Institute Molecular Screening Center (SRIMSC) Affiliation: The Scripps Research Institute, TSRI Assay Provider: Susan Taylor, University of California, San Diego (UCSD) Network: Molecular Library Probe Production Centers Network (MLPCN) Grant Proposal Number: R01 GM34921 Grant Proposal PI: Susan Taylor, University of California, San Diego (UCSD) External Assay ID: PKAR1A_ACT_FP_1536_3XEC50 DRUN Name: Fluorescence polarization-based biochemical high throughput dose response assay for activators of the Protein Kinase A-R1A (PKA-R1A) complex. Description: Protein phosphorylation is one of the most important mechanisms for regulation in mammalian cells, and cAMP is an ancient second messenger signaling molecule. Protein kinase A (PKA) is a ubiquitous cAMP-dependent serine/threonine protein kinase that phosphorylates intracellular protein substrates in response to the secondary messenger, adenosine 3',5'-cyclic monophosphate (cAMP
- Fluorescence polarization-based biochemical high throughput dose response assay for activators of the Protein Kinase A-R2B (PKA-R2B) complex Source (MLPCN Center Name): The Scripps Research Institute Molecular Screening Center (SRIMSC) Affiliation: The Scripps Research Institute, TSRI Assay Provider: Susan Taylor, University of California, San Diego (UCSD) Network: Molecular Library Probe Production Centers Network (MLPCN) Grant Proposal Number: R01 GM34921 Grant Proposal PI: Susan Taylor, University of California, San Diego (UCSD) External Assay ID: PKAR2B_ACT_FP_1536_3XEC50 DRUN Name: Fluorescence polarization-based biochemical high throughput dose response assay for activators of the Protein Kinase A-R2B (PKA-R2B) complex. Description: Protein phosphorylation is one of the most important mechanisms for regulation in mammalian cells, and cAMP is an ancient second messenger signaling molecule. Protein kinase A (PKA) is a ubiquitous cAMP-dependent serine/threonine protein kinase that phosphorylates intracellular protein substrates in response to the secondary messenger, adenosine 3',5'-cyclic monophosphate (cAMP
- IMAP TR-FRET Assay Enzyme Activity. An IMAP TR-FRET assay was used to analyze the enzyme activity (Molecular Devices Corp., Sunnyvale Calif.). 5 uL, of serial diluted PDE10A (BPS Bioscience, San Diego, Calif.) or tissue homogenate was incubated with equal volumes of diluted fluorescein labeled cAMP or cGMP for 60 min in 384-well polystyrene assay plates (Corning, Corning, N.Y.) at room temperature. After incubation, the reaction was stopped by adding 60 uL, of diluted binding reagents and was incubated for 3 hours to overnight at room temperature. The plates were read on an Envision (Perkin Elmer, Waltham, Mass.) for time resolved fluorescence resonance energy transfer. The data were analyzed with GraphPad Prism (La Jolla, Calif.).Enzyme Inhibition. To check the inhibition profile, 5 uL, of serial diluted compounds were incubated with 5 uL, of diluted PDE10 enzyme (BPS Bioscience, San Diego, Calif.) or tissue homogenate in a 384-well polystyrene assay plate (Corning, Corning, N.Y.).
- IMAP TR-FRET assay Enzyme Activity. An IMAP TR-FRET assay was used to analyze the enzyme activity (Molecular Devices Corp., Sunnyvale Calif.). 5 uL of serial diluted PDE10A (BPS Bioscience, San Diego, Calif.) or tissue homogenate was incubated with equal volumes of diluted fluorescein labeled cAMP or cGMP for 60 min in 384-well polystyrene assay plates (Corning, Corning, N.Y.) at room temperature. After incubation, the reaction was stopped by adding 60 uL of diluted binding reagents and was incubated for 3 hours to overnight at room temperature. The plates were read on an Envision (Perkin Elmer, Waltham, Mass.) for time resolved fluorescence resonance energy transfer. The data were analyzed with GraphPad Prism (La Jolla, Calif.).Enzyme Inhibition. To check the inhibition profile, 5 uL of serial diluted compounds were incubated with 5 uL of diluted PDE10 enzyme (BPS Bioscience, San Diego, Calif.) or tissue homogenate in a 384-well polystyrene assay plate (Corning, Corning, N.Y.).
- Luminescent assay for HTS discovery of chemical inhibitors of placental alkaline phosphatase confirmation Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Proposal Number: MH077602-01 Assay Provider: Dr. Jose Luis Millan, Sanford-Burnham Medical Research Institute, San Diego, CA. This PLAP dose response assay is developed and performed to confirm hits originally identified in the PLAP Luminescent HTS assay (AID 690) and to study the structure-activity relationship on analogs of the confirmed hits. Compounds are either acquired from commercial sources or synthesized internally. Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified: three isozymes are tissue-specific and the fourth one is tissue-nonspecific. Placental alkaline phosphatase (PLAP) is high
- SAR analysis of Antagonists of IAP-family anti-apoptotic proteins - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network(MLPCN)Grant Proposal Number: MH081277-01 Assay Provider: John C. Reed, Sanford-Burnham Medical Research Institute, San Diego, CA This XIAP dose response assay is developed and performed to confirm hits originally identified in the XIAP HTS binding assay (AID 1018) and to study the structure-activity relationship on analogs of the confirmed hits. Compounds are either acquired from commercial sources or synthesized internally. The assay was performed in the assay providers' laboratory. Apoptosis plays an essential role in many aspects of normal development and physiology, becoming dysregulated in myriad diseases characterized by insufficient or excessive cell death. Caspases are intracellular proteases that are suppressed by Inhibitor of Apoptosis Proteins (IAPs), a famil
- Biochemical Assay A 500 ml stock volume of 5× Reaction Kinase Buffer was made by mixing 1000 μl of 1M MgCl2, 500 μl of 1M Tris-HCL pH7.4, 0.5 mg/ml (25 mg) of BSA, and 3475 μl of distilled H2O. A 3 ml 2× working stock volume of Reaction Kinase Buffer was made containing a final concentration of 100 μM DTT and 4 mM MnCl2.Components of RIPK1 enzyme (Rigel Pharmaceuticals, South San Francisco, Calif., USA) were thawed on ice. Diluted RIPK1 was prepared in 1× Kinase Reaction Buffer (diluted from 2× buffer) to 31 ng/well. A 166 M working stock ATP assay solution was prepared in 1× Kinase Reaction Buffer (diluted from 2× buffer).
- Confirmation of compounds inhibiting phosphomannose isomerase (PMI) via a fluorescence intensity assay. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Number: R03 MH082386-01 Assay Provider: Dr. Hudson H. Freeze, Sanford-Burnham Medical Research Institute, San Diego, CA Congenital Disorders of Glycosylation (CDG) are autosomal recessive defects in the synthesis of N-linked oligosaccharide chains. CDG group I (CDG-I) defects are defined as those caused by mutations in genes encoding enzymes used for the synthesis and transfer of lipid linked oligosaccharide (LLO) to newly synthesized proteins in the lumen of the ER. The steps in this pathway and the genes encoding them are very similar from yeast to human. It requires 30-40 single gene products, each dependent on the previous step in the linear sequence to produce and transfer the LLO to protein. Therefore, mutations in any step may cause a type of CDG. There is
- HTS identification of compounds inhibiting phosphomannose isomerase (PMI) via a fluorescence intensity assay. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Number: R03 MH082386-01 Assay Provider: Dr. Hudson H. Freeze, Sanford-Burnham Medical Research Institute, San Diego, CA Congenital Disorders of Glycosylation (CDG) are autosomal recessive defects in the synthesis of N-linked oligosaccharide chains. CDG group I (CDG-I) defects are defined as those caused by mutations in genes encoding enzymes used for the synthesis and transfer of lipid linked oligosaccharide (LLO) to newly synthesized proteins in the lumen of the ER. The steps in this pathway and the genes encoding them are very similar from yeast to human. It requires 30-40 single gene products, each dependent on the previous step in the linear sequence to produce and transfer the LLO to protein. Therefore, mutations in any step may cause a type of CDG. There is
- Dose Response confirmation of activators of hexokinase domain containing I (HKDC1) in the hexokinase 1 selectivity assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Proposal Number: R21 NS061703-01S1 Assay Provider: Dr. Jeff Johnson, Metabolex, Inc., Hayward, CA. Since hexokinases (HKs) are the first step in glucose metabolism, they play a major role in regulating the metabolic fate of glucose in the tissues they are expressed. Understanding both the proper and pathological metabolism of glucose is obviously of critical importance to deciphering the dysregulation of glucose that leads to Type 2 diabetes, a disorder of metabolism that morbidly afflicts 35 million Americans and perhaps close to 200 million worldwide. One member of HK enzyme family, Hexokinase IV or glucokinase, has been the subject of intense pharmaceutical development of small molecule activators for treatment of Type 2 diabetes. The other members of
- FASN Inhibition FASN activity of the SKBr3 cell extract was determined by measuring either NADPH oxidation or the amount of thiol-containing coenzyme A (CoA) released during the fatty acid synthase reaction. The dye CPM (7-diethylamino-3-(4′-maleimidyl-phenyl)-4-methylcoumarin) contains a thiol reactive group that increases its fluorescence emission on reaction with the sulfhydryl group of CoA. The biochemical activities shown in TABLE 31 were determined using the fluorescence measurement of CoA release via a procedure described in Chung C. C. et al. (Assay and Drug Development Technologies, 2008, 6(3), 361-374).
- PDE3A Enzyme Inhibition The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of example compounds on the PDE3A reactions 2 μl of the respective example compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3A cell extract from Sf9 cells overexpressing human full length PDE3A (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3A cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:5000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H]adenosine 3′, 5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3A activity vs log compound concentration.
- PDE3A Enzyme Inhibition The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of test substances on the PDE3A reactions 2 μl of the respective test compound solution in DMSO (serial dilutions) were is placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3A cell extract from Sf9 cells overexpressing human full length PDE3A (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3A cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:5000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H] adenosine 3′,5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3A activity vs log compound concentration.
- PDE3B Enzyme Inhibition The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of example compounds on the PDE3B reactions 2 μl of the respective example compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3B cell extract from Sf9 cells overexpressing human full length PDE3B (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3B cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:6000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H]adenosine 3′, 5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3B activity vs log compound concentration.
- PDE3B Enzyme Inhibition The commercially available 3H-cAMP Scintillation Proximity Assay (SPA, Perkin Elmer) system was used for enzyme inhibition studies. For the determination of the in vitro effect of test substances on the PDE3B reactions 2 μl of the respective test compound solution in DMSO (serial dilutions) were placed in wells of microtiter plates (Isoplate-96/200W; Perkin Elmer). 50 μl of a dilution of PDE3B cell extract from Sf9 cells overexpressing human full length PDE3B (SB Drug Discovery, UK) in buffer A (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA) was added. The dilution of the PDE3B cell extract was chosen such that the reaction kinetics was linear and less than 70% of the substrate was consumed (typical dilution 1:6000). The reaction was started by addition of 50 μl (0.025 μCi) of 1:2000 in buffer A w/o BSA diluted substrate [8-3H] adenosine 3′,5′-cyclic phosphate (1 μCi/μl; Perkin Elmer). After incubation at room temperature for 60 min, the reaction was stopped by addition of 25 μl of a suspension containing 18 mg/ml yttrium scintillation proximity beads (Perkin Elmer) in water. The microtiter plates were sealed and measured in a Microbeta scintillation counter (PerkinElmer Wallac). IC50 values were determined from sigmoidal curves by plotting percentage PDE3B activity vs log compound concentration.
- CB1 and CB2 Binding Inhibition by Compounds Purified from Milicia excelsa (African Teak) The organic extract (8 g) from the stem barks of Milicia excelsa, obtained using the methods described in Example 1, was divided and loaded separately onto two pre-packed flash columns (120 g silica, particle size 32-60 μm, 4 cm×19 cm), then the column was eluted with the gradient as described in Example 5. A Diels-Alder adduct of a chalcone and prenylphenyl moiety was isolated from one of the active fractions and identified as Sanggenon C/D/.
- Enzyme Inhibition of Purified HIV-1 Protease Protease inhibitors (PI) are used to treat HIV infection by preventing viral assembly and maturation through the inhibition of HIV-1 protease activity. Biochemical assays were performed to evaluate the potential of PEG-PI conjugates to inhibit HIV-1 protease activity, relative to their respective PI parent molecules. Activity assays were performed at using the SensoLyte 520 HIV-Protease Assay Kit (Anaspec Inc., San Jose, Calif.) and recombinant HIV-1 protease. Protease activity was monitored by the formation of a fluorescent reporter product generated during HIV-1 protease-mediated digestion of a quenched, fluorimetric substrate containing the p17/p24 Prgag cleavage site.
- Flow Cytometric Assay The compounds were tested to determine their ENT1 nucleoside transporter binding ability by a flow cytometric assay using human leukemia K562 cells incubated with fluorescent probe in the presence or absence of varying concentrations of test compounds. Flow cytometric measurements of cell-associated fluorescence were performed with a FACSCalibur instrument (Becton Dickinson, San Jose, CA). Percentage (%) of control (ENT1 transporter specific fluorescence in the presence of SAENTA-fluorescein without test compounds) was calculated for each sample. The results were fed into the GraphPad PRISM to derive concentration-dependent curves. From these curves, the IC50 values were obtained and used to calculate inhibition constants (Ki).
- Dose Response Confirmation of compounds that inhibit VHR1 in Fluorescent Assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084230-01A1 Assay Provider: Dr. Lutz Tautz, Source Affiliation: Sanford-Burnham Medical Research Institute (San Diego, CA) Protein tyrosine phosphatases (PTPs) play vital roles in numerous cellular processes and are implicated in a growing number of human diseases, ranging from cancer to cardiovascular, immunological, infectious, neurological, and metabolic diseases. The Vaccinia H1-related (VHR) PTP is a dual-specific Erk and Jnk phosphatase, the loss of which causes specific cell cycle arrest in HeLa carcinoma cells, suggesting that VHR inhibition may be a useful approach to halt the growth of cancer cells without detrimental effects on normal cells. Recent studies by collaborators and us suggest that VHR is upregulated in several cervix
- Fluorescent assay for identification of compounds that inhibit VHR1 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084230-01A1 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Protein tyrosine phosphatases (PTPs) play vital roles in numerous cellular processes and are implicated in a growing number of human diseases, ranging from cancer to cardiovascular, immunological, infectious, neurological, and metabolic diseases. The Vaccinia H1-related (VHR) PTP is a dual-specific Erk and Jnk phosphatase, the loss of which causes specific cell cycle arrest in HeLa carcinoma cells, suggesting that VHR inhibition may be a useful approach to halt the growth of cancer cells without detrimental effects on normal cells. Recent studies by collaborators and us suggest that VHR is upregulated in several cervix cancer cell lines, in
- MOA VHR1 Fluorescent secondary assay for identification of redox-state modulating compounds Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084230-01A1 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Protein tyrosine phosphatases (PTPs) play vital roles in numerous cellular processes and are implicated in a growing number of human diseases, ranging from cancer to cardiovascular, immunological, infectious, neurological, and metabolic diseases. The Vaccinia H1-related (VHR) PTP is a dual-specific Erk and Jnk phosphatase, the loss of which causes specific cell cycle arrest in HeLa carcinoma cells, suggesting that VHR inhibition may be a useful approach to halt the growth of cancer cells without detrimental effects on normal cells. Recent studies by collaborators and us suggest that VHR is upregulated in several cervix cancer cell lines, in
- SAR analysis of compounds that inhibit VHR1, Fluorescent Assay - Set 2 Data Source: Burnham Center for Chemical Genomics (BCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084230-01A1 Assay Provider: Dr. Lutz Tautz, Source Affiliation: Sanford-Burnham Medical Research Institute (San Diego, CA) Protein tyrosine phosphatases (PTPs) play vital roles in numerous cellular processes and are implicated in a growing number of human diseases, ranging from cancer to cardiovascular, immunological, infectious, neurological, and metabolic diseases. The Vaccinia H1-related (VHR) PTP is a dual-specific Erk and Jnk phosphatase, the loss of which causes specific cell cycle arrest in HeLa carcinoma cells, suggesting that VHR inhibition may be a useful approach to halt the growth of cancer cells without detrimental effects on normal cells. Recent studies by collaborators and us suggest that VHR is upregulated in several cervix cancer ce
- SAR analysis of compounds that potentiate TRAIL-induced apoptosis in MDA-MB-435 cells. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: X01 MH083230-01 Assay Provider: Dr. Dmitri Rozanov, Sanford-Burnham Medical Research Institute, San Diego CA This assay was developed and performed to confirm hits originally identified in "uHTS for the identification of compounds that potentiate TRAIL-induced apoptosis of cancer cells" (AID 1443) and to study the structure-activity relationship on analogs of the confirmed hits. Compounds are either acquired from commercial sources or synthesized internally. The TRAIL-resistant cell line, MDA-MB-435 is used, because we would like to determine if compounds can potentiate TRAIL-mediated cytotoxicity not only in TRAIL-sensitive PPC-1 carcinoma cells(AIDs 1443 and 1624) but also in TRAIL-resistant cells. Cytotoxic chemotherapy induces apoptosis via a pat
- uHTS absorbance assay for the identification of compounds that inhibit VHR1. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084230-01A1 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Protein tyrosine phosphatases (PTPs) play vital roles in numerous cellular processes and are implicated in a growing number of human diseases, ranging from cancer to cardiovascular, immunological, infectious, neurological, and metabolic diseases. The Vaccinia H1-related (VHR) PTP is a dual-specific Erk and Jnk phosphatase, the loss of which causes specific cell cycle arrest in HeLa carcinoma cells, suggesting that VHR inhibition may be a useful approach to halt the growth of cancer cells without detrimental effects on normal cells. Recent studies by collaborators and us suggest that VHR is upregulated in several cervix cancer cell lines, in
- ChEMBL_440740 (CHEMBL888711) Inhibition of human LBD of of ERalpha
- Dose Response confirmation of uHTS hits for Scp-1 phosphatase using a colorimetric assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 DA030556-01A1 Assay Provider: Dr. Yan Jessie Zhang, University of Texas, Austin, TX Human Scp phosphatases are regulators involved in neuronal gene silencing. This family of phosphatases exhibits specificity for phosphoryl-Ser5 in the heptad repeats of the C-terminal domain of RNA polymerase II (RNA Pol II) and thus inhibit initiation of transcription [1, 2]. Scps strongly associate with the REST/NRSF neuronal silencing complex that functions to inhibit transcription of neuronal genes in neuronal stem cells and in nonneuronal tissues [1]. Inhibition of Scps by dominant negative forms of the enzyme or by premature expression of the nervous system-specific microRNA miR-124, results in differentiation of neuronal stem cells into mature neurons [
- GO Biochemical Assay GO biochemical enzymatic reaction was performed in black 384-well low binding plate in a total volume of 25 μL. The reaction mixture contained 5 nM GO, 100 μM glycolate, 0.1 U/mL HRP, 50 μM Amplex Red, and 1:3 serial diluted test compounds in a buffer containing 50 mM Tris pH 7.8, 0.0025% Tween-20, and 0.02% BSA. Twenty five nanoliter of 1000× test compounds were pre-spotted onto 384-well low binding plate by Echo 555 Liquid Handler (Labcyte Inc., San Jose, CA) with starting final concentration of 10 μM, followed by addition of 5 μL/well of 25 nM GO (5× of 5 nM final concentration) and incubated for 15 min. Ten microliter of 2.5× of 0.1 U/mL final concentration HRP was added to each well, followed by addition of 10 μL 2.5× 100 μM final concentration of glycolate substrate and of 2.5× 50 μM final concentration of Amplex Red. The reaction was mixed and incubated at room temperature for 20 min followed by reading the plates by EnVision plate reader (Perkin Elmer, San Jose, CA) with excitation at 570 nm and emission 585 nm. The wells with DMSO were used as negative controls (as 0% inhibition) whereas wells without GO enzymes were used as positive controls (as 100% inhibition). The % inhibition as calculated as 100%×(Well-Negative)/(Positive-Negative
- Omnia Assay Briefly, 10x stocks of EGFR-WT (PV3872) from Invitrogen and EGFR-T790M/L858R (40350) from BPS Bioscience, San Diego, Calif., 1.13xATP (AS001A) and appropriate Tyr-Sox conjugated peptide substrates (KCZ1001) were prepared in 1x kinase reaction buffer consisting of 20 mM Tris, pH 7.5, 5 mM MgCl2, 1 mM EGTA, 5 mM beta-glycerophosphate, 5% glycerol (10x stock, KB002A) and 0.2 mM DTT (DS001A). 5 uL of each enzyme were pre-incubated in a Corning (#3574) 384-well, white, non-binding surface microtiter plate (Corning, N.Y.) for 30 min. at 25C. with a 0.5 L volume of 50% DMSO and serially diluted compounds prepared in 50% DMSO. Kinase reactions were started with the addition of 45 uL of the ATP/Tyr-Sox peptide substrate mix and monitored every 71 seconds for 60 minutes at lamda ex360/lamda em485 in a Synergy4 plate reader from BioTek (Winooski, Vt.). At the conclusion of each assay, progress curves from each well were examined for linear reaction kinetics and fit statistics (R2, 95% confidence interval, absolute sum of squares). Initial velocity (0 minutes to 30 minutes) from each reaction was determined from the slope of a plot of relative fluorescence units vs time (minutes) and then plotted against inhibitor concentration to estimate IC50 from log [Inhibitor] vs Response, Variable Slope model in GraphPad Prism from GraphPad Software (San Diego, Calif.).
- RORgamma Gal4 Reporter Gene Assay (FF) Cells were incubated for additional 16 h before firefly (FF) luciferase activities were measured sequentially in the same cell extract using a Dual-Light-Luciferase-Assay system (Dyer et al., Anal. Biochem. 2000, 282:158). All experiments were done at least in triplicates.
- RORgamma Gal4 Reporter Gene Assay (REN) Cells were incubated for additional 16 h before renilla (REN) luciferase activities were measured sequentially in the same cell extract using a Dual-Light-Luciferase-Assay system (Dyer et al., Anal. Biochem. 2000, 282:158). All experiments were done at least in triplicates.
- In vitro HDACs Inhibition Fluorescence Assay In brief, 10 μL of HeLa nuclear extract was mixed with various concentrations of target compounds (50 μL), SAHA, using 100% and none HDACs groups as control group, and the mixture. After incubation at 37 °C for 10 min, fluorogenic substrate Boc-Lys (acetyl)-AMC (40 μL) was added and then the mixture was incubated at 37 °C for 30 min. The mixture was stopped by addition of 100 μL of developer containing trypsin and TSA afterward. Over the next incubation at 37 °C for 20 min, fluorescence intensity was measured using a microplate reader at excitation and emission wavelengths of 390 and 460 nm, respectively.
- Pharmacological Assay For TLR8 and TLR7 activity testing, HEK-Blue human TLR8 or TLR7 cells (Invivogen, San Diego, Calif., USA) are used, respectively. These cells are designed for studying the stimulation of human TLR8 or TLR7 by monitoring the activation of NF-κB. A SEAP (secreted embryonic alkaline phosphatase) reporter gene is placed under the control of the IFN-b minimal promoter fused to five NF-κB and AP-1-binding sites. Therefore the reporter expression is regulated by the NF-κB promoter upon stimulation of human TLR8 or TLR7 for 20 hours. The cell culture supernatant SEAP reporter activity was determined using Quanti Blue kit (Invivogen, San Diego, Calif., USA) at a wavelength of 640 nm, a detection medium that turns purple/blue in the presence of alkaline phosphatase. EC50 values were determined using Activity Base analysis (ID Business Solution, Limited).The compounds according to formula I have an activity (EC50 value) in the above assay for human TLR8 in the range of 0.01 nM to 0.05 μM, more particularly of 0.001 nM to 0.03 μM, whereas the activity (EC50 value) in the above assay for human TLR7 is greater than 10 μM, in the range of 12 μM to >100 μM, meaning the compounds show high selectivity towards human TLR8.
- Radioligand Labeled Binding Assay and Phosphatidylinositol Hydrolysis Assay for the CCK Receptor Log IC50 values for each test compound were determined from nonlinear regression analysis of data collected from two independent experiments performed in duplicates (40 independent experimental values) using GraphPad Prizm 4 software (GraphPad, San Diego, California). The inhibition constant (Ki) was calculated from the antilogarithmic IC50 value by the Cheng and Prusoff equation. EC50/IC50 values were obtained from measuring the effect of test compounds on agonist-induced phosphatidylinositol hydrolysis.
- MPDE10A7 Enzyme Activity An IMAP TR-FRET assay was used to analyze the enzyme activity (Molecular Devices Corp., Sunnyvale Calif.). 5 L of serial diluted PDE10A (BPS Bioscience, San Diego, Calif.) or tissue homogenate was incubated with equal volumes of diluted fluorescein labeled cAMP or cGMP for 60 min in 384-well polystyrene assay plates (Corning, Corning, N.Y.) at room temperature. After incubation, the reaction was stopped by adding 60 L of diluted binding reagents and was incubated for 3 hours to overnight at room temperature. The plates were read on an Envision (Perkin Elmer, Waltham, Mass.) for time resolved fluorescence resonance energy transfer. The data were analyzed with GraphPad Prism (La Jolla, Calif.). Enzyme Inhibition. To check the inhibition profile, 5 μL of serial diluted compounds were incubated with 5 μL of diluted PDE10 enzyme (BPS Bioscience, San Diego, Calif.) or tissue homogenate in a 384-well polystyrene assay plate (Corning, Corning, N.Y.) for 30 min at room temperature. After incubation, 10 L of diluted fluorescein labeled cAMP or cGMP substrate were added and incubated for 60 min at room temperature. The reaction was stopped by adding 60 μL of diluted binding reagents and plates were read on an Envision (Perkin Elmer, Waltham, Mass.) for time resolved fluorescence resonance energy transfer.
- ChEBML_209063 Inhibition of amidolytic activity of thrombin
- ChEBML_211872 Inhibition of the polymerization of tubulin
- ChEMBL_333145 (CHEMBL865913) Inhibition of reuptake of 5HT
- ChEMBL_333146 (CHEMBL865923) Inhibition of reuptake of Norepinephrine
- Omnia Assay Briefly, 10x stocks of EGFR-WT (PV3872) from Invitrogen and EGFR-T790M/L858R (40350) from BPS Bioscience, San Diego, Calif., 1.13×ATP (AS001A) and appropriate Tyr-Sox conjugated peptide substrates (KCZ1001) were prepared in 1× kinase reaction buffer consisting of 20 mM Tris, pH 7.5, 5 mM MgCl2, 1 mM EGTA, 5 mM β-glycerophosphate, 5% glycerol (10× stock, KB002A) and 0.2 mM DTT (DS001A). 5 μL of each enzyme were pre-incubated in a Corning (#3574) 384-well, white, non-binding surface microtiter plate (Corning, N.Y.) for 30 min. at 27° C. with a 0.5 μL volume of 50% DMSO and serially diluted compounds prepared in 50% DMSO. Kinase reactions were started with the addition of 45 μL of the ATP/Tyr-Sox peptide substrate mix and monitored every 30-90 seconds for 60 minutes at λex360/λem485 in a Synergy4 plate reader from BioTek (Winooski, Vt.). At the conclusion of each assay, progress curves from each well were examined for linear reaction kinetics and fit statistics (R2, 95% confidence interval, absolute sum of squares). Initial velocity (0 minutes to 30 minutes) from each reaction was determined from the slope of a plot of relative fluorescence units vs time (minutes) and then plotted against inhibitor concentration to estimate IC50 from log[Inhibitor] vs Response, Variable Slope model in GraphPad Prism from GraphPad Software (San Diego, Calif.).
- Omnia Assay Protocol for Potency Assessment Against BTK Briefly, 10× stocks of EGFR-WT (PV3872) from Invitrogen and EGFR-T790M/L858R (40350) from BPS Bioscience, San Diego, Calif., 1.13×ATP (AS001A) and appropriate Tyr-Sox conjugated peptide substrates (KCZ1001) were prepared in 1× kinase reaction buffer consisting of 20 mM Tris, pH 7.5, 5 mM MgCl2, 1 mM EGTA, 5 mM 3-glycerophosphate, 5% glycerol (10× stock, KB002A) and 0.2 mM DTT (DS001A). 5 μL of each enzyme were pre-incubated in a Corning (#3574) 384-well, white, non-binding surface microtiter plate (Corning, N.Y.) for 30 min. at 27° C. with a 0.5 μL volume of 50% DMSO and serially diluted compounds prepared in 50% DMSO. Kinase reactions were started with the addition of 45 μL of the ATP/Tyr-Sox peptide substrate mix and monitored every 30-90 seconds for 60 minutes at λex360/λem485 in a Synergy4 plate reader from BioTek (Winooski, Vt.). At the conclusion of each assay, progress curves from each well were examined for linear reaction kinetics and fit statistics (R2, 95% confidence interval, absolute sum of squares). Initial velocity (0 minutes to 30 minutes) from each reaction was determined from the slope of a plot of relative fluorescence units vs time (minutes) and then plotted against inhibitor concentration to estimate IC50 from log[Inhibitor] vs Response, Variable Slope model in GraphPad Prism from GraphPad Software (San Diego, Calif.).
- Omnia Assay Protocol for Potency Assessment Against EGFR (WT) and EGFR (T790M/L858R) Active Enzymes Briefly, 10× stocks of EGFR-WT (PV3872) from Invitrogen and EGFR-T790M/L858R (40350) from BPS Bioscience, San Diego, Calif., 1.13×ATP (AS001A) and appropriate Tyr-Sox conjugated peptide substrates (KCZ1001) were prepared in 1× kinase reaction buffer consisting of 20 mM Tris, pH 7.5, 5 mM MgCl2, 1 mM EGTA, 5 mM β-glycerophosphate, 5% glycerol (10× stock, KB002A) and 0.2 mM DTT (DS001A). 5 μL of each enzyme were pre-incubated in a Corning (#3574) 384-well, white, non-binding surface microtiter plate (Corning, N.Y.) for 30 min. at 25° C. with a 0.5 μL volume of 50% DMSO and serially diluted compounds prepared in 50% DMSO. Kinase reactions were started with the addition of 45 μL of the ATP/Tyr-Sox peptide substrate mix and monitored every 71 seconds for 60 minutes at λex360/λem485 in a Synergy4 plate reader from BioTek (Winooski, Vt.). At the conclusion of each assay, progress curves from each well were examined for linear reaction kinetics and fit statistics (R2, 95% confidence interval, absolute sum of squares). Initial velocity (0 minutes to 30 minutes) from each reaction was determined from the slope of a plot of relative fluorescence units vs time (minutes) and then plotted against inhibitor concentration to estimate IC50 from log [Inhibitor] vs Response, Variable Slope model in GraphPad Prism from GraphPad Software (San Diego, Calif.).
- Recombinant IDH1 Enzyme Assays All reactions were performed in standard enzyme reaction buffer (150 mM NaCl, 20 mM Tris-Cl, pH 7.5, 10% glycerol, 5 mM MgCl2 and 0.03% (w/v) bovine serum albumin). For determination of kinetic parameters, sufficient enzyme was added to give a linear reaction for 1 to 5 seconds. Reaction progress was monitored by observation of the reduction state of the cofactor at 340 nm in an SFM-400 stopped-flow spectrophotometer (BioLogic, Knoxville, Tenn.). Enzymatic constants were determined using curve fitting algorithms to standard kinetic models with the Sigmaplot software package (Systat Software, San Jose, Calif.). It is operated at 1×Km of NADPH.
- SAR VHR1 Fluorescent Assay for In Vitro dose response studies Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084230-01A1 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Protein tyrosine phosphatases (PTPs) play vital roles in numerous cellular processes and are implicated in a growing number of human diseases, ranging from cancer to cardiovascular, immunological, infectious, neurological, and metabolic diseases. The Vaccinia H1-related (VHR) PTP is a dual-specific Erk and Jnk phosphatase, the loss of which causes specific cell cycle arrest in HeLa carcinoma cells, suggesting that VHR inhibition may be a useful approach to halt the growth of cancer cells without detrimental effects on normal cells. Recent studies by collaborators and us suggest that VHR is upregulated in several cervix cancer cell lines, in
- SAR VHR1 Fluorescent Assay for In Vitro dose response studies Set 3 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084230-01A1 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Protein tyrosine phosphatases (PTPs) play vital roles in numerous cellular processes and are implicated in a growing number of human diseases, ranging from cancer to cardiovascular, immunological, infectious, neurological, and metabolic diseases. The Vaccinia H1-related (VHR) PTP is a dual-specific Erk and Jnk phosphatase, the loss of which causes specific cell cycle arrest in HeLa carcinoma cells, suggesting that VHR inhibition may be a useful approach to halt the growth of cancer cells without detrimental effects on normal cells. Recent studies by collaborators and us suggest that VHR is upregulated in several cervix cancer cell lines, in
- SAR VHR1 absorbance Assay for In Vitro dose response studies. Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084230-01A1 Assay Provider: Dr. Lutz Tautz, Sanford-Burnham Medical Research Institute, San Diego CA Protein tyrosine phosphatases (PTPs) play vital roles in numerous cellular processes and are implicated in a growing number of human diseases, ranging from cancer to cardiovascular, immunological, infectious, neurological, and metabolic diseases. The Vaccinia H1-related (VHR) PTP is a dual-specific Erk and Jnk phosphatase, the loss of which causes specific cell cycle arrest in HeLa carcinoma cells, suggesting that VHR inhibition may be a useful approach to halt the growth of cancer cells without detrimental effects on normal cells. Recent studies by collaborators and us suggest that VHR is upregulated in several cervix cancer cell lines, in
- AlphaScreen confirmatory assay for validation of inhibitors of SUMOylation Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084862-01 Assay Provider: Dr. Yuan Chen, Beckman Research Institute, City Of Hope, CA Protein modification by the SUMO (Small Ubiquitin-like MOdifier) family of proteins is an important post-translational modification that plays an essential role in many functions including gene transcription, cell cycle progression, DNA repair, viral infection, and the development of neurodegenerative diseases (1, 2). Recent proteomic studies have found that approximately 10% of the proteins encoded by the yeast genome are substrates for SUMO modification (3-5). The mechanism of how SUMOylation is involved in these cellular functions remains largely unclear. The inhibitors of SUMOylation would be useful to probe the roles of SUMOylation in cellular regulat
- Dose Response confirmation of uHTS activators of Human Intestinal Alkaline Phosphatase using Mouse Intestinal Alkaline Phosphatase Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Proposal Number: X01-MH077602-01 Alkaline phosphatase (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing inorganic phosphate and alcohol. APs are dimeric enzymes found in most organisms. In human, four isozymes of APs have been identified. One isozyme is tissue-nonspecific (designated TNAP) and three other isozymes are tissue-specific and named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) alkaline phosphatases. IAP expression is largely restricted to the gut, especially to the epithelial cells (enterocytes) of the small intestinal mucosa. The exact biological function of IAP is unknown. The goal of this MLPCN probe project is to identify novel and specific activators of
- Enzymatic assay The aim of this in vitro assay was to measure the inhibition of HCV NS3/4A protease complexes by the compounds of the present invention. This assay provides an indication of how effective compounds of the present invention would be in inhibiting HCV NS3/4A proteolytic activity. The inhibition of full-length hepatitis C NS3 protease enzyme was measured essentially as described in Poliakov, 2002 Prot Expression & Purification 25 363 371. Briefly, the hydrolysis of a depsipeptide substrate, Ac-DED(Edans)EEAbu-y-[COO]ASK(Dabcyl)-NH2 (AnaSpec, San Jose, USA), was measured spectrofluorometrically in the presence of a peptide cofactor, KKGSVVIVGRIVLSGK (Ake Engstrom, Department of Medical Biochemistry and Microbiology, Uppsala University, Sweden). [Landro, 1997 #Biochem 36 9340-9348]. The enzyme (1 nM) was incubated in 50 mM HEPES, pH 7.5, 10 mM DTT, 40% glycerol, 0.1% n-octyl-D-glucoside, with 25 uM NS4A cofactor and inhibitor at 30 C. for 10 min.
- HTS Assay The GSK3beta primary screen was conducted in assay ready 1536 plates (Aurora 29847) that contain 2.5 mL/well of 10 mM compound. Human GSK3beta as a GST fusion expressed in baculoviral system was purchased from BPS Bioscience (San Diego, Calif.). The GSK3beta peptide substrate was from American Peptide (Sunnyvale, Calif.; Cat 311153). 1 L/well of CABPE (22.5 nM GSK3beta , 8 uM peptide in AB buffer (12.5 mM DTT, 0.25 mg/mL BSA, 0.5 unit/mL Heparin)), 0.5 L/well of 125 uM of ATP, and 1 L/well of positive control 50 uM of GW8510 (positive control) or AB (DMSO only neutral control) in respective wells according to plate design using BioRAPTR (Beckman, Brea, Calif.). Reactions were incubated at room temperature for 60 minutes. 2.5 uL/well of ADP-Glo (Promega, V9103) was added with BioRAPTR, and incubated at room temperature for 40 minutes followed by addition of 5 uL/well of ADP-Glo detection reagent (Promega, V9103) with Combi nL (Thermo, Waltham, Mass.).
- SAR analysis of GM-Tri-DAP induced IL-8 secretion in MCF-7/NOD1 cells - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. The NOD proteins participate in the signaling events triggered by host recognition of specific motifs (mostly, murope
- SAR analysis of Muramyl dipeptide (MDP) induced IL-8 secretion in MCF-7/NOD2 cells - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. The NOD proteins participate in the signaling events triggered by host recognition of specific motifs (mostly, murope
- SAR analysis of Tumor necrosis factor alpha (TNF-alpha) induced IL-8 secretion in MCF-7/NOD1 cells - Set 2 Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Number: 1 R03 MH084844-01 Assay Provider: Dr. John C. Reed, Sanford-Burnham Medical Research Institute The modulation of immune response activity is one of the major goals in the development of novel therapeutics for auto-immune and inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition, inflammation, and cell death, thereby offering various therapeutic targets. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial peptidoglycans and directly activate NF-kB, a central regulator of immune response, inflammation, and apoptosis. The NOD proteins participate in the signaling events triggered by host recognition of specific motifs (mostly, murope
- SAR analysis of compounds that inhibit Human Immunodeficiency Virus Fusion. Data Source: Burnham Center for Chemical Genomics (BCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Production Centers Network (MLPCN) Grant Number: 1R21NS059403-01 Assay Provider: Dr. Miriam Gochin, Touro University-California, Vallejo, CA The fusion-active conformation of the envelope protein gp41 of HIV-1 consists of an N-terminal trimeric a-helical coiled coil domain, and three anti-parallel C-terminal helices which fold down the grooves of the coiled coil to form a six-helix bundle. Disruption of the six-helix bundle is considered to be a key component of an effective non-peptide fusion inhibitor. This structure forms as a result of a conformational change in gp41, triggered by gp120 and co-receptor binding to host cell receptors. Prevention of six-helix bundle formation has been recognized as an important mechanism for viral fusion inhibition A metallopeptide-based fluorescence assay has been develope
- uHTS HTRF assay for identification of inhibitors of SUMOylation Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Number: 1 R03 MH084862-01 Assay Provider: Dr. Yuan Chen, Beckman Research Institute, City Of Hope, CA Protein modification by the SUMO (Small Ubiquitin-like MOdifier) family of proteins is an important post-translational modification that plays an essential role in many functions including gene transcription, cell cycle progression, DNA repair, viral infection, and the development of neurodegenerative diseases (1, 2). Recent proteomic studies have found that approximately 10% of the proteins encoded by the yeast genome are substrates for SUMO modification (3-5). The mechanism of how SUMOylation is involved in these cellular functions remains largely unclear. The inhibitors of SUMOylation would be useful to probe the roles of SUMOylation in cellular regulati
- uHTS identification of compounds activating TNAP in the absence of phosphate acceptor performed in luminescent assay Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Screening Centers Network (MLSCN) Grant Proposal Number: 1R03 MH082385-01 Alkaline phosphatases (EC 3.1.3.1) (APs) catalyze the hydrolysis of phosphomonoesters, releasing phosphate and alcohol. APs are dimeric enzymes found in the most organisms. In human, four isozymes of APs have been identified. Three isozymes are tissue-specific and the fourth one is tissue-non specific, named TNAP. TNAP deficiency is associated with defective bone mineralization in the form of rickets and osteomalacia. Therefore, there is therapeutic potential of modulating TNAP activity. The goal of this HTS is to identify novel and specific activators of TNAP. The only known to date class of alkaline phosphatases activators are amino-containing alcohols, such as diethanolamine (DEA), that act as phosphoacceptor substrate and exh
- ChEMBL_2267427 Inhibition of human cdc7 incubation of 30 mins in presence of ATP
- ChEMBL_2267428 Inhibition of human CDK9 incubation of 30 mins in presence of ATP
- ChEMBL_66597 (CHEMBL680028) Inhibition of autophosphorylation of cytoplasmic domain of epidermal growth factor receptor
- Assay of ABAD Enzymatic Activity The assay for the inhibition of reduction of S-acetoacetyl-CoA (SAAC) by ABAD was carried out with ABAD (418 ng/ml), SAAC (172 μM), NADH (102 μM), and different concentrations of inhibitors (from 0 to 1000 μM) in 93 mM potassium phosphate buffer (pH 7.3). Before the assay, all the assay components except SAAC were pre-incubated for 5 minutes, and the reaction started with the addition of SAAC into the reaction mix. The reaction was carried out for a total 6 minutes at room temperature under steady-state conditions, and the decrease of NADH absorbance at 340 nm was determined every 10 seconds. Kinetic data were analyzed by PRISM (Scitech, San Diego, Calif.) to determine IC50 values and Ki. One unit of enzyme activity was defined as that which converted 1.0 μmol of substrate to product per minute.
- Biochemical Assay ADP-Glo (Promega, Madison, Wis., USA) reagents were thawed at ambient temperature. Kinase Detection Reagent was prepared by mixing kinase detection buffer with the lyophilized kinase detection substrate.A 500 ml stock volume of 5× Reaction Kinase Buffer was made by mixing 10000 of 1M MgCl2, 500 μl of 1M Tris-HCL pH7.4, 0.5 mg/ml (25 mg) of BSA, and 3475 μl of distilled H2O. A 3 ml 2× working stock volume of Reaction Kinase Buffer was made containing a final concentration of 100 μM DTT and 4 mM MnCl2.Components of RIPK1 enzyme (Rigel Pharmaceuticals, South San Francisco, Calif., USA) were thawed on ice. Diluted RIPK1 was prepared in 1× Kinase Reaction Buffer (diluted from 2× buffer) to 31 ng/well. A 166 μM working stock ATP assay solution was prepared in 1× Kinase Reaction Buffer (diluted from 2× buffer).
- Biological Activity Assay Assaying the inhibition of KDM1A can be determined in vitro, in cultured cells, and in animals. There are a variety of spectrophotometric methods to detect the results of demethylation of methylated lysines, viz., detecting the products of KDM1A demethylase oxidative activity on a peptide fragment of at least 18 amino acid representing the N-terminus of the histone H3 substrate that contains a monomethyl at the fourth lysine residue. Hydrogen peroxide, one product of the KDM1A demethylase reaction, reacts with horseradish peroxidase and dihydroxyphenoxazine (ADHP) to produce the fluorescent compound resorufin (excitation=530-560 nm:emission=590 nm). The KDM1A demethylase enzyme activity can obtained from mammalian cells or tissues expressing KDM1A from an endogenous or recombinant gene and purified or assayed from a whole cell extract. These methods can be used to determine the concentration of the disclosed compounds can inhibit fifty percent of the enzyme activity (IC50). In one aspect, the disclosed compounds exhibit inhibition fifty percent of the KDM1A enzyme activity at a concentration of less than 500 nM, less than 100 nM, less than 50 nM or less than 10 nM.
- ChEMBL_184451 (CHEMBL789405) Inhibition of adenosine stimulated accumulation of cyclic AMP at Adenosine A2 receptor of VA13 fibroblasts of rat
- Dose Response confirmation of uHTS hits for small molecule agonists of the CRF-binding protein and CRF-R2 receptor complex Data Source: Sanford-Burnham Center for Chemical Genomics (SBCCG) Source Affiliation: Sanford-Burnham Medical Research Institute (SBMRI, San Diego, CA) Network: NIH Molecular Libraries Probe Production Centers Network (MLPCN) Grant Proposal Number: 1 R21 DA029966-01 Assay Providers: Selena Bartlett, Ph.D., Ernest Gallo Clinic and Research Center, University of California, San Francisco and Nick Cosford, Ph.D., Sanford-Burnham Medical Research Institute There is accumulating scientific evidence showing that stressors enhance addictive behaviors and are a common cause of relapse to substance abuse. Corticotrophin releasing factor (CRF) is a 41-aa peptide that has been shown to induce various behavioral changes related to adaptation to stress. The CRF system, including the CRF-binding protein (CRF-BP) and the CRF receptors, CRF-R1 and CRF-R2, are thought to contribute, to the physiological adaptations that result from stress. It has been shown that CRF interaction with CRF-BP may positi
- Enzymatic Assay for Mpro The SARS-CoV-2 3CLPro (Mpro), MBP-tagged Assay Kit (BPS Biosciences, San Diego, CA, USA) was used to measure 3CL Protease (Mpro) activity for screening and profiling applications. The 3CL inhibitor GC376 was included as a positive control and also used as a standard. The assay was performed according to the protocol provided by the manufacturer with the concentration of compounds tested being 10−7 M.
- Inhibition of Histone Deacetylase Enzymatic Assay For deacetylase assays, 20,000 cpm of the [3H]-metabolically labeled acetylated histone substrate (M. Yoshida et al., J. Biol. Chem. 265(28): 17174-17179 (1990)) is incubated with 30 ug of H446 nuclear extract or an equivalent amount of the cloned recombinant hHDAC-1 for 10 minutes at 37 C. The reaction is stopped by adding acetic acid (0.04 M, final concentration) and HCl (250 mM, final concentration). The mixture is extracted with ethyl acetate and the released [3H]-acetic acid was quantified by scintillation counting. For inhibition studies, the enzyme is preincubated with compounds at 4 C. for 30 minutes prior to initiation of the enzymatic assay. IC50 values for HDAC enzyme inhibitors are determined by performing dose response curves with individual compounds and determining the concentration of inhibitor producing fifty percent of the maximal inhibition. Alternatively, the following protocol is used to assay the compounds of the invention.
- ChEBML_155363 Inhibition of phosphodiesterase 5 of rabbit platelets
- ChEBML_208905 Evaluation of inhibition of transition state thrombin
- ChEBML_212014 Tested for inhibition of polymerization of tubulin
- ChEBML_305045 Inhibition of Beta-glucosidase of rat intestine
- ChEBML_305098 Inhibition of Beta-galactosidase of bovine liver
- ChEBML_41589 Inhibition of (BChE) Butyrylcholinesterase of horse serum
- ChEMBL_154561 (CHEMBL757896) Inhibition of PDE5 of human platelets
- ChEMBL_154562 (CHEMBL757897) Inhibition of PDE5 of human platelets
- ChEMBL_2277852 Induction of degradation of FKBP12 (unknown origin)
- ChEMBL_2277853 Induction of degradation of USP7 (unknown origin)
- ChEMBL_429830 (CHEMBL915411) Inhibition of dimerization of HIV1 protease
- ChEMBL_440741 (CHEMBL888712) Inhibition of human LBD of ERbeta
- ChEMBL_529187 (CHEMBL974963) Inhibition of peptidase activity of CPP32
- ChEMBL_760063 (CHEMBL1810386) Inhibition of auto-phosphorylation of IGFR1
- ChEMBL_770210 (CHEMBL1833000) Inhibition of first bromodomain of BRD2
- ChEMBL_770211 (CHEMBL1833001) Inhibition of first bromodomain of BRD4