 NINLARO MLN2238 IXAZOMIB CITRATE BDBM50398609 Ixazomib US12054502, Compound Ixazomib CHEMBL2141296
NINLARO MLN2238 IXAZOMIB CITRATE BDBM50398609 Ixazomib US12054502, Compound Ixazomib CHEMBL2141296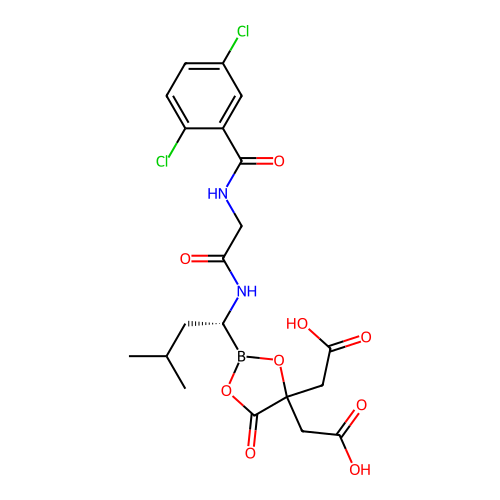 MLN9708 Ixazomib citrate MLN-9708 Ninlaro BDBM50649918
MLN9708 Ixazomib citrate MLN-9708 Ninlaro BDBM50649918 SODIUM CITRATE BDBM92494 Citrate
SODIUM CITRATE BDBM92494 Citrate cid_234845 MLS000736796 ISORESERPILINE, CITRATE SMR000528329 BDBM51257
cid_234845 MLS000736796 ISORESERPILINE, CITRATE SMR000528329 BDBM51257 BDBM50355376 US20250195699, Compound fentanyl Fentanyl FENTANYL CITRATE
BDBM50355376 US20250195699, Compound fentanyl Fentanyl FENTANYL CITRATE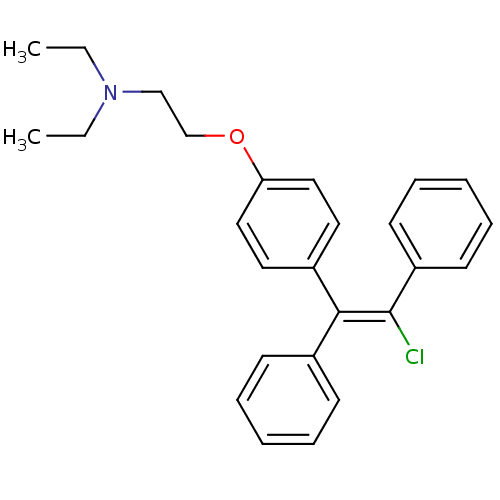 MLS000069760 cid_6420009 CLOMIPHENE SMR000058740 cid_1548953 CLOMIPHENE CITRATE BDBM55354
MLS000069760 cid_6420009 CLOMIPHENE SMR000058740 cid_1548953 CLOMIPHENE CITRATE BDBM55354 SILDENAFIL CITRATE Revatio UK-92480-10 BDBM50359766 Viagra
SILDENAFIL CITRATE Revatio UK-92480-10 BDBM50359766 Viagra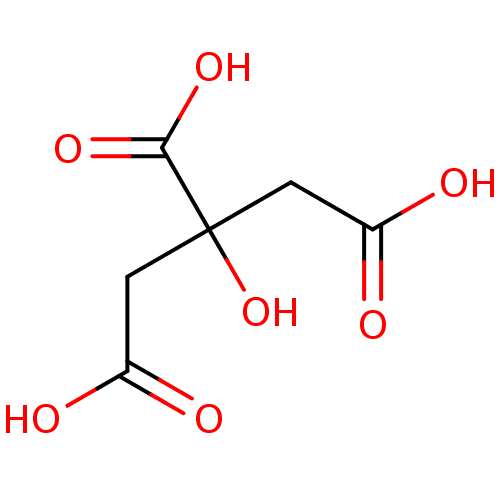 cid_311 CITRIC ACID 2-hydroxypropane-1,2,3-tricarboxylic acid Fragment 2 Citrate CHEMBL1261 BDBM14672
cid_311 CITRIC ACID 2-hydroxypropane-1,2,3-tricarboxylic acid Fragment 2 Citrate CHEMBL1261 BDBM14672 trisodium 2-hydroxypropane-1,2,3-tricarboxylate citric acid trisodium salt BDBM50159799 CHEMBL1355 sodium citrate
trisodium 2-hydroxypropane-1,2,3-tricarboxylate citric acid trisodium salt BDBM50159799 CHEMBL1355 sodium citrate Sildenafil Citrate CHEBI:58987 Revatio UK-9248010 BDBM50238854 UK-92480-10 US12351585, Compound sildenafil Viagra
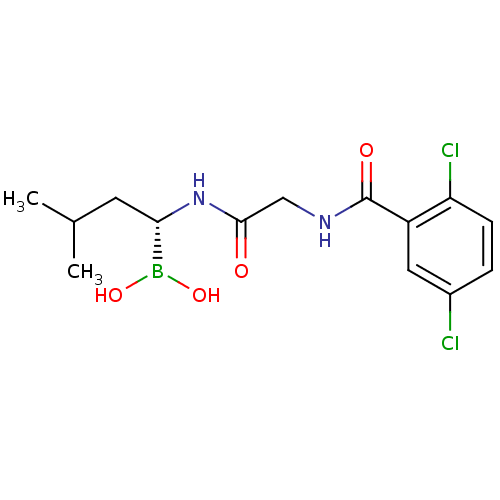
Sildenafil Citrate CHEBI:58987 Revatio UK-9248010 BDBM50238854 UK-92480-10 US12351585, Compound sildenafil Viagra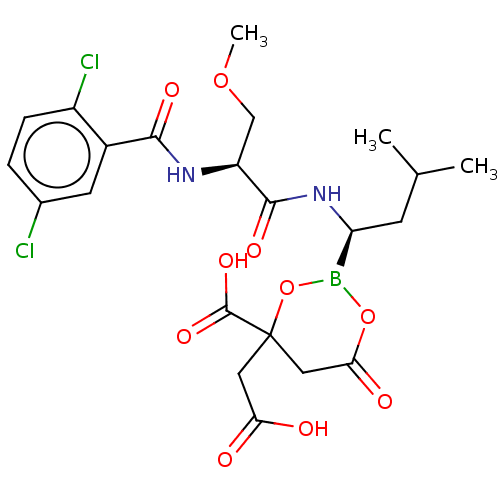 US11542283, Compound V-8B BDBM588480 (S)-N-(2,5-dichlorobenzoyl)-3- methoxypropionamido-D-leucine borate citrate
US11542283, Compound V-8B BDBM588480 (S)-N-(2,5-dichlorobenzoyl)-3- methoxypropionamido-D-leucine borate citrate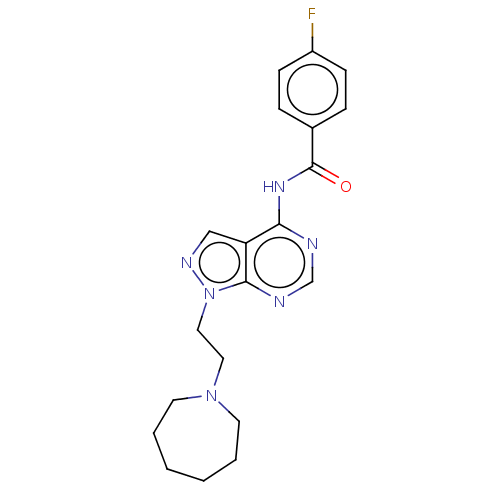 N-(1-(2-(azepan-1- yl)ethyl)-1H-pyrazolo[3,4- d]pyrimidin-4-yl)-4- fluorobenzamide citrate BDBM287110 US9567338, 34
N-(1-(2-(azepan-1- yl)ethyl)-1H-pyrazolo[3,4- d]pyrimidin-4-yl)-4- fluorobenzamide citrate BDBM287110 US9567338, 34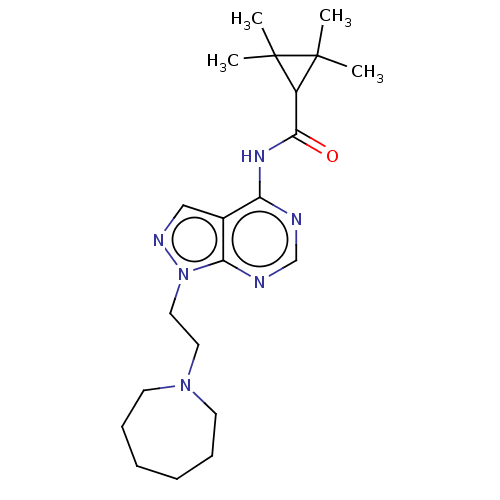 BDBM287111 US9567338, 35 N-(1-(2-(azepan-1- yl)ethyl)-1H-pyrazolo[3,4- d]pyrimidin-4-yl)-2,2,3,3- tetramethylcyclopropane- carboxamide citrate
BDBM287111 US9567338, 35 N-(1-(2-(azepan-1- yl)ethyl)-1H-pyrazolo[3,4- d]pyrimidin-4-yl)-2,2,3,3- tetramethylcyclopropane- carboxamide citrate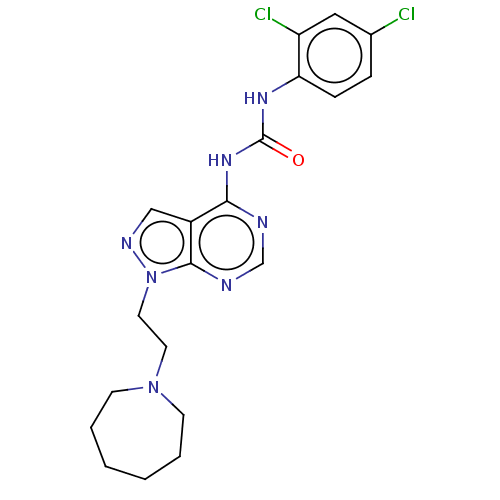 US9567338, 64 BDBM287127 1-(1-(2-(azepan-1- yl)ethyl)-1H- pyrazolo[3,4- d]pyrimidin-4-yl)-3-(2,4- dichlorophenyl)urea citrate
US9567338, 64 BDBM287127 1-(1-(2-(azepan-1- yl)ethyl)-1H- pyrazolo[3,4- d]pyrimidin-4-yl)-3-(2,4- dichlorophenyl)urea citrate US20230391761, Example 9 2-(1-Methyl-1H-imidazol-2-yl)ethyl 3-{[(5-chloro-2-thienyl)carbonyl]amino}-N-({2-ethyl-3-[(3S)-3-hydroxy-2-oxopyrrolidin-1-yl]phenyl}sulfonyl)-S-alaninate citrate BDBM639357
US20230391761, Example 9 2-(1-Methyl-1H-imidazol-2-yl)ethyl 3-{[(5-chloro-2-thienyl)carbonyl]amino}-N-({2-ethyl-3-[(3S)-3-hydroxy-2-oxopyrrolidin-1-yl]phenyl}sulfonyl)-S-alaninate citrate BDBM639357 CHEMBL60889 Mosapride 4-Amino-5-chloro-2-ethoxy-N-[4-(4-fluoro-benzyl)-morpholin-2-ylmethyl]-benzamide; citrate(AS-4370) 4-Amino-5-chloro-2-ethoxy-N-[4-(4-fluoro-benzyl)-morpholin-2-ylmethyl]-benzamide BDBM50011327
CHEMBL60889 Mosapride 4-Amino-5-chloro-2-ethoxy-N-[4-(4-fluoro-benzyl)-morpholin-2-ylmethyl]-benzamide; citrate(AS-4370) 4-Amino-5-chloro-2-ethoxy-N-[4-(4-fluoro-benzyl)-morpholin-2-ylmethyl]-benzamide BDBM50011327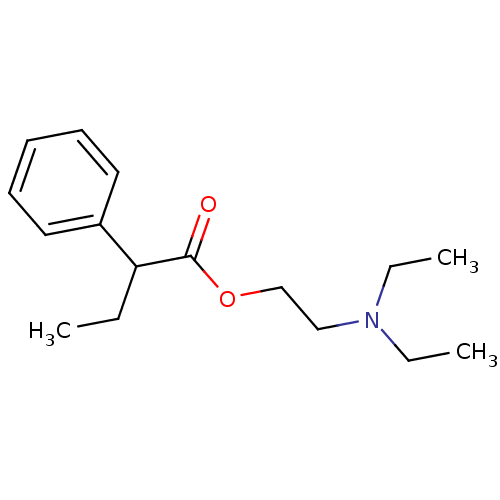 2-(diethylamino)ethyl 2-phenylbutanoate;2-hydroxypropane-1,2,3-tricarboxylic acid BUTETHAMATE CITRATE SMR000058633 citric acid;2-phenylbutyric acid 2-(diethylamino)ethyl ester BDBM94593 2-hydroxypropane-1,2,3-tricarboxylic acid;2-phenylbutanoic acid 2-(diethylamino)ethyl ester cid_117176 MLS001148168 2-(diethylamino)ethyl 2-phenylbutanoate;2-oxidanylpropane-1,2,3-tricarboxylic acid
2-(diethylamino)ethyl 2-phenylbutanoate;2-hydroxypropane-1,2,3-tricarboxylic acid BUTETHAMATE CITRATE SMR000058633 citric acid;2-phenylbutyric acid 2-(diethylamino)ethyl ester BDBM94593 2-hydroxypropane-1,2,3-tricarboxylic acid;2-phenylbutanoic acid 2-(diethylamino)ethyl ester cid_117176 MLS001148168 2-(diethylamino)ethyl 2-phenylbutanoate;2-oxidanylpropane-1,2,3-tricarboxylic acid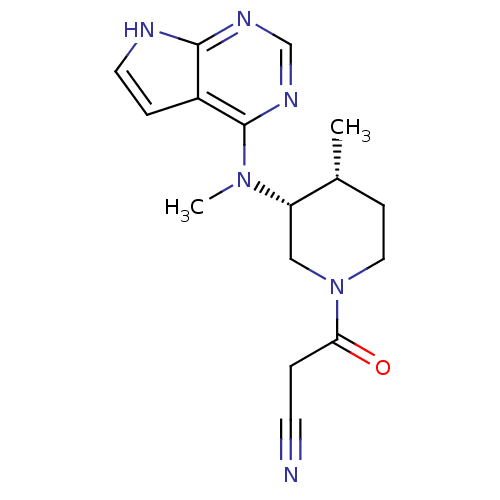 BDBM50193995 US10112907, Example 00035 US10399979, Compound Tofacitinib CHEMBL221959 US20240140952, Compound Tofacitinib 3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile US10875847, Compound Tofacitinib Tofacitinib citrate (1) 3-{(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]piperidin-1-yl}-3-oxopropanenitrile Tofacitinib US10766894, Compound TABLE 1.20 US11203595, TABLE 1.20 US12492204, Compound Tofacitinib TOFACITINIB CITRATE US20250257069, Compound Tofacitinib 3-(4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile US11078206, Example Tofacitinib CP-690550 US11339167, Example Tofacitinib
BDBM50193995 US10112907, Example 00035 US10399979, Compound Tofacitinib CHEMBL221959 US20240140952, Compound Tofacitinib 3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile US10875847, Compound Tofacitinib Tofacitinib citrate (1) 3-{(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]piperidin-1-yl}-3-oxopropanenitrile Tofacitinib US10766894, Compound TABLE 1.20 US11203595, TABLE 1.20 US12492204, Compound Tofacitinib TOFACITINIB CITRATE US20250257069, Compound Tofacitinib 3-(4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile US11078206, Example Tofacitinib CP-690550 US11339167, Example Tofacitinib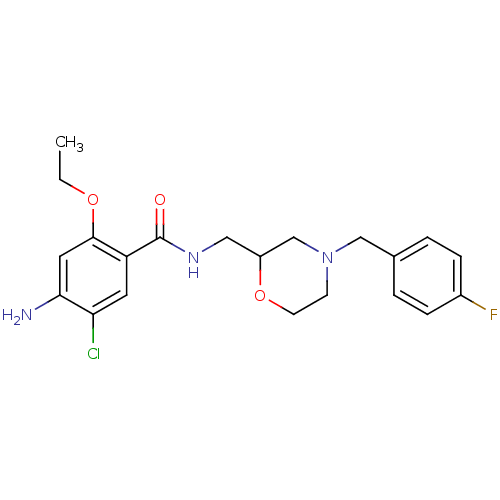 cid_119583 CHEMBL60889 Mosapride SMR000469200 4-Amino-5-chloro-2-ethoxy-N-[4-(4-fluoro-benzyl)-morpholin-2-ylmethyl]-benzamide; citrate(AS-4370) 4-amino-5-chloro-2-ethoxy-N-[[4-[(4-fluorophenyl)methyl]-2-morpholinyl]methyl]benzamide;2-hydroxypropane-1,2,3-tricarboxylic acid MOSAPRIDE CITRATE 4-azanyl-5-chloranyl-2-ethoxy-N-[[4-[(4-fluorophenyl)methyl]morpholin-2-yl]methyl]benzamide;2-oxidanylpropane-1,2,3-tricarboxylic acid 4-amino-5-chloro-2-ethoxy-N-[[4-[(4-fluorophenyl)methyl]morpholin-2-yl]methyl]benzamide;2-hydroxypropane-1,2,3-tricarboxylic acid 4-amino-5-chloro-2-ethoxy-N-[[4-(4-fluorobenzyl)morpholin-2-yl]methyl]benzamide;citric acid BDBM94630 MLS001401439
cid_119583 CHEMBL60889 Mosapride SMR000469200 4-Amino-5-chloro-2-ethoxy-N-[4-(4-fluoro-benzyl)-morpholin-2-ylmethyl]-benzamide; citrate(AS-4370) 4-amino-5-chloro-2-ethoxy-N-[[4-[(4-fluorophenyl)methyl]-2-morpholinyl]methyl]benzamide;2-hydroxypropane-1,2,3-tricarboxylic acid MOSAPRIDE CITRATE 4-azanyl-5-chloranyl-2-ethoxy-N-[[4-[(4-fluorophenyl)methyl]morpholin-2-yl]methyl]benzamide;2-oxidanylpropane-1,2,3-tricarboxylic acid 4-amino-5-chloro-2-ethoxy-N-[[4-[(4-fluorophenyl)methyl]morpholin-2-yl]methyl]benzamide;2-hydroxypropane-1,2,3-tricarboxylic acid 4-amino-5-chloro-2-ethoxy-N-[[4-(4-fluorobenzyl)morpholin-2-yl]methyl]benzamide;citric acid BDBM94630 MLS001401439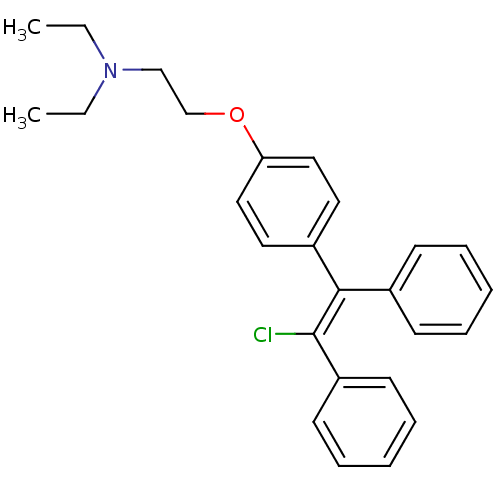 2-[4-[(Z)-2-chloro-1,2-diphenylethenyl]phenoxy]-N,N-diethylethanamine;2-hydroxypropane-1,2,3-tricarboxylic acid cid_3033832 MLS001332630 Clomiphene, 2 Clomiphene citrate salt SMR000875221 CHEMBL167779 2-[4-[(Z)-2-chloranyl-1,2-diphenyl-ethenyl]phenoxy]-N,N-diethyl-ethanamine;2-oxidanylpropane-1,2,3-tricarboxylic acid BDBM71545 2-[4-[(Z)-2-chloro-1,2-diphenyl-vinyl]phenoxy]ethyl-diethyl-amine;citric acid
2-[4-[(Z)-2-chloro-1,2-diphenylethenyl]phenoxy]-N,N-diethylethanamine;2-hydroxypropane-1,2,3-tricarboxylic acid cid_3033832 MLS001332630 Clomiphene, 2 Clomiphene citrate salt SMR000875221 CHEMBL167779 2-[4-[(Z)-2-chloranyl-1,2-diphenyl-ethenyl]phenoxy]-N,N-diethyl-ethanamine;2-oxidanylpropane-1,2,3-tricarboxylic acid BDBM71545 2-[4-[(Z)-2-chloro-1,2-diphenyl-vinyl]phenoxy]ethyl-diethyl-amine;citric acid N-ethyl-3-phenyl-N-(3-phenylpropyl)propan-1-amine;2-oxidanylpropane-1,2,3-tricarboxylic acid citric acid;ethyl-bis(3-phenylpropyl)amine BDBM37636 N-ethyl-3-phenyl-N-(3-phenylpropyl)propan-1-amine;2-hydroxypropane-1,2,3-tricarboxylic acid SMR000058631 MLS000069524 N-ethyl-3-phenyl-N-(3-phenylpropyl)-1-propanamine;2-hydroxypropane-1,2,3-tricarboxylic acid alverine cid_21718 ALVERINE CITRATE SALT
N-ethyl-3-phenyl-N-(3-phenylpropyl)propan-1-amine;2-oxidanylpropane-1,2,3-tricarboxylic acid citric acid;ethyl-bis(3-phenylpropyl)amine BDBM37636 N-ethyl-3-phenyl-N-(3-phenylpropyl)propan-1-amine;2-hydroxypropane-1,2,3-tricarboxylic acid SMR000058631 MLS000069524 N-ethyl-3-phenyl-N-(3-phenylpropyl)-1-propanamine;2-hydroxypropane-1,2,3-tricarboxylic acid alverine cid_21718 ALVERINE CITRATE SALT Acidum citricum Citrate Citric acid monoglyceride Citric acid hydrate E330 CHEBI:30769 Citric acid, anhydrous Citric acid,anhydrous Acidum citricum monohydrate FEMA NO. 2306 Citricum acidum Urologic G NSC-626579 Citric acid, hydrous NSC-30279 Citric acid,hydrous E-330 INS NO.330 BDBM50416759 NSC-112226 Citric acid Citric acid bp Anhydrous citric acid Citric acid anhydrous INS-330 Aciletten B1650
Acidum citricum Citrate Citric acid monoglyceride Citric acid hydrate E330 CHEBI:30769 Citric acid, anhydrous Citric acid,anhydrous Acidum citricum monohydrate FEMA NO. 2306 Citricum acidum Urologic G NSC-626579 Citric acid, hydrous NSC-30279 Citric acid,hydrous E-330 INS NO.330 BDBM50416759 NSC-112226 Citric acid Citric acid bp Anhydrous citric acid Citric acid anhydrous INS-330 Aciletten B1650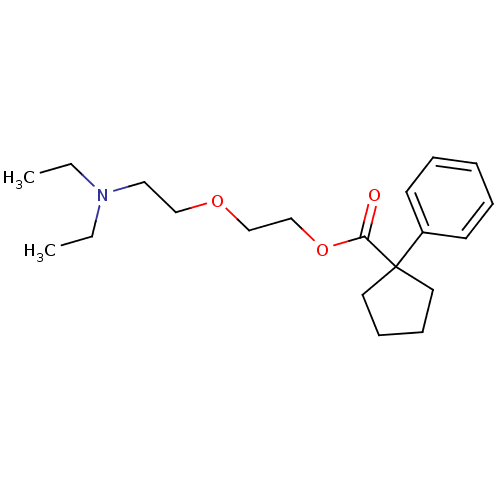 cid_90010 citric acid;1-phenylcyclopentanecarboxylic acid 2-[2-(diethylamino)ethoxy]ethyl ester MLS002222257 BDBM94507 CARBETAPENTANE 2-hydroxypropane-1,2,3-tricarboxylic acid;1-phenyl-1-cyclopentanecarboxylic acid 2-[2-(diethylamino)ethoxy]ethyl ester Carbetapentane citrate SMR000326757 CHEMBL73234 2-[2-(diethylamino)ethoxy]ethyl 1-phenylcyclopentane-1-carboxylate;2-hydroxypropane-1,2,3-tricarboxylic acid 2-[2-(diethylamino)ethoxy]ethyl 1-phenylcyclopentane-1-carboxylate;2-oxidanylpropane-1,2,3-tricarboxylic acid
cid_90010 citric acid;1-phenylcyclopentanecarboxylic acid 2-[2-(diethylamino)ethoxy]ethyl ester MLS002222257 BDBM94507 CARBETAPENTANE 2-hydroxypropane-1,2,3-tricarboxylic acid;1-phenyl-1-cyclopentanecarboxylic acid 2-[2-(diethylamino)ethoxy]ethyl ester Carbetapentane citrate SMR000326757 CHEMBL73234 2-[2-(diethylamino)ethoxy]ethyl 1-phenylcyclopentane-1-carboxylate;2-hydroxypropane-1,2,3-tricarboxylic acid 2-[2-(diethylamino)ethoxy]ethyl 1-phenylcyclopentane-1-carboxylate;2-oxidanylpropane-1,2,3-tricarboxylic acid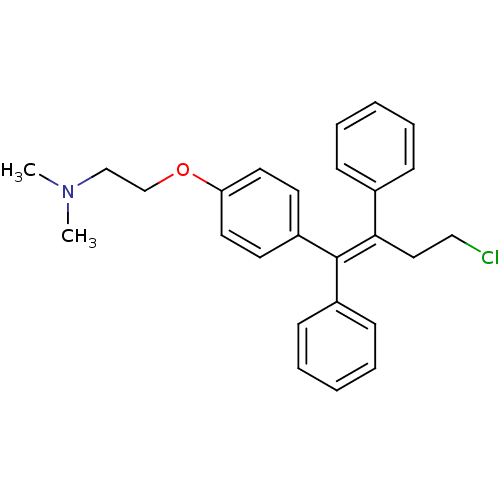 TOREMIFENE cid_3005572 2-[4-[(Z)-4-chloranyl-1,2-diphenyl-but-1-enyl]phenoxy]-N,N-dimethyl-ethanamine;2-oxidanylpropane-1,2,3-tricarboxylic acid SMR000469213 2-[4-[(Z)-4-chloro-1,2-diphenyl-but-1-enyl]phenoxy]ethyl-dimethyl-amine;citric acid BDBM58492 med.21724, Compound Toremifene 2-[4-[(Z)-4-chloro-1,2-diphenylbut-1-enyl]phenoxy]-N,N-dimethylethanamine;2-hydroxypropane-1,2,3-tricarboxylic acid MLS001424189 TOREMIFENE CITRATE
TOREMIFENE cid_3005572 2-[4-[(Z)-4-chloranyl-1,2-diphenyl-but-1-enyl]phenoxy]-N,N-dimethyl-ethanamine;2-oxidanylpropane-1,2,3-tricarboxylic acid SMR000469213 2-[4-[(Z)-4-chloro-1,2-diphenyl-but-1-enyl]phenoxy]ethyl-dimethyl-amine;citric acid BDBM58492 med.21724, Compound Toremifene 2-[4-[(Z)-4-chloro-1,2-diphenylbut-1-enyl]phenoxy]-N,N-dimethylethanamine;2-hydroxypropane-1,2,3-tricarboxylic acid MLS001424189 TOREMIFENE CITRATE US11155558, Compound sildenafil 5-[2-ethoxy-5-(4-methyl-1-piperazinylsulfonyl)phenyl]-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 5-{2-ethoxy-5-[(4-methylpiperazine-1-)sulfonyl]phenyl}-1-methyl-3-propyl-1H,6H,7H-pyrazolo[4,3-d]pyrimidin-7-one BDBM14390 CHEMBL192 SILDENAFIL CITRATE US11242347, Compound sildenafil Sildenafil US12139461, Compound sildenafil US11897890, Compound sildenafil Viagra Sildenafil#
US11155558, Compound sildenafil 5-[2-ethoxy-5-(4-methyl-1-piperazinylsulfonyl)phenyl]-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one 5-{2-ethoxy-5-[(4-methylpiperazine-1-)sulfonyl]phenyl}-1-methyl-3-propyl-1H,6H,7H-pyrazolo[4,3-d]pyrimidin-7-one BDBM14390 CHEMBL192 SILDENAFIL CITRATE US11242347, Compound sildenafil Sildenafil US12139461, Compound sildenafil US11897890, Compound sildenafil Viagra Sildenafil# Duragesic-50 N-(1-Phenethyl-piperidin-4-yl)-N-phenyl-propionamide(Fentanyl) BDBM50008984 Ionsys Duragesic-75 Duragesic-12 FENTANYL CITRATE FENTANYL-HCl Fentora CHEMBL596 US20230399418, Compound Fentanyl N-(1-Phenethyl-piperidin-4-yl)-N-phenyl-propionamide Fentanyl-25 Fentanyl-100 Fentanyl-12 Duragesic-25 Fentanyl-75 Duragesic-100 FENTANYL Fentanyl-50 4-(4-Chloro-benzyl)-2-(1-methyl-azepan-4-yl)-2H-phthalazin-1-one N-(1-phenethylpiperidin-4-yl)-N-phenylpropionamide Innovar
Duragesic-50 N-(1-Phenethyl-piperidin-4-yl)-N-phenyl-propionamide(Fentanyl) BDBM50008984 Ionsys Duragesic-75 Duragesic-12 FENTANYL CITRATE FENTANYL-HCl Fentora CHEMBL596 US20230399418, Compound Fentanyl N-(1-Phenethyl-piperidin-4-yl)-N-phenyl-propionamide Fentanyl-25 Fentanyl-100 Fentanyl-12 Duragesic-25 Fentanyl-75 Duragesic-100 FENTANYL Fentanyl-50 4-(4-Chloro-benzyl)-2-(1-methyl-azepan-4-yl)-2H-phthalazin-1-one N-(1-phenethylpiperidin-4-yl)-N-phenylpropionamide Innovar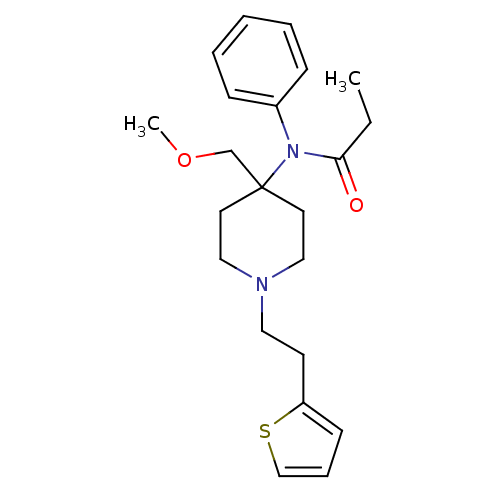 BDBM94503 cid_65494 MLS002320668 SUFENTANIL 2-hydroxypropane-1,2,3-tricarboxylic acid;N-[4-(methoxymethyl)-1-(2-thiophen-2-ylethyl)-4-piperidinyl]-N-phenylpropanamide N-[4-(methoxymethyl)-1-(2-thiophen-2-ylethyl)piperidin-4-yl]-N-phenyl-propanamide;2-oxidanylpropane-1,2,3-tricarboxylic acid citric acid;N-[4-(methoxymethyl)-1-[2-(2-thienyl)ethyl]-4-piperidyl]-N-phenyl-propionamide 2-hydroxypropane-1,2,3-tricarboxylic acid;N-[4-(methoxymethyl)-1-(2-thiophen-2-ylethyl)piperidin-4-yl]-N-phenylpropanamide Sufentanil citrate SMR001338814 Sufentanyl
BDBM94503 cid_65494 MLS002320668 SUFENTANIL 2-hydroxypropane-1,2,3-tricarboxylic acid;N-[4-(methoxymethyl)-1-(2-thiophen-2-ylethyl)-4-piperidinyl]-N-phenylpropanamide N-[4-(methoxymethyl)-1-(2-thiophen-2-ylethyl)piperidin-4-yl]-N-phenyl-propanamide;2-oxidanylpropane-1,2,3-tricarboxylic acid citric acid;N-[4-(methoxymethyl)-1-[2-(2-thienyl)ethyl]-4-piperidyl]-N-phenyl-propionamide 2-hydroxypropane-1,2,3-tricarboxylic acid;N-[4-(methoxymethyl)-1-(2-thiophen-2-ylethyl)piperidin-4-yl]-N-phenylpropanamide Sufentanil citrate SMR001338814 Sufentanyl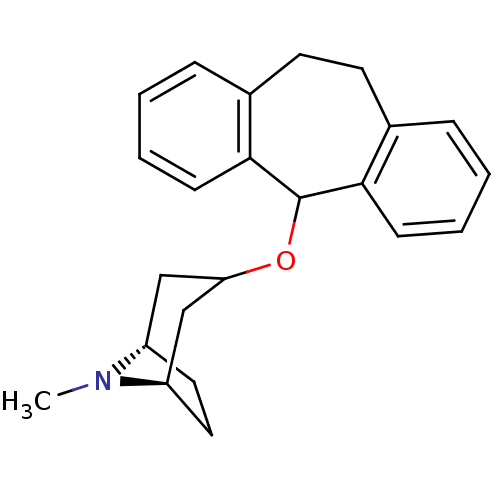 citric acid;(1S,5R)-3-(6,11-dihydro-5H-dibenzo[1,2-a:1',2'-e]cyclohepten-11-yloxy)-8-methyl-8-azabicyclo[3.2.1]octane BDBM79210 SMR001233409 cid_6604492 MLS002154101 (1S,5R)-3-(6,11-dihydro-5H-dibenzo[1,2-a:1',2'-e][7]annulen-11-yloxy)-8-methyl-8-azabicyclo[3.2.1]octane;2-oxidanylpropane-1,2,3-tricarboxylic acid (1S,5R)-3-(6,11-dihydro-5H-dibenzo[1,2-a:1',2'-e][7]annulen-11-yloxy)-8-methyl-8-azabicyclo[3.2.1]octane;2-hydroxypropane-1,2,3-tricarboxylic acid Deptropine citrate
citric acid;(1S,5R)-3-(6,11-dihydro-5H-dibenzo[1,2-a:1',2'-e]cyclohepten-11-yloxy)-8-methyl-8-azabicyclo[3.2.1]octane BDBM79210 SMR001233409 cid_6604492 MLS002154101 (1S,5R)-3-(6,11-dihydro-5H-dibenzo[1,2-a:1',2'-e][7]annulen-11-yloxy)-8-methyl-8-azabicyclo[3.2.1]octane;2-oxidanylpropane-1,2,3-tricarboxylic acid (1S,5R)-3-(6,11-dihydro-5H-dibenzo[1,2-a:1',2'-e][7]annulen-11-yloxy)-8-methyl-8-azabicyclo[3.2.1]octane;2-hydroxypropane-1,2,3-tricarboxylic acid Deptropine citrate BDBM94547 VARDENAFIL CITRATE SMR000469155 MLS001401365 2-[2-ethoxy-5-(4-ethylpiperazin-1-yl)sulfonylphenyl]-5-methyl-7-propyl-1H-imidazo[5,1-f][1,2,4]triazin-4-one;2-hydroxypropane-1,2,3-tricarboxylic acid cid_23581791 citric acid;2-[2-ethoxy-5-(4-ethylpiperazino)sulfonyl-phenyl]-5-methyl-7-propyl-1H-imidazo[5,1-f][1,2,4]triazin-4-one 2-[2-ethoxy-5-[(4-ethyl-1-piperazinyl)sulfonyl]phenyl]-5-methyl-7-propyl-1H-imidazo[5,1-f][1,2,4]triazin-4-one;2-hydroxypropane-1,2,3-tricarboxylic acid 2-[2-ethoxy-5-(4-ethylpiperazin-1-yl)sulfonyl-phenyl]-5-methyl-7-propyl-1H-imidazo[5,1-f][1,2,4]triazin-4-one;2-oxidanylpropane-1,2,3-tricarboxylic acid
BDBM94547 VARDENAFIL CITRATE SMR000469155 MLS001401365 2-[2-ethoxy-5-(4-ethylpiperazin-1-yl)sulfonylphenyl]-5-methyl-7-propyl-1H-imidazo[5,1-f][1,2,4]triazin-4-one;2-hydroxypropane-1,2,3-tricarboxylic acid cid_23581791 citric acid;2-[2-ethoxy-5-(4-ethylpiperazino)sulfonyl-phenyl]-5-methyl-7-propyl-1H-imidazo[5,1-f][1,2,4]triazin-4-one 2-[2-ethoxy-5-[(4-ethyl-1-piperazinyl)sulfonyl]phenyl]-5-methyl-7-propyl-1H-imidazo[5,1-f][1,2,4]triazin-4-one;2-hydroxypropane-1,2,3-tricarboxylic acid 2-[2-ethoxy-5-(4-ethylpiperazin-1-yl)sulfonyl-phenyl]-5-methyl-7-propyl-1H-imidazo[5,1-f][1,2,4]triazin-4-one;2-oxidanylpropane-1,2,3-tricarboxylic acid
- Yang, J Iso-citrate dehydrogenase (IDH) inhibitor US Patent US10752626 (2020)
- Pinkosky, SL; Davenport, AJ; Gleave, LJ; Southey, MW; Ursinyova, NC; Walker, PR; Yarnold, CJ MACROCYCLIC INHIBITORS OF ATP CITRATE LYASE US Patent US20250289793 (2025)
- Li, JJ; Wang, H; Tino, JA; Robl, JA; Herpin, TF; Lawrence, RM; Biller, S; Jamil, H; Ponticiello, R; Chen, L; Chu, CH; Flynn, N; Cheng, D; Zhao, R; Chen, B; Schnur, D; Obermeier, MT; Sasseville, V; Padmanabha, R; Pike, K; Harrity, T 2-hydroxy-N-arylbenzenesulfonamides as ATP-citrate lyase inhibitors. Bioorg Med Chem Lett 17: 3208-11 (2007)
- Yamaguchi, Y; Kato, K; Ichimaru, Y; Jin, W; Sakai, M; Abe, M; Wachino, JI; Arakawa, Y; Miyagi, Y; Imai, M; Fukuishi, N; Yamagata, Y; Otsuka, M; Fujita, M; Kurosaki, H Crystal Structures of Metallo-β-Lactamase (IMP-1) and Its D120E Mutant in Complexes with Citrate and the Inhibitory Effect of the Benzyl Group in Citrate Monobenzyl Ester. J Med Chem 64: 10019-10026 (2021)
- Koerner, SK; Hanai, JI; Bai, S; Jernigan, FE; Oki, M; Komaba, C; Shuto, E; Sukhatme, VP; Sun, L Design and synthesis of emodin derivatives as novel inhibitors of ATP-citrate lyase. Eur J Med Chem 126: 920-928 (2017)
- Zhang, L; Hu, W; Qiu, Z; Li, Z; Bian, J Opportunities and Challenges for Inhibitors Targeting Citrate Transport and Metabolism in Drug Discovery. J Med Chem 66: 9229-9250 (2023)
- Granchi, C ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem 157: 1276-1291 (2018)
- Jernigan, FE; Hanai, JI; Sukhatme, VP; Sun, L Discovery of furan carboxylate derivatives as novel inhibitors of ATP-citrate lyase via virtual high-throughput screening. Bioorg Med Chem Lett 27: 929-935 (2017)
- Huard, K; Gosset, JR; Montgomery, JI; Gilbert, A; Hayward, MM; Magee, TV; Cabral, S; Uccello, DP; Bahnck, K; Brown, J; Purkal, J; Gorgoglione, M; Lanba, A; Futatsugi, K; Herr, M; Genung, NE; Aspnes, G; Polivkova, J; Garcia-Irizarry, CN; Li, Q; Canterbury, D; Niosi, M; Vera, NB; Li, Z; Khunte, B; Siderewicz, J; Rolph, T; Erion, DM Optimization of a Dicarboxylic Series for in Vivo Inhibition of Citrate Transport by the Solute Carrier 13 (SLC13) Family. J Med Chem 59: 1165-75 (2016)
- Bohacek, RS; Dalgarno, DC; Hatada, M; Jacobsen, VA; Lynch, BA; Macek, KJ; Merry, T; Metcalf, CA; Narula, SS; Sawyer, TK; Shakespeare, WC; Violette, SM; Weigele, M X-Ray structure of citrate bound to Src SH2 leads to a high-affinity, bone-targeted Src SH2 inhibitor. J Med Chem 44: 660-3 (2001)
- Harris, GH; Dufresne, C; Joshua, H; Koch, LA; Zink, DL; Salmon, PM; Göklen, KE; Kurtz, MM; Rew, DJ; Bergstrom, JD; Wilson, KE Isolation, structure determination and squalene synthase activity of L-731,120 and L-731,128, alkyl citrate analogs of zaragozic acids A and B Bioorg Med Chem Lett 5: 2403-2408 (1995)
- Beck, J; Vercheval, L; Bebrone, C; Herteg-Fernea, A; Lassaux, P; Marchand-Brynaert, J Discovery of novel lipophilic inhibitors of OXA-10 enzyme (class D beta-lactamase) by screening amino analogs and homologs of citrate and isocitrate. Bioorg Med Chem Lett 19: 3593-7 (2009)
- Gribble, AD; Dolle, RE; Shaw, A; McNair, D; Novelli, R; Novelli, CE; Slingsby, BP; Shah, VP; Tew, D; Saxty, BA; Allen, M; Groot, PH; Pearce, N; Yates, J ATP-citrate lyase as a target for hypolipidemic intervention. Design and synthesis of 2-substituted butanedioic acids as novel, potent inhibitors of the enzyme. J Med Chem 39: 3569-84 (1996)
- McNair, D; Hughes, MJ; Eggelston, D ATP-citrate lyase as a target for hypolipidemic intervention. Sulfoximine and 3-hydroxy-beta-lactam containing analogues of citric acid as potential tight-binding inhibitors. J Med Chem 35: 4875-84 (1992)
- Dolle, RE; Gribble, A; Wilkes, T; Kruse, LI; Eggleston, D; Saxty, BA; Wells, TN; Groot, PH Synthesis of novel thiol-containing citric acid analogues. Kinetic evaluation of these and other potential active-site-directed and mechanism-based inhibitors of ATP citrate lyase. J Med Chem 38: 537-43 (1995)
- Kato, S; Morie, T; Kon, T; Yoshida, N; Karasawa, T; Matsumoto, J Novel benzamides as selective and potent gastrokinetic agents. 2. Synthesis and structure-activity relationships of 4-amino-5-chloro-2-ethoxy-N-[[4-(4-fluorobenzyl)-2- morpholinyl]methyl] benzamide citrate (AS-4370) and related compounds. J Med Chem 34: 616-24 (1991)
- Gribble, AD; Ife, RJ; Shaw, A; McNair, D; Novelli, CE; Bakewell, S; Shah, VP; Dolle, RE; Groot, PH; Pearce, N; Yates, J; Tew, D; Boyd, H; Ashman, S; Eggleston, DS; Haltiwanger, RC; Okafo, G ATP-Citrate lyase as a target for hypolipidemic intervention. 2. Synthesis and evaluation of (3R,5S)-omega-substituted-3-carboxy-3, 5-dihydroxyalkanoic acids and their gamma-lactone prodrugs as inhibitors of the enzyme in vitro and in vivo. J Med Chem 41: 3582-95 (1998)
- ChEMBL_1652714 (CHEMBL4001969) Inhibition of ATP citrate lyase (unknown origin) using sodium citrate as substrate by ADP-Glo luminescence assay
- ChEMBL_29524 (CHEMBL640440) Reversible binding potential for rat ATP-Citrate Lyase as carbon-carbon bond cleavage activity in citrate substrate
- ChEBML_29528 Inhibition against ATP-citrate lyase in liver.
- ChEMBL_2424853 Inhibition of ATP citrate lyase (unknown origin)
- ChEMBL_459068 (CHEMBL925175) Inhibition of human ATP citrate lyase
- ChEMBL_29527 (CHEMBL643514) Inhibition against ATP-citrate lyase in liver
- ChEMBL_455196 (CHEMBL906357) Inhibition of human recombinant ATP-citrate lyase
- ChEMBL_1651959 (CHEMBL4001214) Inhibition of ATP citrate lyase (unknown origin) using sodium citrate as substrate after 60 mins by ADP-Glo luminescence assay
- ChEMBL_1651960 (CHEMBL4001215) Inhibition of Wistar rat liver ATP citrate lyase using tripotassium citrate as substrate by malate dehydrogenase-based oxaloacetate reduction assay
- ChEMBL_29529 (CHEMBL643516) Inhibition against ATP-citrate lyase in rat liver
- ChEMBL_29530 (CHEMBL643517) Inhibition against ATP-citrate lyase in rat liver.
- ChEMBL_29508 (CHEMBL643074) Inhibitory activity against human recombinant ATP-Citrate Lyase (ACL) enzyme
- ChEMBL_219986 (CHEMBL841608) Inhibition of human platelet aggregation collected with sodium citrate as anticoagulant
- ChEMBL_2424854 Binding affinity to ATP citrate lyase (unknown origin) assessed as dissociation constant
- ChEMBL_29522 (CHEMBL640438) Inhibitory activity was tested against rat ATP-Citrate Lyase (ACL) enzyme
- ChEMBL_1803480 (CHEMBL4275772) Non-competitive inhibition of rat liver ACLY using varying levels of citrate
- ChEMBL_2340138 Inhibition of SLC13A5 in human hepatocyte cells assessed as reduction in citrate uptake
- ChEMBL_2340139 Inhibition of SLC13A5 in human HepG2 cells assessed as reduction in citrate uptake
- ChEMBL_29507 (CHEMBL643073) Inhibitory activity was tested against human recombinant ATP-Citrate Lyase (ACL) enzyme
- ChEMBL_29520 (CHEMBL640284) Inhibitory activity was tested against ATP-Citrate Lyase (ACL) enzyme in rat
- ChEMBL_29521 (CHEMBL640437) Inhibitory activity was tested against ATP-Citrate Lyase (ACL) enzyme in rat
- ChEBML_154992 Inhibition of [3H]PAF binding to platelet activating factor receptor citrate-treated dog blood
- ChEMBL_1803477 (CHEMBL4275769) Competitive inhibition of human liver ACLY using varying levels of citrate as substrate
- ChEMBL_857793 (CHEMBL2169036) Competitive inhibition of rat liver ACLY using citrate as substrate by spectrophotometric analysis
- ChEMBL_1803481 (CHEMBL4275773) Non-competitive inhibition of rat liver ACLY using citrate substrate and varying levels of ATP
- ChEMBL_2464283 Inhibition of human SLC13A5 transfected in HEK293T cells incubated for 30 mins by D4-citrate uptake assay
- ChEMBL_29519 (CHEMBL640283) Inhibition of recombinant ATP-Citrate Lyase activity as maleate dehydrogenase catalyzed reduction of oxaloacetate by NADH
- ChEMBL_1557733 (CHEMBL3773092) Inhibition of NaCT in human hepatocytes assessed as [14C]-citrate uptake after 30 mins by scintillation counting method
- ChEMBL_1557738 (CHEMBL3773097) Inhibition of NaCT in mouse hepatocytes assessed as [14C]-citrate uptake after 30 mins by scintillation counting method
- ChEMBL_1557739 (CHEMBL3773098) Inhibition of NaCT in rat hepatocytes assessed as [14C]-citrate uptake after 30 mins by scintillation counting method
- ChEMBL_29518 (CHEMBL640282) Inhibitory activity against rat ATP-citrate lyase activity by the maleate dehydrogenase catalyzed reduction of oxaloacetate by NADH.
- ChEBML_29531 Reversible binding Ki was measured by the inhibition of the carbon-carbon bond cleavage activity against rat ATP-Citrate Lyase
- ChEMBL_945952 (CHEMBL2341026) Binding affinity to GST-tagged human TNF-alpha in 10 mM citrate-phosphate at pH 6.5 by fluorescence assay
- ChEMBL_29505 (CHEMBL642006) Inhibitory activity against human recombinant ATP-Citrate Lyase was measured by the maleate dehydrogenase catalyzed reduction of oxaloacetate by NADH
- ChEMBL_29506 (CHEMBL643072) Inhibitory activity against rat ATP-Citrate Lyase activity was measured by the maleate dehydrogenase catalyzed reduction of oxaloacetate by NADH
- ChEMBL_29531 (CHEMBL643518) Reversible binding Ki was measured by the inhibition of the carbon-carbon bond cleavage activity against rat ATP-Citrate Lyase
- ChEMBL_1557732 (CHEMBL3773091) Inhibition of NaCT (unknown origin) expressed in HEK293 cells assessed as inhibition of [14C]citrate uptake by microbeta plate reader analysis
- ChEMBL_1557734 (CHEMBL3773093) Inhibition of NaDC1 (unknown origin) expressed in HEK293 cells assessed as inhibition of [14C]citrate uptake by microbeta plate reader analysis
- ChEMBL_1557735 (CHEMBL3773094) Inhibition of NaDC3 (unknown origin) expressed in HEK293 cells assessed as inhibition of [14C]citrate uptake by microbeta plate reader analysis
- ChEMBL_29504 (CHEMBL642005) Inhibitory activity against human recombinant ATP-Citrate Lyase activity was measured by the maleate dehydrogenase catalyzed reduction of oxaloacetate by NADH
- ChEMBL_29517 (CHEMBL640281) Inhibitory activity against human recombinant ATP-Citrate Lyase activity was measured by the maleate dehydrogenase catalyzed reduction of oxaloacetate by NADH
- α-Gal A Assay For inhibition assay, 0.1% Triton X-100 extracts were mixed with 4-MU substrates ( 5 mM 4-MU α-ᴅ-galactopyranoside and 0.1 M N-acetyl-ᴅ-galactosamine in 0.1 M citrate buffer (pH 4.5)) in absence or presence of increasing concentrations of DGJ derivatives. For heat-induced degradation, extracts were incubated in 0.1 M citrate buffer (pH 7) at 48 °C for the time indicated. The incubation was terminated by adding 0.1 M citrate buffer (pH 4.5). The absorbance was measured at 340 nm and 460 nm.
- ChEMBL_2272395 Inhibition of Serratia marcescens IMP-1 expressed in Escherichia coli BL21 (DE3) using cephalothin as substrate preincubated for 5 mins followed by substrate addition relative to citrate
- Inhibition Assay Glycosylasparaginase activity was measured in citrate-phosphate buffer at pH 5.8 at 37 C. N-Acetyl-D-glucosamine released during the reaction was measured using the Morgan-Elson assay.
- In Vitro Fucosidase Inhibition Fucosidase inhibition was assessed by preparing a reaction mixture of 50 mM phosphate-citrate pH 4.5, 5 mM MgCl2, 640 nM 4-methylumbelliferyl-alpha-L-fucopyranoside (4MU-FUC), 1 ng/mL rhFUCA1 (R&D Systems), and compound II, III, or IV. Reactions (100 μL) were incubated at 37° C. for 30 minutes and then quenched with the same volume of 600 mM citrate-carbonate buffer, pH 9. Fluorescence was measured by excitation at 360 nm and emission at 450 nm.
- ChEMBL_1900272 (CHEMBL4402387) Inhibition of N-terminal His6-tagged Thermus thermophilus HB8 MqnE expressed in Escherichia coli BL21 (DE3) co-expressing [4Fe-4S]2+ cluster using 3-(1-carboxyvinyloxy)benzoic acid as substrate preincubated for 15 mins in presence of SAM and substrate followed by Ti(3+)citrate addition and measured by discontinuous HPLC analysis
- Inhibition Assay mPGES-1 microsome fractions were prepared from CHO-K1 cells transiently transfected with plasmid encoding the human mPGES-1cDNA. Microsomes were diluted with potassium phosphate buffer containing reduced glutathione (pH7.4), and DMSO containing test compound or DMSO alone was added (such that DMSO final concentration would be 1% in each) and incubated at 4° C. for 20 minutes. Then, the enzymatic reactions were initiated by the addition of PGH2 substrate (final concentration 1 μM) and incubated at 4° C. for 60 seconds. The reaction was terminated by the addition of a citrate solution (final citrate concentration 50 mM) containing ferric chloride (final concentration 1 mg/mL). PGE2 production in the enzyme reaction aliquot was measured using HTRF kit (Cisbio International, catalogue #62P2APEC).
- Inhibition Assay Hsp90 protein is obtained from Stressgen (Cat#SPP-770). Assay buffer: 100 mM Tris-HCl, Ph7.4, 20 mM KCl, 6 mM MgCl2. Malachite green (0.0812% w/v) (M9636) and polyviny alcohol USP (2.32% w/v) (P1097) are obtained from Sigma. A Malachite Green Assay (see Methods Mol Med, 2003, 85:149 for method details) is used for examination of ATPase activity of Hsp90 protein. Briefly, Hsp90 protein in assay buffer (100 mM Tris-HCl, Ph7.4, 20 mM KCl, 6 mM MgCl2) is mixed with ATP alone (negative control) or in the presence of Geldanamycin (a positive control) or a compound of the invention in a 96-well plate. Malachite green reagent is added to the reaction. The mixtures are incubated at 37 C. for 4 hours and sodium citrate buffer (34% w/v sodium citrate) is added to the reaction.
- Caspase Catalytic Activity Assay Experiments were performed in a 384-well format (Greiner no. 781207) as per the conditions noted here. Caspase-1: 2.5 nM enzyme, 6.5 mM WEHD substrate, ECB; Caspase-3: 200 nM enzyme, 3.3 mM DEVD substrate, SCB; Caspase-4: 1 nM enzyme, 10 mM LEHD substrate, HCB; Caspase-5: 20 nM enzyme, 10 mM LEHD substrate, HCB; Caspase-9: 200 nM enzyme, 6.5 mM LEHD substrate, HCB. ECB (Enzo Caspase Buffer): 50 mM HEPES (pH 7.4), 100 mM NaCl, 0.5% Tween 20, 10 mM DTT, and 10% glycerol; SCB (Standard Caspase Buffer): 20 mM PIPES (pH 7.5), 100 mM NaCl, 1 mM EDTA, 10 mM DTT, and 10% sucrose; HCB (High-Citrate Buffer): 50 mM Tris-HCl (pH 7.5), 1 M sodium citrate, 10 mM DTT, and 10% sucrose. Activity was measured as the change in luminescent signal for at least 30 min.
- Enzyme Activity Assay Cells were washed two times with 200 μL dPBS followed by the addition of 70 μL of substrate (2.11 mM 3 mM 4-MU-α-D-glu) in citrate-phosphate buffer (30 mM sodium citrate, 40 mM sodium phosphate dibasic, pH 4.0), and 2.5% DMSO to rows 1-12. Following incubation at 37° C. with 5% CO2 for about 3 h, 70 μL of stop buffer (0.4 M glycine pH 10.8) was added to rows 1-12. The plate was read in a Victor2 multilabel counter-Wallac fluorescent plate reader and the fluorescence at F460 nm was determined b at an excitation of 355 nm and emission of 460 nm using 1 second read time per well. Enzyme activity per μg of protein in the supernatant was calculated from the amount of fluorescence emitted, which is directly proportional to the amount of substrate hydrolyzed, and hence, the amount of Gaa activity in the lysate.
- Inhibition Assay Hsp90 protein is obtained from Stressgen (Cat#SPP-770). Assay buffer: 100 mM Tris-HCl, Ph7.4, 20 mM KCl, 6 mM MgCl2. Malachite green (0.0812% w/v) (M9636) and polyvinyl alcohol USP (2.32% w/v) (P1097) are obtained from Sigma. A Malachite Green Assay (see Methods Mol Med, 2003, 85:149 for method details) is used for examination of ATPase activity of Hsp90 protein. Briefly, Hsp90 protein in assay buffer (100 mM Tris-HCl, Ph7.4, 20 mM KCl, 6 mM MgCl2) is mixed with ATP alone (negative control) or in the presence of Geldanamycin (a positive control) or a compound of the invention in a 96-well plate. Malachite green reagent is added to the reaction. The mixtures are incubated at 37° C. for 4 hours and sodium citrate buffer (34% w/v sodium citrate) is added to the reaction. The plate is read by an ELISA reader with an absorbance at 620 nm.
- Inhibition Assay Hsp90 protein is obtained from Stressgen (Cat#SPP-770). Assay buffer: 100 mM Tris-HCl, Ph7.4, 20 mM KCl, 6 mM MgCl2. Malachite green (0.0812% w/v) (M9636) and polyvinyl alcohol USP (2.32% w/v) (P1097) are obtained from Sigma. A Malachite Green Assay (see Methods Mol Med, 2003, 85:149 for method details) is used for examination of ATPase activity of Hsp90 protein. Briefly, Hsp90 protein in assay buffer (100 mM Tris-HCl, Ph7.4, mM KCl, 6 mM MgCl2) is mixed with ATP alone (negative control) or in the presence of Geldanamycin (a positive control) or a compound of the invention in a 96-well plate. Malachite green reagent is added to the reaction. The mixtures are incubated at 37° C. for 4 hours and sodium citrate buffer (34% w/v sodium citrate) is added to the reaction. The plate is read by an ELISA reader with an absorbance at 620 nm.
- Malachite Green Assay Hsp90 protein is obtained from Stressgen (Cat#SPP-770). Assay buffer: 100 mM Tris-HCl, Ph7.4, 20 mM KCl, 6 mM MgCl2. Malachite green (0.0812% w/v) (M9636) and polyvinyl alcohol USP (2.32% w/v) (P1097) are obtained from Sigma. A Malachite Green Assay (see Methods Mol Med, 2003, 85:149 for method details) is used for examination of ATPase activity of Hsp90 protein. Briefly, Hsp90 protein in assay buffer (100 mM Tris-HCl, Ph7.4, 20 mM KCl, 6 mM MgCl2) is mixed with ATP alone (negative control) or in the presence of Geldanamycin (a positive control) or a compound of the invention in a 96-well plate. Malachite green reagent is added to the reaction. The mixtures are incubated at 37° C. for 4 hours and sodium citrate buffer (34% w/v sodium citrate) is added to the reaction. The plate is read by an ELISA reader with an absorbance at 620 nm.
- Human Platelet Adhesion Assay Flat bottom microtiter plates (96-well) were coated with soluble type I collagen. Unoccupied protein binding sites on the wells were blocked with bovine serum albumin. Human platelets were isolated from blood anticoagulated with sodium citrate by gel-filtration. The gelfiltered platelet suspension was added to the protein-coated wells in the absence or presence of an inhibitor. Following incubation for 30 min, the plates were washed, and the number of adherent platelets measured using the colorimetric assay.
- Inhibition Assay mPGES-1 microsome fractions were prepared from CHO-K1 cells transiently transfected with plasmid encoding the human mPGES-1cDNA. Microsomes were diluted with potassium phosphate buffer containing reduced glutathione (pH7.4), and DMSO containing test compound or DMSO alone was added (such that DMSO final concentration would be 1% in each) and incubated at 4° C. for 20 minutes. Then, the enzymatic reactions were initiated by the addition of PGH2 substrate (final concentration 1 μM) and incubated at 4° C. for 60 seconds. The reaction was terminated by the addition of a citrate solution (final citrate concentration 50 mM) containing ferric chloride (final concentration 1 mg/mL). PGE2 production in the enzyme reaction aliquot was measured using HTRF kit (Cisbio International, catalogue #62P2APEC). The solution free of test compound was used as positive control, and the solution free of test compound and microsome sample was used as negative control. 100% activity was defined as PGE2 production in the positive control minus PGE2 production in negative control. IC50 value was calculated by standard method.
- NS5B RNA Polymerase Assay NS5B RNA polymerase reactions for IC50 determination were performed in 96-well microtiter plates in 20 μL reactions containing assay buffer (50 mM Na+ HEPES, 1 mM MgCl2, 0.75 mM MnCl2, 2 mM DTT, pH 7.5), 1 U/μL SUPERase. In (Life Technologies), 20 ng/μL IRES RNA template, 1 μM each ATP, CTP, GTP, and UTP (Life Technologies) including [α-32P]-CTP at a final specific activity of 50 Ci/mmol (PerkinElmer), test compounds in 10-point half-log dilution series, and NS5B polymerase. Reactions were incubated at 27° C. for 60 minutes and terminated by dilution to 100 μL in 1× stop solution (final concentrations 144 mM Na+ citrate, 1.44 M NaCl, 10 mM EDTA, pH 7.0). 80 μL stopped samples were applied by vacuum to nylon membrane filtermats (PerkinElmer), washed 4× in 60 mM Na+ citrate, 600 mM NaCl (pH 7.0), rinsed sequentially in H2O and EtOH, dried, and counted in a MicroBeta2 counter with scintillation cassette (PerkinElmer).
- HCV Replicon Assay The HCV NS5B reaction was performed in a 20 μL mixture containing varying concentrations of the test compound, 1 μM of all four natural ribonucleotides, [α-32P]UTP, 20 ng/μL of genotype 1b (−) IRES RNA template, 1 unit/μL of SUPERase·In (Ambion, Austin, Tex.), 40 ng/μL of wild type or S282T NS5B Genotype 1b, 1 mM MgCl2, 0.75 mM MnCl2, and 2 mM DTT in 50 mM Hepes buffer (pH 7.5). The reaction was quenched by adding 80 μL of stop solution (12.5 mM EDTA, 2.25 M NaCl, and 225 mM sodium citrate) after incubating at 27° C. for 30 minutes. The radioactive RNA products were separated from unreacted substrates by passing the quenched reaction mixture through a Hybond N+ membrane (GE Healthcare, Piscataway, N.J.) using a dot-blot apparatus. The RNA products were retained on the membrane and the free nucleotides were washed out. The membrane was washed 4 times with a solution containing 0.6 M NaCl and 60 mM sodium citrate. After rinsing the membrane with water followed by ethanol, the membrane was exposed to a phosphorscreen and the products were visualized and quantified using a phosphorimager.
- Enzymatic Assay All enzymatic assays are carried out in triplicate at 37° C. using a stopped assay procedure by measuring the amount of 4-nitrophenolate liberated as determined by absorption measurements at 400 nm. Reactions (50 μL) are initiated by the addition, via syringe, of enzyme (3 μL). Time-dependent assay of β-hexosaminidase has revealed that the enzyme is stable in the buffer over the period of the assay: 50 mM citrate, 100 mM NaCl, 0.1% BSA, pH 4.25. β-hexosaminidase is used at a concentration of 0.036 mg/mL with pNP-GlcNAc as a substrate at a concentration of 0.5 mM.
- Enzyme Assay To 2.5 μl of supernatant (in 96-well plates) was added 17.5 μl reaction buffer (citrate phosphate buffer, pH 4.5, no Triton X-100), and 50 μl of 4-methyl umbelliferone (4-MU)-labeled substrate, β-glucopyranoside, or a labeled negative controls α-glucopyranoside or α-galacatopyranoside). Plates were incubated at 370 for 1 hour, followed by the addition of 70 μl stop buffer (0.4 M glycine-NaOH, pH 10.6). Activity of GCase was determined by measuring the emission at 460 nm by exciting at 355 nm using a 1 second read time per well (Victor2 multilabel counter-Wallac).
- PTP activity assay The enzyme activity was measured using p-nitrophenyl phosphate (pNPP) as substrate in a 96-well plate and by detecting the absorbance at 405 nm for the amount of produced p-nitrophenol. Purified human recombinant PTP1B or TCPTP (0.05 µg) in 50 µL buffer containing 50 mM citrate (pH 6.0), 0.1 M NaCl, 1 mM EDTA, and 1 mM DTT and test compounds were added to each well. Each mixture was pre-incubated for 15 min at room temperature, followed by the addition of 50 µL of reaction buffer containing 2 mM pNPP and incubated at 37°C for 30 min.
- Fluorogenic Assay The assay was run in Optiplate 96-wells black plates, in a total reaction volume of 200 μL. NAAA protein preparation (4.0 μg) was pre-incubated for 10 min with various concentrations of test compounds or vehicle control (5% DMSO) in 100 mM citrate/phosphate buffer (pH 4.5) containing 3.0 mM DTT, 0.1% Triton X-100, 0.05% BSA, 150 mM NaCl. N-(4-methyl-2-oxo-chromen-7-yl)-hexadecanamide was used as a substrate (5.0 μM) and the reaction carried over for 30 min at 37° C. The samples were then read in a Perkin Elmer Envision plate reader using an excitation wavelength of 360 nm and emission 460 nm.
- Inhibition Assay The degree of inhibition activity for PTP1B was investigated by using 2 mM p-nitrophenyl phosphate (p-NPP) as a substrate to measure the dephosphorylation degree. First, PTP1B diluted with distilled water was reacted with 2 mM p-nitrophenyl phosphate {p-NPP, 0.1 M NaCl, 1 mM EDTA, 50 mM citrate pH 6.0, and 1 mM dithiothreitol (DTT)} and Compound 1 at various concentrations at 30° C. for 30 minutes, and then the reaction was terminated with a 1 N-sodium hydroxide (NaOH) solution. The absorbance of the samples thus prepared was measured to confirm the inhibition degree (IC50: The half maximal inhibitory concentration) for PTP1B activity according to the concentration of Compound 1.
- PTP1B Assay In each 96-well plates (total 200 μL of volume), there were 2 mM p-NPP and PTP1B (0.05-0.1 μg) in a buffer containing 50 mM citrate (pH 6.0), 0.1 M NaCl, 1 mM EDTA, and 1 mM dithiothreitol (DTT) with or without test compound, following by pre-incubated at 37 °C for 10 min, and then implemented with 50 μL of p-NPP. The reaction was finally added with 10 M NaOH. The amount of product (p-NP) was measured the absorbance at 405 nm. The excess amounts of 2mM p-NPP were determined with absorbance at 405 nm obtained in the absence of PTP1B enzyme.
- hACLY ADP-Glo Activity Assay Table 3: Test compounds were 3-fold serially diluted in DMSO over 11-point concentration range and dispensed onto a 384-well plate. Recombinant human ACLY full length protein was purified. Concentrations of ACLY protein, sodium citrate, coenzyme A, and ATP in the reaction were optimized for standardized homogenous enzyme assay using ADP-Glo™ Kinase (Promega Inc.). The assay measured ADP formed from the enzymatic reaction.The reaction buffer consisted of the assay buffer (50 mM HEPES pH 8.0, 10 mM MgCl2, 4 mM 1,4-Dithiothreitol, 0.01% Brij® 35).ACLY protein (0.5 nM) was added to the prepared reaction buffer, and the mixture was dispensed into the assay plate and incubated for 30 minutes at room temperature. Next, 15 μM sodium citrate, 1 μM coenzyme A, and 80 μM ATP were added into the assay plate and incubated for 60 minutes at room temperature. The final reaction volume for each well was 5 μL.5 μL of ADP-Glo™ reagent was added, and the mixture was incubated for 40 minutes at room temperature. 10 μl ADP-Glo™ detection reagent was added, and the mixture was incubated for 30 minutes at room temperature. Luminescence was determined using EnVision microplate reader with ultra-luminescence module (Perkin Elmer Inc). Concentration-response curve-fitting and IC50 determination was performed using Xlfit Software (version 5.5.0) with a four-parameter logistic regression fit model.
- β-Gal Inhibitory Assay β-Gal activity was measured by using 4-methylumbelliferyll-β-D-galactopyranoside in buffer B (0.15 M sodium citrate, pH 4.5, and 0.2 M NaCl) as a substrate, and incubating at 37 °C for 6 min. The reaction was terminated by adding 0.2 M glycine-NaOH buffer (pH 10.7). The liberated 4-methylumbelliferyll was measured with a fluorescence plate reader (excitation 355 nm; emission 460 nm; fluoroskan ascent, Thermo electron Corp.). For inhibition assays, the ligand compounds (galactose, 25 mM; DGJ, 250 μM; NOEV, 2.5 μM; 6S-NBI-DGJ, 250 μM; 6S-NBI-GJ, 2.5 mM; and NBT-DGJ, 2.5mM) were added to the purified β-Gal, and then the activityof β-Gal was measured as described above.
- Fluorescence Resonance Energy Transfer (FRET) Assay Recombinant CathepsinD was expressed in CHO cells. The assay buffer for CathepsinD is 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The CathepsinD enzyme (9 nM) is pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays are effectively started by the addition of different FRET substrates (20 nM for CathepsinD) and the combination is incubated for one hour. The FRET assay is terminated with by addition of Tris buffer, which raises the pH to neutrality, and the fluorescence is determined. The FRET substrate is a peptide with commercially available fluorophore and quencher, on opposite sides of the BACE cleavage site.
- GCase IC50 Assay The assays were performed at 37°C with 4-MU-β-ᴅ-Glu as the substrate in Mcllvaine buffer (sodium citrate (100mM) and sodium phosphate (200mM); pH 5.2 or 7.0) containing sodium taurocholate (0.25% w/v) and Triton X-100 (0.10% v/v). Enzyme solution (12.5µL, 0.1mg/mL) and inhibitor (7.5µL, various concnetrations) were mixed and incubated at 37°C for 30 min, followed by the addition of substrates (30µL, 4.0mM, in Mcllvaine buffer, pH 5.2 or 7.0) and further incubated for 10 min. The assay was terminated by adding glycine/NaOH buffer (150µL, 100mM, pH 10.6). The amount of released 4-metylumbelliferone (4-MU) was determined by fluorimetry (excitation 355nm, emmision 460nm).
- PTP1B Inhibitory Assay PTP1B activity was measured by adding 2mM p-NPP and PTP1B in a 50 mM citrate buffer (pH 6.0, 0.1 M NaCl, 1 mMEDTA, and 1 mM dithiothreitol), with or without tested compounds. The plate was pre-incubated at 37 °C for 10 min, and then 50 μL of p-NPP in buffer was added. After incubating at 37 °C for 30 min, the reaction was then terminated with 1 N NaOH. The amount of produced p-nitrophenyl after enzymatic dephosphorylation was obtained by measuring the absorbance at 405 nm using a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA, USA). The non-enzymatic reactions of 2 mM p-NPP were determined by measuring the increase in absorbance at 405 nm without PTB1B enzyme.
- Spectrophotometric 384 Well Assay Assay reactions are then carried out in 384-well plates, with hACC2 in an appropriate dilution and at final assay concentrations (f.c.) of 100 mM Tris (pH 7.5), 10 mM trisodium citrate, 25 mM KHCO3, 10 mM MgCl2, 0.5 mg/ml BSA, 3.75 mM reduced L-glutathione, 15 U/ml lactate dehydrogenase, 0.5 mM phosphoenolpyruvate, 15 U/ml pyruvate kinase, compounds at different concentrations at final DMSO concentrations of 1%.The enzymatic reaction is then started by addition of a mixture of NADH, acetyl Coenzyme A (both 200 μM f.c.) and ATP (500 uM f.c.). The decrease of the optical density (slope S) is then determined at 25° C. at a wavelength of 340 nm over 15 minutes in a spectrophotometric reader.
- ADP-Glo Assay Materials and Instruments Used in the ADP-Glo AssayVendor Cat No.Materials ACL Sino Biological 11769-H07BCoANa2 Sigma C3144Potassium Citrate Sigma 89306ATP Promega V915BADP-GloTM Kinase Assay Promega V9102kitHEPES Life Technologies 15630080MgCl2 Sigma M1028Brij 35 detergent Merck 203728DTT Sigma 646563DMSO MP 196055Optiplate-384, White 384- Perkin Elmer 6007290well384 dilution plate Corning 3657Topseal A Perkin Elmer E534196-well plate Nunc 249944InstrumentPlate reader Perkin Elmer Envision 2104Centrifuge Eppendorf 5810RCompounds of the present invention were evaluated in an ADP-GLO assay as follows:a) Dilute cpd 1:3 in succession in DMSO by hand for each cpds for 12 ptsb) Add 0.1 μL diluted cpd solution to assay plate, each dose with 2 replicatesc) Centrifuge 1000 RPM for 1 mind) Add 5 μL ACL working solution to 384-well assay plate, centrifuge 1000 RPM for 1 mine) Incubate at 25° C. for 15 minf) Add 5 μL substrate working solution to initiate reactiong) Final ACL reaction concentrations: 3 nM ACL, 15 μM ATP, 3 μM CoA, 300 μM Citrate, 0.01% Brij35, 4 mM DTT, 1% DMSO;h) Reference final conc: 30 uM starting Conc, 3× dilution, 11+0 points. Test cpd conc: 30/100 uM starting Conc, 3× dilution, 11+0 points.i) Incubate at 25° C. for 60 minj) Add 10 μL ADP Glo reagent, centrifuge 1000 RPM for 1 mink) Incubate at 25° C. for 40 minl) Add 20 μL kinase detection reagent, centrifuge 1000 RPM for 1 minm) Incubate at 25° C. for 40 minn) Read on Envision for US LUM as RLU
- Enzyme Assay To 2.5 μl of supernatant (in 96-well plates) was added 17.5 μl reaction buffer (citrate phosphate buffer, pH 4.5, no Triton X-100), and 50 μl of 4-methyl umbelliferone (4-MU)-labeled substrate, β-glucopyranoside, or a labeled negative controls (α-glucopyranoside or α-galacatopyranoside). Plates were incubated at 37° for 1 hour, followed by the addition of 70 μl stop buffer (0.4 M glycine-NaOH, pH 10.6). Activity of GCase was determined by measuring the emission at 460 nm by exciting at 355 nm using a 1 second read time per well (Victor2 multilabel counter-Wallac) Enzyme activity was normalized to the amount in μl of lysate added, and enzyme activity per μl of lysate was estimated.
- MALT1 Protease Assay 1 The final assay buffer includes 2 nM of MALT1 full-length protein, 50 μM Ac-LRSR-AMC substrate, 50 mM Tris pH 7.5, 600 mM Sodium Citrate, 1 mM DTT, 1 mM EDTA, and 0.05% BSA in 384-well plate format using black microtiter square well plates (Optiplate 384-F, Perkin Elmer). Test compounds were dissolved in 100% DMSO at stock of 10 mM, with final DMSO concentration 0.1%. Test compounds were pre-incubated with MALT1 protein for 2 h at room temperature. Substrate was added after the pre-incubation and fluorescence signal was measured using Envision at excitation 355 nm and emission 460 nm after 8 hr incubation at RT. Increase in the assay signal was linear over this period and proportional with increase in the enzyme content.
- MALT1 Protease Assay 2 The final assay buffer includes 1 nM of MALT1 full-length protein, 50 μM Ac-LRSR-AMC substrate, 50 mM Tris pH 7.5, 600 mM Sodium Citrate, 1 mM DTT, 1 mM EDTA, and 0.05% BSA in 384-well plate format using black microtiter square well plates (Optiplate 384-F, Perkin Elmer). Test compounds were dissolved in 100% DMSO at stock of 10 mM, with final DMSO concentration 0.1%. Test compounds were pre-incubated with MALT1 protein for 2 h at room temperature. Substrate was added after the pre-incubation and fluorescence signal was measured using Envision at excitation 355 nm and emission 460 nm after 8 hr incubation at RT. Increase in the assay signal was linear over this period and proportional with increase in the enzyme content.
- RNA-Dependent RNA HCV NS5B (polymerase) Assay and IC50 Determination The reaction mixtures consisted of 50 mM Hepes-KOH, pH 7.5, 5 mM MgCl2, 5 mM DTT, 2% glycerol, 0.01% Triton X-100, 0.5 uM polyA:U16 substrate, purified HCV RNA-dependent RNA polymerase, 10 μM UTP, and 32P-UTP (Perkin Elmer). The reaction mixtures incubated at 30° C. for 60 minutes, and then filtered through Zeta probe membrane (BioRad). The filter was washed with 5×SSC (75 mM sodium citrate, pH 7 and 750 mM NaCl), and the radiolabeled RNA products were quantitated by microbeta (Perkin Elmer). For IC50 determination, different concentrations of inhibitors were added to the polymerase reaction mixtures, and incubated at 37° C. for 60 minutes. IC50 values were determined using GraFit (Erithaus software).
- Biochemical Assay The potency of compounds to inhibit MALT-1 (Isoform1) enzyme was tested in a Fluorescent assay using recombinant MALT-1 (Isoform1, aa-840) generated in-house. The assay buffer was 50 mM HEPES (pH 7.5), 100 mM NaCl, 10 mM DTT, 1 mM EDTA, 0.9 M sodium citrate, 0.01% CHAPS. 1.5 nM of MALT-1 enzyme was incubated with various concentrations of test compounds (1 M) (1% DMSO) in the presence of buffer for 30 minutes at 30° C. The protease reaction was initiated by adding 50 μM of AC-LRSR-MAC (Peptide International, USA) substrate and incubated for 240 min. After 240 min incubation, fluorescence emission of the samples at 460 nm was measured at an excitation of 355 nm. The percent inhibition was calculated using the following formula: (Control GD−(Sample GD/Control GD))×100.
- In Vitro Evaluation in ATP-Citrate Lyase (ACLY) Enzymatic Assay Table 4: ATP citrate lyase was obtained from Sino Biological Inc. Cat #11769-H07B, Lot #LC08DE1701. It was prepared from a DNA sequence encoding the human ACLY (P53396) (Met 1-Met 1101) expressed in Baculovirus-Insect Cells, with a polyhistidine tag at the N-terminus. The recombinant human ACLY consists of 1120 amino acids and has a calculated molecular mass of 123 kDa. It migrates as an approximately 110 kDa band in SDS-PAGE under reducing conditions. The enzyme came as a lyophilized powder in sterile 20 mM Tris, 500 mM NaCl, pH 8.0, 10% glycine. Normally 5%-8% trehalose and mannitol are added as protectants before lyophilization. The recombinant human ACLY from Sino Biological was formulated in 45 mM Tris-HCl, pH 8.0, 124 mM NaCl, 2.4 mM KCl, 18 mM glutathione, 10% glycerol, and 3 mM DTT.To detect ADP produced from ACL assay, the ADP-Glo™ assay format from Promega, Madison, WI, was used, to detect the conversion of ATP to ADP. In the ADP-Glo™ (Promega, Cat #V9101) method, as shown in FIG. 1 , the protocol provided by the manufacturer was followed. This assay is performed in two steps: i) after the ACL-mediated enzymatic reaction that utilizes ATP and produces ADP, ADP-Glo™ Reagent is added to terminate the kinase reaction and deplete the remaining ATP, and ii) the Kinase Detection Reagent is added to convert ADP to ATP and allow the newly synthesized ATP to be measured using a luciferase/luciferin reaction. The light generated, measured in a luminometer, correlates to the amount of ADP generated in the ACL assay, which is indicative of ACL activity.
- Inhibitory Activity on Human ACC1 and the ACC2 Recombinant human ACC1 and recombinant human ACC2, which were prepared by the method mentioned above, were preincubated with assay buffer solution (50 mM HEPES-KOH (pH 7.4), 10 mM magnesium chloride, 6-10 mM potassium citrate, 4 mM reduced form of glutathione, 1.5 mg/ml bovine serum albumin) for one hour. Then, 0.2 μL of each this invention compound solution (in DMSO) were dispensed to 384-well microplate, 5 μL of the preincubated enzyme solution and 5 μL of substrate solution (50 mM HEPES-KOH (pH 7.4), 1 mM ATP, 0.8 mM acetyl CoA and 25-50 mM potassium bicarbonate) were added to microplate. After centrifugation and shaking, the reaction mixtures were incubated in a humidified box at room temperature for 1 to 3 hours. After the incubation, the enzyme reactions were stopped by the addition of EDTA. Then, after the samples were cocrystallized with CHCA (α-cyano-4-hydroxy cinnamic acid) matrices on MALDI target plate, by using the matrix assist laser deionization time-of-flight mass spectrometer (MALDI-TOF MS), samples were measured in reflector negative mode. Deprotonated ions of acetyl CoA (AcCoA) of substrate and malonyl CoA (MalCoA) of the reaction product were detected, then, the conversion rates of acetyl CoA to malonyl CoA was calculated by the intensity of [MalCoA-H]−/(Intensity of [MalCoA-H]−+Intensity of [AcCoA-H]−) using each signal strength. The 50% inhibitory concentration (IC50) was calculated from the inhibition rate of the enzymatic reaction at each concentration of the compounds. In addition, potassium citrate concentrations in assay buffer solution, potassium hydrogen carbonate concentrations in substrate solution and incubation time were adjusted by each lot of enzyme.
- The Measurement of Inhibitory Activity on Human ACC1 and the ACC2 Recombinant human ACC1 and recombinant human ACC2, which were prepared by the method mentioned above, were preincubated with assay buffer solution (50 mM HEPES-KOH (pH 7.4), 10 mM magnesium chloride, 6-10 mM potassium citrate, 4 mM reduced form of glutathione, 1.5 mg/ml bovine serum albumin) for one hour. Then, 0.2 μL of each this invention compound solution (in DMSO) were dispensed to 384-well microplate, 5 μL of the preincubated enzyme solution and 5 μL of substrate solution (50 mM HEPES-KOH (pH 7.4), 1 mM ATP, 0.8 mM acetyl CoA and 25-50 mM potassium bicarbonate) were added to microplate. After centrifugation and shaking, the reaction mixtures were incubated in a humidified box at room temperature for 1 to 3 hours. After the incubation, the enzyme reactions were stopped by the addition of EDTA. Then, after the samples were cocrystallized with CHCA (α-cyano-4-hydroxy cinnamic acid) matrices on MALDI target plate, by using the matrix assist laser deionization time-of-flight mass spectrometer (MALDI-TOF MS), samples were measured in reflector negative mode. Deprotonated ions of acetyl CoA (AcCoA) of substrate and malonyl CoA (MalCoA) of the reaction product were detected, then, the conversion rates of acetyl CoA to malonyl CoA was calculated by the intensity of [MalCoA-H]−/(Intensity of [MalCoA-H].+Intensity of [AcCoA-H] ) using each signal strength. The 50% inhibitory concentration (IC50) was calculated from the inhibition rate of the enzymatic reaction at each concentration of the compounds. In addition, potassium citrate concentrations in assay buffer solution, potassium hydrogen carbonate concentrations in substrate solution and incubation time were adjusted by each lot of enzyme.
- Fluorogenic Assay h-NAAA: The assay was run in 96-well microplates (Black OptiPlate-96 F; PerkinElmer, Massachusetts, USA), in a total reaction volume of 200 μL. h-NAAA protein preparation (4.0 μg) was pre-incubated for 30 min with various concentrations of test compounds or vehicle control (DMSO 5%) in 100 mM citrate/phosphate buffer (pH 4.5) containing 3.0 mM DTT, 0.1% NP40 0.1%, 0.05% BSA, 150 mM NaCl. N-(4-methyl-2-oxo-chromen-7-yl)-hexadecanamide (PAMCA) was used as a substrate (2.0 μM) and the reaction carried for 50 min at 37° C. Fluorescence was measured with EnVision 2014 Multilabel Reader (PerkinElmer, Massachusetts, USA) using an excitation wavelength of 355 nm and emission 460 nm. IC50 values were calculated by non-linear regression analysis of log [concentration]/inhibition curves using GraphPad Prism 5 (GraphPad Software Inc., CA, USA) applying a standard slope curve fitting.
- Inhibition Assay Various concentrations of the substances to be tested are dissolved in dimethyl sulphoxide and diluted with water. In white 96-well plates having a flat bottom, 20 μl of substance dilution are mixed with 20 μl of ecarin solution (ecarin reagent, from Sigma E-0504, final concentration 20 mU per batch) in Ca buffer (200 mM Hepes+560 mM sodium chloride+10 mM calcium chloride+0.4% PEG) or with 20 μl of Ca buffer (as unstimulated control). Furthermore, 20 μl of fluorogenic thrombin substrate (from Bachem I-1120, final concentration 50 μmol/l) and 20 μl of citrate plasma (from Octapharma) are added and homogenized thoroughly. The plate is measured in a SpectraFluorplus Reader using an excitation filter of 360 nm and an emission filter of 465 nm each minute over a period of 20 minutes.
- ACCase-Coupled Enzyme Assay Assays were performed in 384-well clear bottom plates (Corning; catalog no. 3702), that contained inhibitor solvated in DMSO. To each well of the plate consisting of 50 mM Hepes (pH 8.0), 100 mM KCl, 1 mM TCEP, 5 mM MgCl2, 0.1 mg/mL BSA, 0.005% (vol/vol) Tween 20, 30 nM E. coli BC, 50 nM E. coli CT, 50 nM biotinylated E. coli BCCP, and 0.5 unit/mL E. coli purine nucleoside phosphorylase was added. After 5 min of coincubation of inhibitors with solution 1, the ACCase reaction was initiated by addition of 40 uL of solution 2, which consisted of 500 uM citrate (pH 4.2), 150 uM 7-methyl-6-thioguanosine (MESG; Berry and Associates), and 0.005% (vol/vol) Tween 20. Reaction progress was monitored by the increase in absorbance at 360 nm. Inhibitor potency was assessed by duplicate 20-point titrations of inhibitor from 96 uM to 9.6 nM. Inhibition data were fit to the standard IC50 equation.
- Dundee MALDI-TOF Mass Spectrometry Assay (ICs) USP30 (25 ng/μl) tested against K48-linked diubiquitin (5.6 μM). USP30 was diluted in a buffer containing 40 mM Tris, 0.01% BSA, 1 mM DTT and K48 in 40 mM Tris, 0.01% BSA. The compounds were pre-incubated with the USP30 for 5 mins at room temp before the K48 dimer addition. The assay mixture was then incubated for 45 mins at room temp. The assay was stopped by the addition of TFA to a final concentration of 2% (v/v). Acidified samples of the DUB assays were mixed with 0.5 mM 15N-ubiquitin and then with one part of 2% (v/v) TFA and one part of 2.5 DHAP matrix solution (7.6 mg of 2.5 DHAP in 375 ml ethanol and 125 ml of an aqueous 12 mg ml 1 diammonium hydrogen citrate). Then 250 nl of these solutions were spotted onto an MTP AnchorChip 1,536 TF and this is analysed on the Bruker rapifleX MALDI-TOF.
- Enzymatic Assay r-AC protein samples were pre-incubated with various concentrations of test compounds or vehicle control in 100 mM NaH2PO4/citrate buffer pH 4.5, 0.1% Nonidet P-40, 3 mM DTT for 30 min at 37° C. Samples were incubated with 100 μM N-lauroyl ceramide (Nu-Chek Prep, Elysian, Minn.) at 37° for 30 min. The reaction was stopped by addition of a mixture of chloroform/methanol (2:1 vol/vol) containing 1 nmol of heptadecanoic acid (HDA; NuChek Prep). The organic phases were collected, dried under nitrogen, and analyzed by LC/MS in the negative-ion mode using heptadecanoic acid (HDA) as internal standard (m/z=199 for lauric acid, m/z=269 for HDA). HDA was eluted on an XDB Eclipse C18 column isocratically at 2.2 mL/min for 1 min with a solvent mixture of 95% methanol and 5% water, both containing 0.25% acetic acid, and 5 mM ammonium acetate. The column temperature was 50° C.
- Human KLK7 Fluorescence-Lifetime Assay Recombinant human KLK7 (5 nM concentration) was pre-incubated with inhibitor at various concentrations for 1 h at room temperature in 50 mM sodium citrate buffer at pH 5.6, containing 150 mM NaCl and 0.05% (w/v) CHAPS. The enzyme reaction was initiated by addition of the substrate Ac-Glu-Phe-Lys-Pro-Ile-Leu-Trp-Arg-Leu-Gly-Cys(PT14)-Glu-NH2 (0.8 μM; Biosyntan, Berlin, Germany, product BS-#7599). Fluorescence-lifetime measurements were conducted on an Ultra Evolution fluorescence lifetime reader (TECAN, M nnedorf, Switzerland). The excitation light source was a semiconductor laser at 405 nm, producing picosecond light pulses with a selected repetition frequency of 10 MHz. The emission was collected through a 450 nm bandpass filter with 25 nm bandwidth. The measurement time per well was set to 1 s, yielding approximately 1,000 counts in the peak channel. The parameters used as assay readout were the mono-exponential lifetimes.
- In Vitro Enzymatic FRET Assay Recombinant CatD was expressed in CHO cells. The assay buffer for CatD was 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The CatD enzyme (9 nM) was pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays was effectively started by the addition of different FRET substrates (20 nM for CatD) and the combination was incubated for one hour. The FRET assay was terminated with by addition of tris buffer, which raises the pH to neutrality, and the fluorescence was determined. The FRET substrate was a peptide with commercially available fluorophore and quencher, on opposite sides of the CatD cleavage site. The CatD substrate peptide sequence was based on sequence #1 of Table 1 from Gulnik et al., FEBS Lett. 413(2):379-384 (1997). Proteolytic cleavage of the FRET substrate released quenching of fluorescence (CatD excitation 500 nm and emission 580 nm).
- In Vitro Enzymatic FRET Assay Recombinant CatD was expressed in CHO cells. The assay buffer for CatD was 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The CatD enzyme (9 nM) was pre-incubated for one hour with inhibitors, typically in about luL of DMSO according to a serial dilution, is added thereto. The assays was effectively started by the addition of different FRET substrates (20 nM for CatD) and the combination was incubated for one hour. The FRET assay was terminated with by addition of tris buffer, which raises the pH to neutrality, and the fluorescence was determined. The FRET substrate was a peptide with commercially available fluorophore and quencher, on opposite sides of the CatD cleavage site. The CatD substrate peptide sequence was based on sequence #1 of Table 1 from Gulnik et al., FEBS Lett. 413(2):379-384 (1997). Proteolytic cleavage of the FRET substrate released quenching of fluorescence (CatD excitation 500 nm and emission 580 nm).
- Inhibition Assay Various concentrations of the substances to be tested are dissolved in dimethyl sulphoxide and mixed with an aqueous refludan solution (10 μg/ml). In clear 96-well plates having a flat bottom, 30 μl of citrate plasma (Octapharma) are mixed with 10 μl of the substance dilution. Then, either 20 μl of a solution of a rattlesnake toxin (Russel viper venom (RVV); RVV reagent: Pentapharm 121-06, final concentration 0.6 mU) in an aqueous calcium chloride solution buffer (final concentration of calcium chloride 0.05 M) or 20 μl of the aqueous calcium chloride solution (final concentration of calcium chloride 0.05 M) without RVV reagent (as reference for an unstimulated sample) are added. After addition of 20 μl of ChromozymX substrate (final concentration 1.6 mmol/l, Bachem L-1565, diluted in water) the samples are measured in a SpectraFluor Reader using a measurement filter of 405 nm each minute over a period of 20 minutes.
- Spectrophotometric 384 Well Assay Assay reactions are then carried out in 384-well plates, with hACC2 in an appropriate dilution and at final assay concentrations (f.c.) of 100 mM Tris (pH 7.5), 10 mM trisodium citrate, 25 mM KHCO3, 10 mM MgCl2, 0.5 mg/ml BSA, 3.75 mM reduced L-glutathione, 15 U/ml lactate dehydrogenase, 0.5 mM phosphoenolpyruvate, 15 U/ml pyruvate kinase, compounds at different concentrations at final DMSO concentrations of 1%.The enzymatic reaction is then started by addition of a mixture of NADH, acetyl Coenzyme A (both 2000 f.c.) and ATP (500 uM f.c.). The decrease of the optical density (slope S) is then determined at 25° C. at a wavelength of 340 nm over 15 minutes in a spectrophotometric reader.Each assay microtiter plate contains wells with vehicle instead of compound as controls for the non-inhibited enzyme (100% CTL; HIGH) and wells without acetyl-CoA as controls for non-specific NADH degradation (0% CTL; LOW).
- Spectrophotometric 384 Well Assay Malonyl CoA formation by acetyl CoA carboxylases is stoichometrically linked to the consumption of ATP. ACC2 activity is measured in a NADH-linked kinetic method measuring ADP generated during the ACC reaction using a coupled lactate dehydrogenase/pyruvate kinase reaction. Assay reactions are then carried out in 384-well plates, with hACC2 in an appropriate dilution and at final assay concentrations (f.c.) of 100 mM Tris (pH 7.5), 10 mM trisodium citrate, 25 mM KHCO3, 10 mM MgCl2, 0.5 mg/ml BSA, 3.75 mM reduced L-glutathione, 15 U/ml lactate dehydrogenase, 0.5 mM phosphoenolpyruvate, 15 U/ml pyruvate kinase, compounds at different concentrations at final DMSO concentrations of 1%.The enzymatic reaction is then started by addition of a mixture of NADH, acetylCoenzyme A (both 2000 f.c.) and ATP (500 uM f.c.). The decrease of the optical density (slope S) is then determined at 25° C. at a wavelength of 340 nm over 15 minutes in a spectrophotometric reader.
- Spectrophotometric 384 Well Assay Malonyl CoA formation by acetyl CoA carboxylases is stoichometrically linked to the consumption of ATP. ACC2 activity is measured in a NADH-linked kinetic method measuring ADP generated during the ACC reaction using a coupled lactate dehydrogenase/pyruvate kinase reaction.For biological testing, a human ACC2 construct which lacks the 128 amino acids at the N-terminus for increased solubility (nt 385-6966 in Genbank entry AJ575592) is cloned. The protein is then expressed in insect cells using a baculoviral expression system. Protein purification is performed by anion exchange.All compounds are dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM.Assay reactions are then carried out in 384-well plates, with hACC2 in an appropriate dilution and at final assay concentrations (f.c.) of 100 mM Tris (pH 7.5), 10 mM trisodium citrate, 25 mM KHCO3, 10 mM MgCl2, 0.5 mg/mL BSA, 3.75 mM reduced L-glutathione, 15 U/mL lactate dehydrogenase, 0.5 mM phosphoenolpyruvate.
- Spectrophotometric 384 Well Assay Malonyl CoA formation by acetyl CoA carboxylases is stoichometrically linked to the consumption of ATP. ACC2 activity is measured in a NADH-linked kinetic method measuring ADP generated during the ACC reaction using a coupled lactate dehydrogenase/pyruvate kinase reaction.For biological testing, a human ACC2 construct which lacks the 128 amino acids at the N-terminus for increased solubility (nt 385-6966 in Genbank entry AJ575592) is cloned. The protein is then expressed in insect cells using a baculoviral expression system. Protein purification is performed by anion exchange.All compounds are dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM.Assay reactions are then carried out in 384-well plates, with hACC2 in an appropriate dilution and at final assay concentrations (f.c.) of 100 mM Tris (pH 7.5), 10 mM trisodium citrate, 25 mM KHCO3, 10 mM MgCl2, 0.5 mg/ml BSA, 3.75 mM reduced L-glutathione, 15 U/ml lactate dehydrogenase, 0.5 mM phosphoenolpyruvate.
- pH-Based Assay Two days prior to performing this assay, cells are seeded on poly-D-lysine-coated 96-well clear-bottom black plates (commercially available from Becton-Dickinson) at 75,000 cells/well in growth media containing 5 uM PonA (commercially available from Invitrogen) to induce expression of TRPV1. On the day of the assay, the plates are washed with 0.2 mL 1x Hank's Balanced Salt Solution (commercially available from Life Technologies) containing 1.6 mM CaCl2 and 20 mM HEPES, pH 7.4 (wash buffer), and loaded using 0.1 mL of wash buffer containing Fluo-4 (3 uM final concentration, commercially available from Molecular Probes). After 1 h, the cells are washed twice with 0.2 mL wash buffer and resuspended in 0.05 mL 1x Hank's Balanced Salt Solution (commercially available from Life Technologies) containing 3.5 mM CaCl2 and 10 mM Citrate, pH 7.4 (assay buffer). Plates are then transferred to a FLIPR for assay.
- pH-Based Assay Two days prior to performing this assay, cells are seeded on poly-D-lysine-coated 96-well clear-bottom black plates (commercially available from Becton-Dickinson) at 75,000 cells/well in growth media containing 5 uM PonA (commercially available from Invitrogen) to induce expression of TRPV1. On the day of the assay, the plates are washed with 0.2 mL 1x Hank's Balanced Salt Solution (commercially available from Life Technologies) containing 1.6 mM CaCl2 and 20 mM HEPES, pH 7.4 (wash buffer), and loaded using 0.1 mL of wash buffer containing Fluo-4 (3 uM final concentration, commercially available from Molecular Probes). After 1 h, the cells are washed twice with 0.2 mL wash buffer and resuspended in 0.05 mL 1x Hank's Balanced Salt Solution (commercially available from Life Technologies) containing 3.5 mM CaCl2 and 10 mM Citrate, pH 7.4 (assay buffer). Plates are then transferred to a FLIPR for assay. The test compound is diluted in assay buffer.
- ACLY Enzyme Activity Assay Experimental method: ADP Glo luminescence method was used for determination. It reflects the activity of ACLY enzyme by quantitatively detecting the amount of ADP, and the enzymatic reaction catalyzed by ACLY is proportional to the amount of ADP detected by the luminescent signal. Firstly, the compound is diluted with 10% DMSO, and then 11 of compound dilution is added to a 5 μl reaction system, so that the final reaction system has a DMSO content of 2%. The enzymatic reaction catalyzed by ACLY was carried out at 37° C. for 30 minutes. 5 μl reaction mixture contained the following components: 40 mM Tris, pH 8.0, 10 mM MgCl2, 5 mM DTT, ATP, CoA, sodium citrate, and ACLY. After the enzymatic reaction was complete, 2.5 μl of ADP Glo reagent was added to each reaction system and incubated at room temperature for 1 hour. Afterwards, 5 μl of kinase detection reagent was added and incubated at room temperature for 30 minutes. The luminous signal was detected using Envision (PerkinElmer, USA).
- Acid Ceramidase Activity Assay A hAC protein preparation (10 μg) was preincubated with inhibitors (final DMSO concentration 1%) in assay buffer (100 mM sodium phosphate, 0.1% Nonidet P-40, 150 mM NaCl, 3 mM DTT, 100 mM sodium citrate, pH 4.5) for 30 min at 37° C. Reactions were started by the addition of 50 μM N-lauroyl ceramide (Nu-Chek Prep, Elysian, Minn.) and carried on for 30 min at 37° C. Reactions were stopped by addition of a mixture of chloroform/methanol (2:1, vol/vol) containing 1 nmol 11-lauroleic acid (NuChek Prep). The organic phases were collected, dried under nitrogen and analyzed by UPLC/MS (Acquity, Waters) in the negative-ion mode monitoring the reaction product (lauric acid, m/z=199) using 11-lauroleic acid as internal standard.Lipids were eluted on an Acquity UPLC BEH C18 column (50 mm length, 2.1 mm i.d., 1.7 μm pore size, Waters) column at 0.5 mL-min−1 for 1.5 min with a gradient of acetonitrile (CH3CN) and water, both containing 0.25% acetic acid and 5 mM ammonium acetat
- Biochemical Assay Inhibitor potency was evaluated by measuring enzymatic activity of full length MALT1 at varying concentrations of compound. The enzymatic assay consists of a single substrate reaction that monitors the release of a fluorescent dye upon cleavage of the peptide substrate. The peptide substrate has the following sequence: Ac-Leu-Arg-Ser-Arg-Rh110-dPro (custom synthesis from WuXi AppTec, Shanghai, China). The assay buffer consists of 50 mM Hepes, pH 7.5, 0.8 M sodium citrate, 1 mM DTT, 0.004% tween-20, and 0.005% bovine serum albumin (BSA). Steady-state kinetic analysis of peptide substrate binding resulted in a Michaelis-Menten constant (KM) of 150 μM. The assay was performed in a 384-well F-bottom polypropylene, black microplate (Greiner Bio_One, Catalog no. 781209) at 15 nM enzyme and 30 μM peptide substrate. The reaction was quenched after 60 minutes with the addition of iodoacetate at a final concentration of 10 mM. Total fluorescence was measured using an Envision (PerkinElmer) with fluorescence excitation at 485 nm and emission at 520 mm.
- Cytopathic Effect Assay with RSV A2 in HEp-2 Cells Test compounds were dissolved in DMSO and then prepared in eight half-log dilutions in MEM medium with 50 μg/mL gentamicin and 2% FBS (MA-104 cells only). Each dilution was added to 5 wells of a 96-well plate with 80-100% HEp-2 cells. Three wells of each dilution were infected with respiratory syncytial virus A2 (ATCC VR-1540) and two wells remained uninfected as toxicity controls. After maximum cytopathic effect (CPE) was observed microscopically in untreated virus control wells, plates were stained with neutral red dye for approximately 2 hours, then supernatant dye was removed and the incorporated dye was extracted in 50:50 Sorensen citrate buffer/ethanol and read on a spectrophotometer. Optical density values were normalized based on cell and virus controls, then the concentration of test compound required to inhibit CPE by 50% (EC50) was calculated by regression analysis. The reported EC50 value is the mean of at least two experiments.
- Cytopathic Effect Assay with RSV A2 in MA-104 Test compounds were dissolved in DMSO and then prepared in eight half-log dilutions in MEM medium with 50 μg/mL gentamicin and 2% FBS (MA-104 cells only). Each dilution was added to 5 wells of a 96-well plate with 80-100% MA-104 cells. Three wells of each dilution were infected with respiratory syncytial virus A2 (ATCC VR-1540) and two wells remained uninfected as toxicity controls. After maximum cytopathic effect (CPE) was observed microscopically in untreated virus control wells, plates were stained with neutral red dye for approximately 2 hours, then supernatant dye was removed and the incorporated dye was extracted in 50:50 Sorensen citrate buffer/ethanol and read on a spectrophotometer. Optical density values were normalized based on cell and virus controls, then the concentration of test compound required to inhibit CPE by 50% (EC50) was calculated by regression analysis. The reported EC50 value is the mean of at least two experiments.
- Enzyme Inhibition Assay Plasmepsin-2 (PM-2; Plm II) and Plasmepsin (PM-4; Plm IV) expression and purification was performed following the published protocols (Istvan E S and Goldberg D E, 2005). The final purified protein was activated by diluting the protein to 0.3 mg/mL in activating buffer (0.1 M citrate pH 4.5, 0.1% Tween-20, 50 mM dithiothreitol) and incubated at room temperature for 40 min, then the activated enzyme was diluted in assay buffer (50 mM sodium acetate pH 4.7, 0.01% Tween-20). The enzymatic inhibition reaction was performed in 384 well plates with a total volume of 20 μl. 10 μl of diluted PM-2 or PM-4 enzyme was added to the 384-well plate except blank wells (blank wells add 10 μL of assay buffer) and 20 nL of serials of diluted 1000× compounds were added to the wells with 520 Echo Liquid Handling System (Labcyte Inc.). 10 μL PM-2 peptide substrate (AnaSpec, Cat#, 62050) with assay buffer was then added to final concentration of 20 μM to start the reaction.
- HumanCHIT1 Activity Assay An enzymatic assay with recombinant human CHIT1 was used in order to establish inhibitory activity of the compounds (Boot et al., 2001, JBC: 276). The assay was run in the 96-well plate format, each reaction in the total volume of 100 μL. 4-methylumbelliferyl β-D-N,N′,N″-triacetylchitotriose was used as a substrate for the enzyme. Upon hydrolysis by CHIT1, the substrate releases 4-methylumbelliferyl (4MU) that, when ionized in basic pH, emits fluorescence at 460 nm.Briefly, 40 μL of a substrate was added to each well, followed by 10 μL of compound dilution and 50 μL of CHIT1 recombinant enzyme solution. The reaction was carried out in citrate buffer, pH 5.2, in the dark, at 37° C. for 60 minutes with shaking. After that time the reaction was stopped by adding 195 μL of Stop Solution (pH 10.5) to each well. The fluorescence of the reaction product was measured in Perkin Elmer Envision fluorescent plate reader at an excitation wavelength of 355 nm.
- In Vitro Binding Assay I. Master Stock SolutionUnless specified otherwise, the sample compounds were diluted in 100% anhydrous DMSO including all dilutions. The compounds were tested as the citrate salt (1 equivalent per molecule). If the sample compound is provided in a different solvent all master stock dilutions are performed in the specified solvent. All control wells contained identical solvent final concentrations as the sample compound wells.II. Compound Plate AssayThe sample compounds were transferred from a master stock solution into a daughter plate that was used in the assay. Each sample compound was diluted into an assay buffer (1×HBSS with 20 mM HEPES, 2.5 mM Probenecid, and 0.4% Free Fatty Acid BSA) at an appropriate concentration to obtain final specified concentrations.III. Antagonist Assay FormatUsing the EC80 values that were determined real-time, stimulated all pre-incubated sample compounds and reference antagonists (if applicable) were compared with the EC80 values of reference agonist. These were read for 180 seconds using the FLIPRTETRA (This assay added reference agonist to respective wells-then fluorescences measurements were collected to calculate IC50 values).
- In Vitro Enzymatic Cathepsin D (Cat D) FRET Assay Recombinant Cat D was expressed in CHO cells. The assay buffer for CathepsinD is 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The Cat D enzyme (9 nM) is pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays are effectively started by the addition of different FRET substrates (20 nM for Cat D) and the combination is incubated for one hour. The FRET assay is terminated with by addition of Tris buffer, which raises the pH to neutrality, and the fluorescence is determined. The FRET substrate is a peptide with commercially available fluorophore and quencher, on opposite sides of the BACE cleavage site. The Cat D substrate peptide sequence is based on sequence #1 of Table 1 from Gulnik et al. FEBS Letters v413 p 379-384 1997. Proteolytic cleavage of the FRET substrate releases quenching of fluorescence (Cat D excitation 500 nm and emission 580 nm).
- In Vitro Enzymatic Cathepsin D (CatD) FRET Assay Recombinant CatD was expressed in CHO cells. The assay buffer for CatD was 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The CatD enzyme (9 nM) was pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays was effectively started by the addition of different FRET substrates (20 nM for CatD) and the combination was incubated for one hour. The FRET assay was terminated with by addition of tris buffer, which raises the pH to neutrality, and the fluorescence was determined. The FRET substrate was a peptide with commercially available fluorophore and quencher, on opposite sides of the CatD cleavage site. The CatD substrate peptide sequence was based on sequence #1 of Table 1 from Gulnik et al., FEBS Lett. 413(2):379-384 (1997). Proteolytic cleavage of the FRET substrate released quenching of fluorescence (CatD excitation 500 nm and emission 580 nm).
- In Vitro Enzymatic Cathepsin D FRET Assay Recombinant Cat D was expressed in CHO cells. The assay buffer for CathepsinD is 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The Cat D enzyme (9 nM) is pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays are effectively started by the addition of different FRET substrates (20 nM for Cat D) and the combination is incubated for one hour. The FRET assay is terminated with by addition of Tris buffer, which raises the pH to neutrality, and the fluorescence is determined. The FRET substrate is a peptide with commercially available fluorophore and quencher, on opposite sides of the BACE cleavage site. The Cat D substrate peptide sequence is based on sequence #1 of Table 1 from Gulnik et al. FEBS Letters v413 p 379-384 1997. Proteolytic cleavage of the FRET substrate releases quenching of fluorescence (Cat D excitation 500 nm and emission 580 nm).
- In Vitro Enzymatic FRET Assay Recombinant Cat D was expressed in CHO cells. The assay buffer for CathepsinD is 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The Cat D enzyme (9 nM) is pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays are effectively started by the addition of different FRET substrates (20 nM for Cat D) and the combination is incubated for one hour. The FRET assay is terminated with by addition of Tris buffer, which raises the pH to neutrality, and the fluorescence is determined. The FRET substrate is a peptide with commercially available fluorophore and quencher, on opposite sides of the BACE cleavage site. The Cat D substrate peptide sequence is based on sequence #1 of Table 1 from Gulnik et al. FEBS Letters v413 p 379-384 1997. Proteolytic cleavage of the FRET substrate releases quenching of fluorescence (Cat D excitation 500 nm and emission 580 nm).
- In Vitro Enzyme Assay The proteolytic enzyme, Dipeptidyle Peptidase IV (DppIV) was monitored in vitro by the substrate Gly-Pro 4-methoxy-β-naphthylamide while in the presence of compound 7. 20 ul of 1.3 mM DppIV was pipetted into twenty-four micro centrifuge tubes containing various concentrations of compound 7 in a total volume of 100 uL. The samples were thoroughly vortexed and centrifuged at 5,000 RPM for 30 seconds. The micro centrifuge tube were then placed in a 37° C. water bath and incubated for 30 minutes. After incubation, 0.625 uM of substrate was added and the volume was adjusted to 130 uM with incubation buffer. Samples were thoroughly mixed and centrifuged at 5,000 RPM for 30 seconds before placed in a 37° C. water bath for an additional 30 minutes. The reaction was terminated by adding 1 ml of 100 mM Citrate buffer pH 4.0 and vortexing thoroughly for 1 minute. The excitation and emission spectrum of each sample was measured at 340 nm and 425 nm respectfully on a Fluoromax-II Fluorometer.
- MALT1 Enzyme Activity Assay 1.1 Experimental PurposeAc-LRSR-AMC was used as the substrate to test the IC50 of the inhibitory activity of compounds on MALT1 enzyme activity at their Km concentration.1.2 Experimental Instruments and ReagentsInstruments:Instrument name Manufacturer Specification and modelCentrifuge Eppendorf 5810RMicrocentrifuge DALB SCIENTIFIC D1008CO., LTD.Microplate reader Bio Tek H1MFDReagents:Reagent materials Manufacturer Item No.Recombinant human Abcam ab271604MALT1/MLT proteinAc-LRSR-AMC SM biochemicals ab2716041.3 Test MethodAc-LRSR-AMC was used as the substrate which was digested by MALT1 to produce products, which produced fluorescence that could be detected at 460 nm. The highest concentration of the compound detected was 1000 nM, 3-fold dilution, a total of 11 concentrations (1000 nM-0.017 nM). The reaction system is 10 nM FL MALT1 protein, 200 mM Ac-LRSR-AMC, 50 mM Tris pH 7.5, 0.6M Citrate, 1 mM DTT, 1 mM EDTA, 0.05% BSA and 1.5% DMSO. The compound and the enzyme were pre-incubated at room temperature for 50 min. The substrate was then added and the reaction was carried out for 4 h.
- h-NAAA Assay HEK293 cells stably transfected with the human NAAA coding sequence cloned from a human spleen cDNA library (catalog no. 639124, Clontech, Mountain View, Calif., USA) were used as enzyme source. The assay was run in 96-well microplates (Black OptiPlate -96 F; PerkinElmer, Massachusetts, USA), in a total reaction volume of 200 μL. hNAAA protein preparation (4.0 μg) was pre-incubated for 10 minutes with various concentrations of test compounds or vehicle control (5% DMSO) in 100 mM citrate/phosphate buffer (pH 4.5) containing 3.0 mM DTT, 0.1% NP40 0.1%, 0.05% BSA, 150 mM NaCl. N-(4-methyl-2-oxo-chromen-7-yl)-hexadecanamide (PAMCA) was used as a substrate (5.0 μM) and the reaction carried for 50 min at 37° C. Fluorescence was measured with EnVision 2014 Multilabel Reader (PerkinElmer, Massachusetts, USA) using an excitation wavelength of 340 nm and emission 450 nm. IC50 values were calculated by non-linear regression analysis of log[concentration]/inhibition curves using GraphPad Prism 5 (GraphPad Software Inc., CA, USA) applying a standard slope curve fitting.
- ADP-Glo Assay Compounds of the present invention were evaluated in an ADP-GLO assay as follows:a) Dilute cpd 1:3 in succession in DMSO by hand for each cpds for 12 ptsb) Add 0.1 μL diluted cpd solution to assay plate, each dose with 2 replicatesc) Centrifuge 1000 RPM for 1 mind) Add 5 μL ACL working solution to 384-well assay plate, centrifuge 1000 RPM for 1 mine) Incubate at 25° C. for 15 minf) Add 5 μL substrate working solution to initiate reactiong) Final ACL reaction concentrations: 3 nM ACL, 15 μM ATP, 3 μM CoA, 300 μM Citrate, 0.01% Brij35, 4 mM DTT, 1% DMSO;h) Reference final conc: 30 uM starting Conc, 3× dilution, 11+0 points. Test cpd conc: 30/100 uM starting Conc, 3× dilution, 11+0 points.i) Incubate at 25° C. for 60 minj) Add 10 μL ADP Glo reagent, centrifuge 1000 RPM for 1 mink) Incubate at 25° C. for 40 minl) Add 20 μL kinase detection reagent, centrifuge 1000 RPM for 1 minm) Incubate at 25° C. for 40 minn) Read on Envision for US LUM as RLU.
- AMCase Activity Assay An enzymatic assay with recombinant human AMCase was used in order to establish inhibitory activity of the compounds (Boot et al., 2001, JBC:276). The assay was run in the 96-well plate format, each reaction in the total volume of 100 μL. 4-Methylumbelliferyl B-D-N,N′-diacetylchitobioside hydrate was used as a substrate for the enzyme. Upon hydrolysis by AMCase, the substrate releases 4-methylumbelliferyl (4MU) that, when ionized in basic pH, emits fluorescence at 460 nm. Briefly, 40 μL of a substrate was added to each well, followed by 10 μL of compound dilution and 50 μL of hAMCase recombinant enzyme solution. The reaction was carried out in citrate buffer, pH 5.2, in the dark, at 37° C. for 60 minutes with shaking. After that time the reaction was stopped by adding 195 μL of Stop Buffer (pH 10.5) to each well. The fluorescence of the reaction product was measured in Tecan Spark multimode plate reader at an excitation wavelength of 355 nm. The IC50 values were calculated using GraphPad Prism.
- CHIT1 Activity Assay An enzymatic assay with recombinant human CHIT1 was used in order to establish inhibitory activity of the compounds (Boot et al., 2001, JBC:276). The assay was run in the 96-well plate format, each reaction in the total volume of 100 μL. 4-methylumbelliferyl β-D-N,N′,N″-triacetylchitotriose was used as a substrate for the enzyme. Upon hydrolysis by CHIT1, the substrate releases 4-methylumbelliferyl (4MU) that, when ionized in basic pH, emits fluorescence at 460 nm. Briefly, 40 μL of a substrate was added to each well, followed by 10 μL of compound dilution and 50 μL of CHIT1 recombinant enzyme solution. The reaction was carried out in citrate buffer, pH 5.2, in the dark, at 37° C. for 60 minutes with shaking. After that time the reaction was stopped by adding 195 μL of Stop Solution (pH 10.5) to each well. The fluorescence of the reaction product was measured in Tecan Spark multimode plate reader at an excitation wavelength of 355 nm. The IC50 values were calculated using GraphPad Prism.
- Human AMCase Activity Assay An enzymatic assay with recombinant human AMCase was used in order to establish inhibitory activity of the compounds (Boot et al., 2001, JBC: 276). The assay was run in the 96-well plate format, each reaction in the total volume of 100 μL. 4-Methylumbelliferyl B-D-N,N′-diacetylchitobioside hydrate was used as a substrate for the enzyme. Upon hydrolysis by AMCase, the substrate releases 4-methylumbelliferyl (4MU) that, when ionized in basic pH, emits fluorescence at 460 nm.Briefly, 40 μL of a substrate was added to each well, followed by 10 μL of compound dilution and 50 μL of hAMCase recombinant enzyme solution. The reaction was carried out in citrate buffer, pH 5.2, in the dark, at 37° C. for 60 minutes with shaking. After that time the reaction was stopped by adding 195 μL of Stop Buffer (pH 10.5) to each well. The fluorescence of the reaction product was measured in Perkin Elmer Envision fluorescent plate reader at an excitation wavelength of 355 nm. The IC50 values were calculated using GraphPad Prism.
- Human CHIT1 Activity Assay An enzymatic assay with recombinant human CHIT1 was used in order to establish inhibitory activity of the compounds (Boot et al., 2001, JBC: 276). The assay was run in the 96-well plate format, each reaction in the total volume of 100 μL. 4-methylumbelliferyl β-D-N,N′,N″-triacetylchitotriose was used as a substrate for the enzyme. Upon hydrolysis by CHIT1, the substrate releases 4-methylumbelliferyl (4MU) that, when ionized in basic pH, emits fluorescence at 460 nm.Briefly, 40 μL of a substrate was added to each well, followed by 10 μL of compound dilution and 50 μL of CHIT1 recombinant enzyme solution. The reaction was carried out in citrate buffer, pH 5.2, in the dark, at 37° C. for 60 minutes with shaking. After that time the reaction was stopped by adding 195 μL of Stop Solution (pH 10.5) to each well. The fluorescence of the reaction product was measured in Perkin Elmer Envision fluorescent plate reader at an excitation wavelength of 355 nm. The IC50 values were calculated using GraphPad Prism.
- Aromatase Inhibition Activity Assay This fluorescence-based assay measures the rate at which recombinant human aromatase (baculovirus/insect cell-expressed) converts the substrate 7-methoxy-trifluoromethylcoumarin (MFC) into a fluorescent product 7-ethynyl-trifluoromethylcoumarin (HFC; λex = 409 nm, λem = 530 nm) in a NADPH regenerating system. Briefly, concentrated stock solutions of test compounds were prepared in acetonitrile. Hundred microliters of samples containing serial dilutions of test compounds (dilution factor of 3 between samples) and cofactor mixture (0.4 U/mL glucose-6-phosphate dehydrogenase; 16.2 μm NADP+; 825 μm MgCl2; 825 μm glucose-6- phosphate; 50 μm citrate buffer, pH 7.5) was prepared in a 96-well plate. After incubating the plate for 10 min at 37 °C, 100 μL of an aromatase/P450 reductase/substrate solution (105 μg protein/mL enzyme; 50 μm MFC; 20 mm phosphatebuffer, pH 7.4) was added to each well. The plate was covered and incubated for 30 min at 37 °C. Seventy-five microliters of 0.5 m Tris base was then added to stop the reaction, and the fluorescence of the formed de-methylated MFC was measured with a plate reader (Spectra Max Gemini; Molecular Devices, Sunnyvale, CA, USA).
- Human ACLY (hACLY) RF/MS Activity Assay Table 2: Compounds of the disclosure were evaluated for their efficacy in inhibiting hACLY using rapid fire mass spectrometry (RF/MS). The final reaction volume for each compound was 20 μl and consisted of buffer (50 mM Hepes pH 8.0, 10 mM MgCl2, 0.003% BSA, 0.01% Brij35, 50 mM NaCl, 4 mM DTT) and 1 nM hACLY, EV12992, PP6692) using Greiner, 384 well small volume, deep well plates (Cat #784201). A two-fold dilution series with a top concentration of 10 μM was used to record a concentration response curve. Both the substrate (CoA) and product (Acetyl-CoA) were quantified, and given a ratio. The ratio was normalized using both a negative (0% inhibition) and positive (100% inhibition) control to determine the % inhibition. The final DMSO-concentration was 1% (v/v). Compounds were pre-incubated for 30 min with the buffered enzyme solution at RT (20° C.), and substrate solution was added (final concentrations: 15 μM Coenzyme A, 50 μM ATP and 50 μM citrate) to initiate the enzyme reaction. The enzyme reaction was incubated for additional 30 min at RT. The reaction was quenched upon addition of 40 μl of 5% Formic acid in H2O and centrifuged (4350 rpm at 20° C. for 10 min).
- Human Platelet Aggregation Inhibition Test Blood collected from healthy human volunteers in aqueous trisodium citrate solution was centrifuged at 150 g for 15 min and the upper layer was recovered to obtain platelet-rich plasma (PRP). The residual blood was centrifuged at 3000 g for 10 min and the supernatant was collected as platelet-poor plasma (PPP). Platelet concentration in the PRP was determined using the Z series Beckman Coulter particle counter (Beckman, Fullerton, Calif.) and adjusted to 250,000 platelets/μL using PPP. 480 μL of PRP was pre-incubated at 37° C. and stirred at 1200 rpm with 10 μL aqueous test compound solution for 1 min prior to induction of aggregation by the addition of 10 μL of aqueous adenosine diphosphate (ADP) solution to adjust the final ADP concentration in the PRP to 1×10−5 M. The maximal amplitude of aggregation response within 3 min was determined and measured in triplicate using the Chronolog model 490 aggregometer (Chrono-log Corp., Havertown, Pa.). Percent inhibition of aggregation was calculated from the maximum decrease in optical density of the control (addition of water in place of the test compound solution) sample and of the samples containing test compound
- In Vitro Enzymatic Cathepsin D (CatD) FRET Assay Recombinant CatD was expressed in CHO cells. The assay buffer for CatD was 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The CatD enzyme (9 nM) was pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays was effectively started by the addition of different FRET substrates (20 nM for CatD) and the combination was incubated for one hour. The FRET assay was terminated with by addition of tris buffer, which raises the pH to neutrality, and the fluorescence was determined. The FRET substrate was a peptide with commercially available fluorophore and quencher, on opposite sides of the CatD cleavage site. The CatD substrate peptide sequence was based on sequence #1 of Table 1 from Gulnik et al., FEBS Lett. 413(2):379-384 (1997). Proteolytic cleavage of the FRET substrate released quenching of fluorescence (CatD excitation 500 nm and emission 580 nm).Alternatively, a CatD assay may also be run according to the procedure described in Yasuda et al., J. Biochem. 125(6):1137-1143 (1999). In addition, the CatD and Cathepsin E assays are described in International Patent Application Publication No. WO2011069934.
- In Vitro Enzymatic Recombinant Cat D was expressed in CHO cells. The assay buffer for CathepsinD is 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The Cat D enzyme (9 nM) is pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays are effectively started by the addition of different FRET substrates (20 nM for Cat D) and the combination is incubated for one hour. The FRET assay is terminated with by addition of Tris buffer, which raises the pH to neutrality, and the fluorescence is determined. The FRET substrate is a peptide with commercially available fluorophore and quencher, on opposite sides of the BACE cleavage site. The Cat D substrate peptide sequence is based on sequence #1 of Table 1 from Gulnik et al. FEBS Letters v413 p 379-384 1997. Proteolytic cleavage of the FRET substrate releases quenching of fluorescence (Cat D excitation 500 nm and emission 580 nm).Alternatively, a Cat D assay may also be run according to the procedure described in the article, Characterization of new fluorgenic substrates for the rapid and sensitive assay of cathepsin E and cathepsin D, J. Biochem., 125:1137, 1999.
- Luminometric Assay The reaction mixture contained 50 mM tris acetate (Merck, #1.08382.1000), pH 7.5, 250 μM acetyl coenzyme A (Sigma, #A2181),16 mM NaHCO3 (Merck, #1.06329.0500), 0.9 mg/ml bovine serum albumin (Sigma, #A8806), 25 μM ATP (Sigma, #A7699), 1.1 μM β-mercaptoethanol (Roth, #4227,1), 4.3 mM magnesium acetate ((Merck, #1.05819.0250), 10 mM sodium citrate (Merck, #1.06448.0500) and 200-400 ng of the ACC preparations. The mixture also contained a test compound. The test compounds were each dissolved in 10 mM DMSO and were assayed at final concentrations of 0 μM, 0.01 μM, 0.03 μM, 0.1 μM, 0.3 μM, 1 μM, 3 μM, 10 μM, 30 μM and 100 μM. The final concentration of the DMSO in the assay was 0.1% (v/v). The reaction was started by adding ACC and was carried out at 37° C. for 90 min. The reaction was then stopped by adding 150 μl of 0.5 M tris acetate. 10 μl of the mixture were removed and mixed with 90 μl of ATP monitoring reagent (Cambrex, #LT27-002). After incubation for 10 min, the luminescence was measured in a luminometer (Tecan Genios Pro).
- MALT-1 Biochemical Assay A Quenched Fluorescence Resonance Energy Transfer (Quench-FRET) enzyme assay was used to test the ability of exemplary MALT-1 inhibitors Example 1 to Example 39 to inhibit the cleavage of a MALT1 specific quenched fluorescent substrate of sequence [TAMRA-PEG2-Leu-Val-Ser-Arg-Gly-Ala-Ala-Ser-PEG2-K(QSY7) by human MALT-1 protein; where TAMRA is 6-carboxytetramethylrhodamine, PEG2 is 2-(2-(2-aminoethoxy)ethoxyamide linker, QSY7 is a non-fluorescent quenching dye (Thermo-Fisher catalog #Q10193). The assay was performed by pre-dispensing compounds into 384 well Proxiplates (PE 6008289). Human MALT-1 (6 nM) was prepared using the assay buffer (25 mM HEPES pH 7.5, 1 mM ethylenediaminetetraacetic acid, 0.8 M sodium citrate, 0.005% Bovine serum albumin (BSA), 2 mM dithiothreitol (DTT)) and added to the compound plates. The plates were incubated 40 minutes at room temperature. The reaction was carried out by adding the substrate (2 μM) to the plates and incubating for 60 minutes at room temperature. A fluorescence readout was then measured using a PE EnVision reader (Ex.535/Em.590). The results of the enzyme assay are provided subsequently in Table 2 and demonstrate the ability of the compounds of the present disclosure to inhibit MALT-1 protease activity.
- Biochemical FRET Based Assay This assay is a Fluorescence Resonance Energy Transfer Assay (FRET) based assay. The substrate for this assay contains the Swedish Lys-Met/Asn-Leu mutation of the amyloid precursor protein (APP) beta-secretase cleavage site. This substrate also contains two fluorophores: (7-methoxycoumarin-4-yl) acetic acid (Mca) is a fluorescent donor with excitation wavelength at 320 nm and emission at 405 nm and 2,4-Dinitrophenyl (Dnp) is a proprietary quencher acceptor. The distance between those two groups has been selected so that upon light excitation, the donor fluorescence energy is significantly quenched by the acceptor, through resonance energy transfer. Upon cleavage by the beta-secretase, the fluorophore Mca is separated from the quenching group Dnp, restoring the full fluorescence yield of the donor. The increase in fluorescence is linearly related to the rate of proteolysis.Briefly in a 384-well format recombinant BACE2 protein in a final concentration of 0.4 μg/ml is incubated for 450 minutes at room temperature with 10 μM substrate in incubation buffer (50 mM Citrate buffer pH 5.0, 0.05% PEG, no DMSO) in the absence or presence of compound. Next the amount of proteolysis is directly measured by fluorescence measurement at T=0 and T=450 (excitation at 320 nm and emission at 405 nm). Results are expressed in RFU (Relative Fluorescence Units), as difference between T450 and TO.
- Inhibition of Compounds on PARP1/7 Enzyme Activity (1) Pre-coating: Add 100 μL of a PBS buffer (10 mM NaH2PO4, 10 mM Na2HPO4, 150 mM NaCl, pH 7.4) containing 20 g/mL of histone to each well of a 96-well plate, and incubate at 4° C. overnight.(2) Add 30 μL of reaction buffer (50 mM Tris, 2 mM MgCl2, pH 8.0) containing 100 μM NAD+, 25 μM biotinylated NAD+, and 200 nM slDNA to each well.(3) Add 5 μL of the test substance or solvent to each well.(4) Add 20 μL of(PARP1 or PARP7 (50 ng/well), and incubate at 30° C. for 1 hour.(5) Add 50 μL of streptavidin-HRP to the reaction mixture, and incubate at 30° C. for 30 minutes.(6) Finally, add 100 μL of a citrate buffer containing H2O2 and luminol (0.1 M, pH 5.4), and measure the luminescent signal using a microplate reader (Molecular Devices SpectraMax M5).(7) Calculate the inhibition rate of PARP1 or PARP7 enzyme activity as [(control group−treatment group)/control group]×10000. Fit the dose-response data with standard dilutions using Prism GraphPad software and calculate the concentration required to achieve 5000 inhibition of PARP1 or PARP7 enzyme activity (IC50).
- Inhibition of Mast Cell Degranulation Buffers and solutions: Phosphate buffered saline (PBS) and 1 M HEPES were provided by the in-house service facility. Tyrode's buffer (TyB) consisted of Minimum Essential Medium without Phenol Red supplemented with 2 mM L-glutamine and 20 mM HEPES. Lysis buffer consisted of 25 mM Tris-HCl, pH 7.5, 150 mM NaCl. 5 mM EDTA and 0.1% (w/v) Triton X-100. DNP-HSA was dissolved to 1 mg/ml in water. MUG substrate solution consisted of 2.5 mM 4-methylumbelliferyl-N-acetyl-β-D-glucosaminide in 0.05 M citrate, pH 4.5; stop solution was 0.1 M NaHCO3/0.1 M Na2CO3, pH 10.Consumables and equipment: For small-volume liquid handling procedures, Rainin LTS electronic pipettes were routinely used (Mettler-Toledo). Costar-Corning 24-well plates (3337) were centrifuged in an Eppendorf 5804 R centrifuge. A Heraeus B15 table top incubator was used for incubations at 37° C. under non-sterile conditions. Fluorescence was measured in black Nunc 96-well plates (237105) using a microplate reader (Tecan Safire) or FlexStation 3 (Molecular Devices) multi-mode plate reader. Cells were maintained in Hera Cell 240 CO2 incubators (Thermo Scientific). Serological pipettes (4487, 4488 and 4489) and cell culture flasks (431080) were from Corning-Costar, 1.5 and 2 ml microcentrifuge tubes (0030 120.086 and 0030 120.094) were from Eppendorf.
- SARS-CoV-2, SARS-CoV-1, MERS, 0C43 and 229E Mpro Biochemical Protease Activity Assay SARS-CoV-2 Mpro biochemical assays were performed in 8 μL in 384-well Proxi-Plus plate (PerkinElmer, Waltham, MA USA) at ambient temperature in the optimized assay buffer (50 mM Hepes, pH 7.1, 800 mM sodium citrate, 1 mM ethylenediaminetetraacetic acid, 1 mM TCEP ((tris(2-carboxyethyl)phosphine)), 0.005% BSA (bovine serum albumin)) with 1 nM of recombinant Mpro and 5 μM substrate. After 10 minutes of preincubation of inhibitor with Mpro in assay buffer, reactions were initiated by the addition of a FRET-compatible peptide substrate (TAMRA)-dPEG2-S-A-V-L-Q-S-G-dPEG2-K(QSY7)-amide. Fluorescence was measured at 20 minutes using a 531/579 excitation/emission filters on EnVision (PerkinElmer, Waltham, MA USA). Data were normalized to DMSO minus no enzyme controls. IC50 and Ki of inhibitor was analyzed using Dotmatics software on 11 points of inhibitor with varies concentration. Each inhibitor was tested at least twice at different dates. 2 nM Mpro and 5 μM substrate for SARS-CoV-1, 5 nM Mpro and 5 μM for MERS, 1 nM of Mpro and 2 μM substrate for 0C43, 3 nM Mpro and 2 μM substrate for 229E were used in respective Mpro biochemical protease activity assays.
- MALT1 Inhibition Assay The standard reagent formulations used in this assay were prepared and stored as follows. A 1.1 M solution of sodium citrate (161.3 g, 500 mL) was prepared and stored at room temperature. A 10% (w/v) CHAPS solution (2.0 g, 20 mL) was prepared and stored at 4 C. A 500 mM HEPES solution having pH of 6.89 was prepared from 200 mL of a 1 M HEPES solution having a pH of 7.5 using concentrated hydrochloric acid and brought to a final volume of 400 mL with MILLI-Q H2O. The substrate, 10 mM Ac-LRSR-AMC Peptide, was prepared (10 mg, 1.370 mL DMSO) and stored at −20 C.[1801]Compounds were plated to provide a 2% DMSO final concentration using a ProxiPlate-384 Plus F Black 384-shallow well microplate. The assay-ready plates were equilibrated to room temperature. A reaction buffer (30 mL total volume) was prepared by combining HEPES (pH 6.89, 25 mM, 1.5 mL), sodium citrate (660 mM, 18.0 mL), T-CEP (1 mM, 0.06 mL), EDTA (0.1 mM, 0.06 mL), CHAPS (0.05%, 0.15 mL), DMSO (2%, 0.6 mL), and MILLI-Q H2O (10.23 mL; 9.63 mL when backfilled to 2% DMSO) followed by thorough mixing. DMSO was added only when compound plates had not been DMSO-backfilled to 2%.[1802]MALT-1 was thawed and kept on ice. Peptide substrate was thawed on the bench under ambient conditions. A MALT-1 enzyme working stock was prepared from Avi-tagged FL MALT-1 (40 nM in a prepared reagent volume of 16.5 μL) and the reaction buffer (13.0 mL). MALT1 working stock (5 μL) was added to each well of the microplate. MALT-1 was pre-incubated with compounds for 30 minutes at room temperature.Two substrate working stocks (Km and 10 Km) were prepared. The 1 Km substrate was prepared from Ac-LRSR-AMC Peptide (50 μM in a prepared reagent volume of 35.0 μL) and the reaction buffer (6.965 mL). The 10 Km substrate was prepared from Ac-LRSR-AMC Peptide (280 μM in a prepared reagent volume of 196.0 μL) and the reaction buffer (6.804 mL). The reaction was initiated by the addition of substrate working stock (5 μL, 50 μM) to Km plates and the addition of substrate working stock (5 μL 280 μM) to 10 Km plates. The plates were covered and incubated on the bench at room temperature for 90 minutes.Fluorescence intensity was determined using a CLARIOstar microplate reader (BMG LABTECH) using the optimised AMC mode. Before taking readings, a 70% gain was applied on the neutral control well.
- In Vitro Assay MALT1 protease activity was assessed in an in vitro assay using a tetrapeptide as substrate and full-length MALT1 protein (Strep-MALT1(1-824)-His) purified from baculovirus-infected insect cells. The tetrapeptide LRSR is coupled to AMC (7-amino-4-methylcoumarin) and provides a quenched, fluorescent substrate for the MALT1 protease (SM Biochemicals). Cleavage of AMC from the Arginine residue results in an increase in coumarin fluorescence measured at 460 nm (excitation 355 nm). The final assay buffer consisted of 10 nM FL MALT1 protein, 200 μM Ac-LRSR-AMC, 50 mM Tris pH 7.5, 0.6 M Citrate, 1 mM DTT, 1 mM EDTA, 0.05% BSA and 1.5% DMSO. Test compounds were spotted at 50 nL in 100% DMSO per well of a black 384-Proxiplate (Perkin Elmer). Test compound concentrations ranged from 30 μM to 0.5 nM using 11 dilution steps (1:3). Background signal was measured from control wells containing assay buffer without enzyme which functions as low control (LC). High control (HC) values were generated using the reaction with enzyme but no compound treatment. Compounds were pre-incubated with MALT1 enzyme for 50 minutes at RT. Substrate was added subsequently and fluorescence was measured in Labsystems fluoroskan at excitation 355 nm and emission 460 nm to determine time 0. The reaction was subsequently incubated for 4 h at RT and fluorescence was measured.
- Kinase Assay Kinase assays were performed in 96 well microtiter plates that were coated overnight with 30 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.2-7.4. The plates were incubated with 1% BSA and then washed four times with PBS prior to starting the reaction. Reactions were carried out in 120 μL reaction volumes containing 3.6 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 0.5 ng of purified protein. Following a ten minute incubation at 25 °C., the reactions were washed four times with PBS containing 0.05% Tween-20. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate was diluted 1:10000 in PBS-Tween-20 and added to the wells for 30 minutes. Following four washes with PBS-Tween-20, 100 μl of 0-phenylenediamine Dihydrochloride in Phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2 SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- PDGFRbeta Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 36 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase.
- PDGFRbeta Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 36 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase.
- cAMP Assay Cells were seeded at a density of 10,000 cells per well in poly-L-lysine treated 96-well culture plates (BD Biosciences). The following day wells were incubated with 40 μl DMEM/F12 containing 0.5% (w/v) BSA and 0.5 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich) for 30 minutes prior to 15 min stimulation with 50 μM forskolin (Tocris Cookson) and varying concentrations of indicated compounds at 37° C., 5% CO2. Assays were stopped by removal of media and addition of 100% ice cold ethanol. Plates were then frozen for a minimum of two hours before complete evaporation of ethanol. The well contents were then reconstituted in 50 μl cAMP assay buffer (20 mM HEPES pH 7.5 and 5 mM EDTA). Half of the reconstituted sample was transferred to round bottom 96-well plates (Greiner Bio-One GmbH) with 50 μl 0.01% w/v PKA (cAMP dependent protein kinase (Sigma-Aldrich) in 1 mM Na citrate pH 6.5 with 2 mM dithiothreitol) and 25 μl [3H]-cAMP (at 22 nM in cAMP assay buffer) (GE Healthcare, Life Sciences). This was allowed to equilibrate for 3-18 hours. Following this a charcoal slurry (5% (w/v) activated charcoal and 0.2% (w/v) BSA in cAMP assay buffer) was added to the samples and the plates centrifuged at 3000×g, 4° C. for 5 min.
- pH-Based Assay Two days prior to performing this assay, cells are seeded on poly-D-lysine-coated 96-well clear-bottom black plates (commercially available from Becton-Dickinson) at 75,000 cells/well in growth media containing 5 uM PonA (commercially available from Invitrogen) to induce expression of TRPV1. On the day of the assay, the plates are washed with 0.2 mL 1x Hank's Balanced Salt Solution (commercially available from Life Technologies) containing 1.6 mM CaCl2 and 20 mM HEPES, pH 7.4 (wash buffer), and loaded using 0.1 mL of wash buffer containing Fluo-4 (3 uM final concentration, commercially available from Molecular Probes). After 1 h, the cells are washed twice with 0.2 mL wash buffer and resuspended in 0.05 mL 1x Hank's Balanced Salt Solution (commercially available from Life Technologies) containing 3.5 mM CaCl2 and 10 mM Citrate, pH 7.4 (assay buffer). Plates are then transferred to a FLIPR for assay. The test compound is diluted in assay buffer, and 50 uL of the resultant solution is added to the cell plates and the solution is monitored for two minutes. The final concentration of the test compound is adjusted to range from about 50 picoM to about 3 uM. Agonist buffer (wash buffer titrated with 1N HCl to provide a solution having a pH of 5.5 when mixed 1:1 with assay buffer) (0.1 mL) is then added to each well, and the plates are incubated for 1 additional minute.
- Biochemical Assay The assay employs a short peptide substrate labeled with the single fluorophore PT14 as a fluorescence lifetime probe sensitive to the cleavage state of the substrate (PT14: 6-(9-oxo-9H-acridin-10-yl)-hexanoate, AssayMetrics, UK). The peptide substrate has the following sequence: Ac-Trp-Leu-Arg-Ser-Arg{circumflex over ( )}Cys(PT14)-NH2 (Product number BS-9117, Biosyntan, Germany, N-terminus to C-terminus from left to right in three letter code, Ac: acetyl group, Cys(PT14): cysteine residue with the fluorophore PT14 conjugated to the cysteine sulfhydryl group via a maleimide group; C-terminus of the peptide is amidated; within the substrate sequence written above, {circumflex over ( )} indicates the scissile bond). The assay buffer consists of 200 mM Tris/HCl at pH 7.5, 0.8 M Na citrate, 100 μM EGTA, 100 μM DTT and 0.05% (w/v) CHAPS. The kinetic characterization of the enzymatic reaction led to the determination of a Michaelis Constant (KM) of 40 μM and a kcat value of 34 s−1. The assay was established for the 384-well plate format using black microtiter round well plates (Product number 95040020, Thermo Electron Oy, Finland). Test compounds were dissolved in 100% (v/v) DMSO or a mixture containing 90% (v/v) DMSO and 10% (v/v) H2O at a stock concentration of 100 mM. Serial dilutions of test compounds were prepared using either 100% (v/v) DMSO or a mixture containing 90% (v/v) DMSO and 10% (v/v) H2O.
- Inhibition of Test Compounds NAAA Assay In order to have an assay method more conducive to high-throughput screening than those published for measuring the NAE hydrolyzing activity of NAAA, the fluorogenic PEA analog N-(4-methyl coumarin)palmitamide (PAMCA), which is hydrolyzed to fluorescent 7-amino-4-methyl coumarin (AMC) and palmitic acid, was developed. For three point concentration inhibition assays with hNAAA the following procedure was used. Purified activated NAAA (final concentration of 0.25 μg/mL) was incubated in assay buffer (100 mM citrate-phosphate buffer, pH 4.5, 3 mM DTT, 0.1% Triton X-100, 0.05% BSA, and 150 mM NaCl) made up to a total volume of 180 μL, followed by addition of the compound dissolved in 10 μL DMSO (along with DMSO neat for the control sample) with the final concentrations for each compound of 100, 10, and 1 μM, in triplicate on a 96 well plate. These samples were allowed to incubate for 15 min at room temperature and then 10 μL of a PAMCA stock solution in DMSO (final PAMCA concentration [5 μM]) was added. After 5 minutes of agitation on a shaking plate, the reaction was allowed to proceed at 37° C. for 120 minutes, with fluorescence readings taken every 10 minutes at a wavelength of 460 nm (using an excitation wavelength of 360 nm) on a Synergy HT Plate Reader using Gen5 software from Bio-Tek. The enzyme activity was calculated by converting the relative fluorescence units to AMC formed, using a standard curve of AMC.
- Inhibition of rhGAA Determined by Fluopol-ABPP The inhibiting activity on recombinant human acid α-glucosidase (rhGAA) of the compounds is implemented using the Fluopol-ABPP method (Fluorescence Polarization Activity Based Protein Profiling) described in the publication of Lahav et al., 2017, J. Am. Chem. Soc. 139: 14192-14197. This technique, based on the competition between an inhibitor and a fluorescent probe capable of binding covalently to the active site of an enzyme, makes it possible to measure the affinity of this inhibitor for the active site of the rhGAA enzyme used for these experiments by the laboratory of Professors Herman S. Overkleeft and Johanes M. F. G. Aerts, Leiden Institute of Chemistry, Leiden University (NL), is the enzyme marketed under the name Myozyme . The median inhibiting concentrations (IC50) are determined in the Mcllvaine buffer (citrate-phosphate) 150 mM at pH 5.0, in the presence of 0.1% bovine gamma-globulin (p/v) and 0.5 mg/mL of Chaps detergent (Sigma) in 96-well plates (Griener). The rhGAA enzyme (10 μg/mL) is pre-incubated with solutions of inhibitor (containing 2.5% DMSO that was used to prepare the mother solutions of the compounds) at various concentrations [I] in the buffer, for 45 minutes at 37 C. The tetraaminomethylrhodamine (TAMRA) fluorescent probe in solution (25 nM) in the buffer is next added to the mixture. After 4 hours, the samples are irradiated with a polarized light (λ=530 nm) and the fluorescence emitted (λ=580 nm) is measured by means of an Infinite M1000Pro (Tecan) spectrofluorimeter.
- MALT-1 Inhibition Assay MALT-1 paracaspase activity was measured using the fluorogenic substrate Ac-LRSR-Rh110-DP (purchased from Biosantan GmbH). Proteolytic cleavage of the peptide—rhodamine conjugate results in an increase of rhodamine fluorescence which is inhibited by test compounds. Test compounds were diluted in DMSO in a series of 10 semi-log step doses, 15 nL of each compound dose were dispensed in 384 well polypropylene plates (HiBase non-binding, Greiner Bio-One cat #784900). All other assay components were diluted to appropriate working concentrations in assay buffer composed of: 200 mM Tris-HCl (pH 7.5; Sigma-Aldrich cat #T2663-1L), 0.1 mM EGTA (Sigma-Aldrich cat #E3889-10G), 0.05% CHAPS—Sigma-Aldrich cat #C9426-1G), 1 mM TCEP (Sigma-Aldrich cat #646547-10×1 mL), 0.8 M sodium citrate (Sigma-Aldrich cat #S1804-500G). Recombinant human MALT-1 (amino acids 340-824, accession NP_006776.1) was added to compound doses and equilibrated for 40 minutes at rt. The reaction was initiated by addition of substrate. Final concentrations of MALT-1 and substrate were 3 nM and 10 μM respectively. Reactions were incubated in the dark for 60 minutes at 25° C. Fluorescence was measured in a PHERAstar FSX plate reader (BMG LABTECH) with optical setup for excitation at 485 nM and emission at 520 nM, focal height of 11.8 mm, 20 flashes, gain 300. Percent inhibition values were calculated from relative fluorescence units at different doses and fitted to a 4-parameter logistic curve to determine IC50 values.
- PDGFRbeta Kinase Assay Biochemical PDGFRbeta kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 36 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 ul of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- PDGFRbeta Kinase Assay Biochemical PDGFRbeta kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per wellPBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 36 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 ul of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- pH-Based Assay Two days prior to performing this assay, cells are seeded on poly-D-lysine-coated 96-well clear-bottom black plates (commercially available from Becton-Dickinson) at 75,000 cells/well in growth media containing 5 μM PonA (commercially available from Invitrogen) to induce expression of TRPV1. On the day of the assay, the plates are washed with 0.2 mL 1× Hank's Balanced Salt Solution (commercially available from Life Technologies) containing 1.6 mM CaCl2 and 20 mM HEPES, pH 7.4 ("wash buffer"), and loaded using 0.1 mL of wash buffer containing Fluo-4 (3 μM final concentration, commercially available from Molecular Probes). After 1 h, the cells are washed twice with 0.2 mL wash buffer and resuspended in 0.05 mL 1× Hank's Balanced Salt Solution (commercially available from Life Technologies) containing 3.5 mM CaCl2 and 10 mM Citrate, pH 7.4 ("assay buffer"). Plates are then transferred to a FLIPR for assay. The test compound is diluted in assay buffer, and 50 μL of the resultant solution is added to the cell plates and the solution is monitored for two minutes. The final concentration of the test compound is adjusted to range from about 50 picoM to about 3 μM. Agonist buffer (wash buffer titrated with 1N HCl to provide a solution having a pH of 5.5 when mixed 1:1 with assay buffer) (0.1 mL) is then added to each well, and the plates are incubated for 1 additional minute.
- Malachite Green ATPase Assay (1) Greiner 384-well (Greiner 781101) or Costar 384-well flat-bottomed polystyrene multiwell plates (VWR). (2) Assay buffer of (a) 100 mM Tris-HCl, pH 7.4, (b) 150 mM KCl, (c) 6 mM MgCl2. Stored at room temperature. (3) 0.0812% (w/v) malachite green (M 9636, Sigma Aldrich Ltd., Poole, UK). Stored at room temperature. (4) 2.32% (w/v) polyvinyl alcohol USP (P 1097, Sigma Aldrich Ltd, Poole, UK) in boiling water (see Comment 1), allowed to cool, and stored at room temperature. (5) 5.72% (w/v) ammonium molybdate in 6 M hydrochloric acid. Stored at room temperature. (6) 34% (w/v) sodium citrate. Stored at room temperature. (7) 100 mM ATP, disodium salt, special quality (47699, Sigma Aldrich). Stored at −20° C. (8) E. coli expressed yeast HSP90 protein, purified >95% (see, e.g., Panaretou et al., 1998) and stored in 50 uL aliquots at −80° C. Method 1. Dilute test compounds to 500 μM in AR water (DMSO concentration will be 2.5%). Transfer 2.5 μl of these compounds directly from the daughter plate to the assay plate, giving a final assay concentration of 100 μM. To obtain 12 point IC50 values, perform serial dilutions 1:2 to produce a range of assay concentrations from 100 μM to 97.6 nM (2.5% DMSO), and transfer 2.5 μl of each concentration into the assay plate. Column 1 in the assay plate contains no compound, as a negative control. An additional row with no compound is also used as a background. 2. Prepare ATP by diluting 100 mM stock to 925 μM with assay buffer, and aliquot 5 μl of diluted ATP to each well including controls (final assay concentration 370 μM). 3. Add 5 μl of buffer to background row. 4. Dilute enzyme preparation to 1.05 μM with assay buffer, and aliquot 5 μl into each compound well and to the negative control column. 5. Collect the reagents to the bottom of the well, cover plate with plate seal and incubate overnight at 37 deg C. 6. First thing in the morning prepare the Malachite Green Reagent. Add 2 parts of Malachite Green Solution, 1 part of Polyvinyl Alcohol Solution, 1 part of Ammonium Molybdate Solution, and 2 parts of AR water. 7. Invert to mix, and leave for approximately 1 hour until the colour turns from brown to golden yellow. 8. Add 40 μl of Malachite Green Reagent to each well, allow 5 mins for colour to develop. 9. Add 5 μl of Sodium Citrate Reagent to each well (see comment 2) 10. Re-cover with plate seal and shake on plate shaker for at least 15 mins. 11. Measure Absorbance at 620 nM using a suitable plate reader (e.g. Victor, Perkin Elmer Life Sciences, Milton Keynes, UK). Under these conditions, the control absorbance is 0.9 to 1.4, and the background is 0.2-0.35 giving a signal to noise ratio of 12. The Z′ factor calculated from data obtained using these conditions is between 0.6 and 0.9. Comments
- Inhibitory Activity of Human ACC1 and ACC2 The recombinant human ACC1 and the recombinant human ACC2 obtained from the above Preparation Examples were preincubated in an assay buffer (50 mM HEPES-KOH (pH 7.4), 10 mM magnesium chloride, 6 to 10 mM potassium citrate, 4 mM glutathione reduced form, and 1.5 mg/ml bovine serum albumin) for 1 hour. Then, 5 μL of the pre-incubated enzyme solution and 5 μL of a substrate solution (50 mM HEPES-KOH (pH 7.4), 1 mM ATP, 0.8 mM acetyl-CoA, and 25 to 50 mM potassium bicarbonate) were added to a 384-well microplate into which 0.2 μl of each of solutions of the compound of the present invention (DMSO had been dispensed, and the mixture was centrifuged and shaken, and then incubated in a wet box at room temperature for 1 to 3 hours. After incubation, the enzymatic reaction was stopped by addition of EDTA, and then the sample was co-crystallized with an α-cyano-4-hydroxy cinnamic acid (CHCA) matrix on a MALDI target plate, and measurement was performed in a reflector negative mode using a matrix-assisted laser desorption ionization-time-of-flight mass spectrometer (MALDI-TOF MS). Deprotonated ions of the substrate acetyl-CoA (AcCoA) and the reaction product malonyl-CoA (MalCoA) were detected, and the intensity of each signal was used to calculate a conversion rate to malonyl-CoA or succinyl-CoA, i.e., intensity of [MalCoA-H]—/(intensity of [MalCoA-H]-+intensity of [AcCoA.H].). A 50% inhibitory concentration (IC50 value) was calculated from the inhibition rate of the enzyme reaction at each compound concentration.
- ACC1 Enzyme Assay B1 For the assay, 50 nl of a 100-times concentrated solution of the test substance in DMSO were pipetted into a white low-volume 384-well microtitre plate (Greiner Bio-One, Frickenhausen, Germany), 2.5 ul of a solution of hACC1 in assay buffer [50 mM HEPES/NaOH pH 7.5, 2 mM MgCl2, 2 mM potassium citrate, 12 mM NaHCO3, 2 mM dithiothreitol (DTT), 0.005% (w/v) bovine serum albumin (BSA)] were added and the mixture was incubated for 15 min to allow prebinding of the substances to the enzyme prior to the enzyme reaction. The enzyme reaction was then started by addition of 2.5 ul of a solution of adenosine triphosphate (ATP, 100 uM=>final concentration in 5 ul of assay volume: 50 uM) and acetyl-CoA (20 uM=>final concentration in 5 ul assay volume: 10 uM) in assay buffer, and the resulting mixture was incubated at 22° C. for the reaction time of 45 min. The concentration of the hACC1 was adapted to the respective activity of the enzyme and adjusted such that the assay operated in the linear range. Typical concentrations were in the range of 1.75 ng/ul. The reaction was stopped by addition of 2.5 ul of the "ADP-GLO reagent" (1:1.5-times diluted), and the resulting mixture was incubated at 22° C. for 1 h to convert the unreacted ATP completely into cAMP. 2.5 ul of the "kinase detection reagent" were then added (1.2-times more concentrated than rec mmended by the manufacturer), the resulting mixture was incubated at 22° C for 1 h.
- ACC2 Enzyme Assay B2 For the assay, 50 nl of a 100-times concentrated solution of the test substance in DMSO were pipetted into a white low-volume 384-well microtitre plate (Greiner Bio-One, Frickenhausen, Germany), 2.5 ul of a solution of hACC2 in assay buffer [50 mM HEPES/NaOH pH 7.5, 2 mM MgCl2, 2 mM potassium citrate, 12 mM NaHCO3, 2 mM dithiothreitol (DTT), 0.005% (w/v) bovine serum albumin (BSA)] were added and the mixture was incubated for 15 min to allow prebinding of the substances to the enzyme prior to the enzyme reaction. The enzyme reaction was then started by addition of 2.5 ul of a solution of adenosine triphosphate (ATP, 100 uM=>final concentration in 5 ul of assay volume: 50 uM) and acetyl-CoA (20 uM=>final concentration in 5 ul assay volume: 10 uM) in assay buffer, and the resulting mixture was incubated at 22° C. for the reaction time of 45 min. The concentration of the hACC2 was adapted to the respective activity of the enzyme and adjusted such that the assay operated in the linear range. Typical concentrations were in the range of 2 ng/ul. The reaction was stopped by addition of 2.5 ul of the ADP-GLO reagent (1:1.5-times diluted), and the resulting mixture was incubated at 22° C. for 1 h to convert the unreacted ATP completely into cAMP. 2.5 ul of the kinase detection reagent were then added (1.2-times more concentrated than recommended by the manufacturer), the resulting mixture was incubated at 22° C for 1 h.
- In Vitro c-Met and VEGFR-2 Enzyme Assay Briefly, 20 μg/mL poly (Glu,Tyr) 4:1 (Sigma) was pre-coated in 96-well plates as a substrate. A 50 μL aliquot of 10 μmol/L ATP solution diluted in kinase reaction buffer (50 mmol/L HEPES [pH 7.4], 50 mmol/L MgCl2, 0.5 mmol/L MnCl2, 0.2 mmol/L Na3VO4, and 1 mmol/L DTT) was added to each well; 1 μL of various concentrations of indicated compounds diluted in 1% DMSO (v/v) (Sigma) was then added to each reaction well. DMSO (1%, v/v) was used as the negative control. The kinase reaction was initiated by the addition of purified tyrosine kinase proteins diluted in 49 μL of kinase reaction buffer. After incubation for 60 min at 37 °C, the plate was washed three times with phosphate-buffered saline (PBS) containing 0.1% Tween 20 (T-PBS). Anti-phosphotyrosine (PY99) antibody was then added. After a 30-min incubation at 37 °C, the plate was washed three times, and horseradishperoxidase-conjugated goat anti-mouse IgG was added. The plate was then incubated at 37 °C for 30 min and washed 3 times. A 100 μL aliquot of a solution containing 0.03% H2O2 and 2 mg/ml ophenylenediamine in 0.1 mol/L citrate buffer (pH 5.5) was added. The reaction was terminated by the addition of 50 μL of 2 mol/L H2SO4 as the color changed, and the plate was analyzed using a multi-well spectrophotometer (SpectraMAX 190, Molecular Devices) at 490 nm.
- Kinase Assay Biochemical KDR kinase assays were performed in 96 well microliter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mLs per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mLs per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mLs per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mLs per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microliter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- Kinase Assay PDGFRβ: Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- Kinase Assay VEGFR2: Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- PDGFRβ Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- PDGFRβ Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microliter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-β protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- PDGFRβ Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFRβ protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mis per well wash with PBS-Tween-20, 100 μl of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- PDGFRβ Kinase Assay Biochemical PDGFR3 kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30 °C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microliter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 M ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30 °C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mis per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30 μC., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mis per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 2.7 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 ul of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm.
- Biochemical Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values.
- Biochemical Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values.
- Biochemical Assay IC50 values of test compounds were determined with an enzyme activity assay using the C-domain of MALT1 (amino acids 329-824). The readout parameter is the increase of fluorescence lifetime over time, proportional to enzyme activity.The assay employs a short peptide substrate labeled with the single fluorophore PT14 as a fluorescence lifetime probe sensitive to the cleavage state of the substrate (PT14: 6-(9-oxo-9H-acridin-10-yl)-hexanoate, AssayMetrics, UK). The peptide substrate has the following sequence: Ac-Trp-Leu-Arg-Ser-Arg^Cys(PT14)-NH2 (Product number BS-9117, Biosyntan, Germany, N-terminus to C-terminus from left to right in three letter code, Ac: acetyl group, Cys(PT14): cysteine residue with the fluorophore PT14 conjugated to the cysteine sulfhydryl group via a maleimide group; C-terminus of the peptide is amidated; within the substrate sequence written above, ^ indicates the scissile bond). The assay buffer consists of 200 mM Tris/HCl at pH 7.5, 0.8 M Na citrate, 100 μM EGTA, 100 μM DTT and 0.05% (w/v) CHAPS. The kinetic characterization of the enzymatic reaction led to the determination of a Michaelis Constant (KM) of 40 μM and a kcat value of 34 s−1. The assay was established for the 384-well plate format using black microtiter round well plates (Product number 95040020, Thermo Electron Oy, Finland). Test compounds were dissolved in 100% (v/v) DMSO or a mixture containing 90% (v/v) DMSO and 10% (v/v) H2O at a stock concentration of 100 mM. Serial dilutions of test compounds were prepared using either 100% (v/v) DMSO or a mixture containing 90% (v/v) DMSO and 10% (v/v) H2O.
- Biochemical Protease Assay MALT1 protease activity was assessed in an in vitro assay using a tetrapeptide as substrate and full-length MALT1 protein (Strep-MALT1(1-824)-His) purified from baculovirus-infected insect cells. The tetrapeptide LRSR is coupled to AMC (7-amino-4-methylcoumarin) and provides a quenched, fluorescent substrate for the MALT1 protease (SM Biochemicals). Cleavage of AMC from the Arginine residue results in an increase in coumarin fluorescence measured at 460 nm (excitation 355 nm). The final assay buffer consisted of 10 nM FL MALT1 protein, 200 μM Ac-LRSR-AMC, 50 mM Tris pH 7.5, 0.6 M Citrate, 1 mM DTT, 1 mM EDTA, 0.05% BSA and 1.5% DMSO. Test compounds were spotted at 50 nL in 100% DMSO per well of a black 384-Proxiplate (Perkin Elmer). Test compound concentrations ranged from 30 μM to 0.5 nM using 11 dilution steps (1:3). Background signal was measured from control wells containing assay buffer without enzyme which functions as low control (LC). High control (HC) values were generated using the reaction with enzyme but no compound treatment. Compounds were pre-incubated with MALT1 enzyme for 50 minutes at RT. Substrate was added subsequently and fluorescence was measured in Labsystems fluoroskan at excitation 355 nm and emission 460 nm to determine time 0. The reaction was subsequently incubated for 4 h at RT and fluorescence was measured. For ICso calculations, timepoint 0 was subtracted from the 4 h timepoint to correct for any potential autofluorescence of the compounds. The enzyme reaction was linear during the 4 h incubation period. Characterization of the substrate Ac-LRSR-AMC determined the Michaelis constant KM at 200 μM.
- Human ACC1 Enzyme Assay For the assay, 50 nl of a 100-times concentrated solution of the test substance in DMSO were pipetted into a white low-volume 384-well microtitre plate (Greiner Bio-One, Frickenhausen, Germany), 2.5 μl of a solution of hACC1 in assay buffer [50 mM HEPES/NaOH pH 7.5, 2 mM MgCl2, 2 mM potassium citrate, 12 mM NaHCO3, 2 mM dithiothreitol (DTT), 0.005% (w/v) bovine serum albumin (BSA)] were added and the mixture was incubated for 15 min to allow prebinding of the substances to the enzyme prior to the enzyme reaction. The enzyme reaction was then started by addition of 2.5 μl of a solution of adenosine triphosphate (ATP, 100 μM=>final concentration in 5 μl of assay volume: 50 μM) and acetyl-CoA (20 μM=>final concentration in 5 μl assay volume: 10 μM) in assay buffer, and the resulting mixture was incubated at 22 °C. for the reaction time of 45 min. The concentration of the hACC1 was adjusted to the respective activity of the enzyme and set such that the assay was carried out in the linear range. Typical concentrations were in the range of 1.75 μl. The reaction was stopped by addition of 2.5 μl of the "ADP-GLO reagent" (1:1.5-times diluted), and the resulting mixture was incubated at 22 °C. for 1 h to convert the unreacted ATP completely into cAMP. 2.5 μl of the "kinase detection reagent" were then added (1.2-times more concentrated than recommended by the manufacturer), the resulting mixture was incubated at 22 °C. for 1 h and the luminescence was then measured using a suitable measuring instrument (Viewlux or Topcount from Perkin-Elmer or Pherastar from BMG Labtechnologies).
- Human ACC2 Enzyme Assay For the assay, 50 nl of a 100-times concentrated solution of the test substance in DMSO were pipetted into a white low-volume 384-well microtitre plate (Greiner Bio-One, Frickenhausen, Germany), 2.5 μl of a solution of hACC2 in assay buffer [50 mM HEPES/NaOH pH 7.5, 2 mM MgCl2, 2 mM potassium citrate, 12 mM NaHCO3, 2 mM dithiothreitol (DTT), 0.005% (w/v) bovine serum albumin (BSA)] were added and the mixture was incubated for 15 min to allow prebinding of the substances to the enzyme prior to the enzyme reaction. The enzyme reaction was then started by addition of 2.5 μl of a solution of adenosine triphosphate (ATP, 100 μM=>final concentration in 5 μl of assay volume: 50 μM) and acetyl-CoA (20 μM=>final concentration in 5 μl assay volume: 10 μM) in assay buffer, and the resulting mixture was incubated at 22 °C. for the reaction time of 45 min. The concentration of the hACC2 was adjusted to the respective activity of the enzyme and set such that the assay was carried out in the linear range. Typical concentrations were in the range of 2 ng/μl. The reaction was stopped by addition of 2.5 μl of the "ADP-GLO reagent" (1:1.5-times diluted), and the resulting mixture was incubated at 22 °C. for 1 h to convert the unreacted ATP completely into cAMP. 2.5 μl of the "kinase detection reagent" were then added (1.2-times more concentrated than recommended by the manufacturer), the resulting mixture was incubated at 22 °C. for 1 h and the luminescence was then measured using a suitable measuring instrument (Viewlux or Topcount from Perkin-Elmer or Pherastar from BMG Labtechnologies).
- In Vitro Evaluation in ACC1/ACC2 Enzymatic Assay Table 3: Human ACC1 (Cat #50202 Lot #120830) and ACC2 (Cat #50201 Lot #160217) were obtained from BPS Biosciences, San Diego, CA 92121. Human ACC1 had C-terminal flag and His-tags with a MW of 270 KDa after purification from Baculovirus infected Sf9 cell expression system and came as a solution in 50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol, 1 mM DTT, 100 μg/ml FLAG peptide with 70% purity. The stock concentration was 0.25 mg/ml corresponding to 0.926 μM. Human ACC2 was also purified from Baculovirus infected Sf9 expression system. It had a MW of 277 KDa with C-terminal flag and His-tags and came as a solution in 40 mM Tris pH 8.0, 110 mM NaCl, 2.2 mM KCl, 0.04% Tween-20, 20% glycerol, and 3 mM DTT with 56% purity. The stock concentration was 0.1 mg/ml corresponding to 0.36 μM. The assay buffer for measuring the activity of ACC contained: 30 mM HEPES (pH 7.4), 2 mM MgCl2, 0.01% Brij35, 2 mM DTT, 1% DMSO (solvent for compound). For ACC1 and ACC2 assays, the following concentrations of substrate and cofactors were used: 12 mM NaHCO3, 10 μM acetyl CoA, 10 μM ATP, and 2 mM K-citrate. The recombinant human enzyme was used at 5 nM for ACC1 and 1 nM for ACC2. In brief, enzyme alone in the above buffer without substrate was used for background. All test materials were dissolved in 100% DMSO using acoustic technology (Echo550). The prepared solution was preincubated for 15 min followed by the addition of 5 μL/well of ADP standard. ATP was then added to start the reaction.
- Kinase Assay PDGFRβ: Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 300° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values.
- Kinase Assay The cytoplasmic domain of the human VEGF receptor (VEGFR-2) was expressed as a Histidine-tagged fusion protein following infection of insect cells using an His engineered baculovirus. His-VEGFR-2 was purified to homogeneity, as determined by SDS-PAGE, using nickel resin chromatography. Kinase assays were performed in 96 well microtiter plates that were coated overnight with 30 .mu g of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.2-7.4. The plates were incubated with 1% BSA and then washed four times with PBS prior to starting the reaction. Reactions were carried out in 120 .mu L reaction volumes containing 3.6 .mu M ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl.sub.2, 0.1 mM MnCl.sub.2 and 0.2 mM Na.sub.3 VO.sub.4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 0.5 ng of purified protein. Following a ten minute incubation at 25.degree. C., the reactions were washed four times with PBS containing 0.05% Tween-20. 100 .mu l of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate was diluted 1:10000 in PBS-Tween-20 and added to the wells for 30 minutes. Following four washes with PBS-Tween-20, 100 .mu l of O-phenylenediamine Dihydrochloride in Phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 .mu l of 2.5N H.sub.2 SO.sub.4 to each well and read using a microplate ELISA reader set at 492 nm.
- Kinase Assay VEGFR2: Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values.
- PDGFRβ Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values.
- PDGFRbeta Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS +0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values.
- UPLC/MS Assay NAAA protein preparation (10 ug) was pre-incubated with various concentrations of test compound or vehicle control in 100 mM NaH2PO4, 100 mM Tri Sodium Citrate Dehydrate, 0.1% Triton-X 100, 3 mM DTT, pH 4.5 for 30 min at 37° C. Duplicate samples were then incubated with 50 uM C17:1 10-cis-heptadecenoylethanolamide (Avanti Polar Lipids, Alabaster, Ala. USA) at 37° C. for 30 minutes. The reaction was terminated by the addition of 0.2 mL of cold methanol containing 1 nmol of heptadecanoic acid (NuChek Prep, Elysian, Minn. USA) as internal standard. Samples were then analyzed by UPLC/MS. Heptadecenoic and heptadecanoic acids were eluted on an Acquity UPLC BEH C18 column (50 mm length, 2.1 mm i.d., 1.7 um pore size, Waters) isocratically at 0.5 mL/min for 1.5 min with a solvent mixture of 95% methanol and 5% water, both containing 0.25% Acetic Acid and 5 mM Ammonium Acetate. The column temperature was 40° C. Electrospray ionization was in the negative mode, capillary voltage was 0.5 kV, cone voltage was 25 kV, desolvation temperature was 500° C. N2 was used as drying gas at a flow rate of 1000 L/hour and a temperature of 500° C. The [M-H]- ion was monitored in the selected-ion monitoring mode (m/z values: heptadecenoic acid 267.37, heptadecanoic acid 269.37). Calibration curves were generated using commercial heptadecenoic acid (NuCheck Prep). Inhibition of NAAA activity was calculated as reduction of heptadecenoic acid in the samples compared to vehicle controls. IC50 values were calculated by non-linear regression analysis of log [concentration]/inhibition curves using GraphPad Prism 5 (GraphPad Software Inc., CA-USA) applying a standard slope curve fitting.
- VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS +0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values.
- VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values.
- VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 2.7 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 ul of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values.
- etermination of IC50 of Inhibition of Enzymatic Activity of ACC1 and ACC2 by the Compound of the Present Disclosure The degree of inhibition of the enzymatic activity of recombinant human ACC1, ACC2 proteins under in-vitro conditions by the preferred compounds of the present disclosure is determined by the following method.The principle of the method was based on the reaction of ACC protein-catalyzed acetyl-CoA to form malonyl-CoA. ATP was consumed during this reaction and ADP was produced. The produced ADP was reconverted into ATP by using ADP-Glo Kinase Kit from Promega. This part of ATP reacted with the luciferase-fluorescence in the kit, and generated chemiluminescent signals. Thus, by measuring the intensity of chemiluminescent signal, the amount of ADP produced in the catalytic reaction was reflected, thereby indirectly determining the enzymatic activity of ACC protein and the effect of the tested compound on the enzymatic activity. The main reagents used included ACC1, ACC2 proteins (purchased from BPS bioscience, ACC1 Art. No. 50200, ACC2 Art. No. 50201), acetyl-CoA (acetyl-CoA, purchased from Sigma, Art. No. A2056), NaHCO3 (purchased from Sigma, Art. No. S6014), ADP-Glo Kinase assay kit (purchased from Promega, Art. No. V9102).The test procedure was briefly described as follows: firstly, a 1× buffer solution required for the reaction was prepared, comprising: 50 mM HEPES (pH7.4 purchased from Invitrogen, Art. No. 15630), 2 mM magnesium chloride (MgCl2, purchased from Sigma, Art. No. M1028), 2 mM potassium citrate (Potassium citrate, purchased from Sigma, Art. No. 89306), 0.01% Brij-35 detergent (purchased from Merck, Art. No. 203728), 2 mM DTT (purchased from Sigma, Art. No. D0632). The test compound powder was dissolved in DMSO to prepare a stock solution having a concentration of 10 mM, followed by three-fold dilution to prepare the concentration required for the test, and each compound was set at 10 concentration points ranging from 10 μm to 0.5 nM. Firstly, appropriate amount of ACC protein (2 nM) was added to a 384-well microplate, and then diluted test compound solutions were added to each well. A duplicate well was provided at each concentration, and a solution control group (blank group) and a negative control group (DMSO group) were set at the same time. The 384-well plates were then shaken on a microwell plate oscillator and incubated for 15 minutes at room temperature. Thereafter, a substrate mixture containing ATP, acetyl-CoA and NaHCO3 which was diluted with the aforementioned buffer solution was added to each well to start the reaction, and the final concentrations of the three components were respectively ATP 20 μM, acetyl-CoA 10 μM, and NaHCO3 30 mM. After reacting for 30 minutes at room temperature, the corresponding reaction solution and detection solution were added to each well according to the method in the specification of ADP-Glo Kinase assay kit (referring to the specification of the kit for specific methods). Finally, the relative light unit (RLU) values of each well were tested using an Envision 2104 multi-function microplate reader (Perkin Elmer). The percentage inhibition rate of a compound at a concentration on ACC enzyme activity was calculated by the following formula:Inhibition rate %=[(RLU average value of negative control wells−RLU average value of blank control wells)−(RLU average values of test wells−RLU average values of blank control wells)]/(RLU average values of negative control wells−RLU average values of blank control wells)*100Finally, nonlinear regression analysis of the logarithm of the concentration of the compound and the percentage inhibition rate of the corresponding concentration was carried out in the GraphPad Prism5 software to obtain the half maximal inhibitory concentration (IC50) of the compound.
- ACC1 Enzyme Assay A1 For the assay, 50 nl of a 100-fold concentrated solution of the test substance in DMSO were pipetted into a black low-volume 384-well microtitre plate (Greiner Bio-One, Frickenhausen, Germany), 2 ul of a solution of ACC1 in assay buffer [50 mM HEPES/NaOH pH 7.5, 12 mM sodium bicarbonate, 2 mM MgCl2, 2 mM potassium citrate, 0.005% (w/v) bovine serum albumin (BSA)] were added and the mixture was incubated for 15 min to allow pre-binding of the substances to the enzyme prior to the enzyme reaction. The enzyme reaction was then started by addition of 3 ul of a solution of adenosine triphosphate (ATP, 83.5 uM=>the final concentration in 5 ul assay volume is 50 uM, Amersham Pharmacia Biotech #27-2056-01) and acetyl-CoA (33.4 uM=>the final concentration in 5 ul assay volume is 20 uM, Roche Bioscience #10101893001) in assay buffer, and the resulting mixture was incubated at 22° C. for a reaction time of 20 min. The concentration of the ACC1 was adjusted to the respective activity of the enzyme and set such that the assay was carried out in the linear range. Typical concentrations were in the range of 2.5 ng/ul. The reaction was stopped by successive addition of 2.5 ul of a solution of d2-labelled ADP (HTRF Transscreener ADP kit, C is biointernational, Marcoule, France) in EDTA-containing HTRF Transscreener ADP detection buffer (contained in the HTRF Transscreener ADP kit, 50 mM HEPES pH 7.0, 60 mM EDTA, 0.1% (w/v) BSA, 0.02% sodium azide, 400 mM potassium fluoride) and 2.5 ul of a solution of europium cryptate-labelled anti-ADP antibody (HTRF Transscreener ADP kit) in HTRF Transscreener ADP detection buffer. The resulting mixture was incubated at 22° C. for 1 h to allow binding of the europium cryptate-labelled anti-ADP antibody to the ADP formed by the enzyme reaction and the d2-labelled ADP.
- ACC2 Enzyme Assay A2 For the assay, 50 nl of a 100-fold concentrated solution of the test substance in DMSO were pipetted into a black low-volume 384-well microtitre plate (Greiner Bio-One, Frickenhausen, Germany), 2 ul of a solution of ACC2 in assay buffer [50 mM HEPES/NaOH pH 7.5, 12 mM sodium bicarbonate, 2 mM MgCl2, 2 mM potassium citrate, 0.005% (w/v) bovine serum albumin (BSA)] were added and the mixture was incubated for 15 min to allow pre-binding of the substances to the enzyme prior to the enzyme reaction. The enzyme reaction was then started by addition of 3 ul of a solution of adenosine triphosphate (ATP, 83.5 uM=>the final concentration in 5 ul assay volume is 50 uM, Amersham Pharmacia Biotech #27-2056-01) and acetyl-CoA (33.4 uM=>the final concentration in 5 ul assay volume is 20 uM, Roche Bioscience #10101893001) in assay buffer, and the resulting mixture was incubated at 22° C. for a reaction time of 20 min. The concentration of the ACC2 was adjusted to the respective activity of the enzyme and set such that the assay was carried out in the linear range. Typical concentrations were in the range of 0.6 ng/ul. The reaction was stopped by successive addition of 2.5 ul of a solution of d2-labelled ADP (HTRF Transscreener ADP kit, C s biointernational, Marcoule, France) in EDTA-containing HTRF Transscreener ADP detection buffer (contained in the HTRF Transscreener ADP kit, 50 mM HEPES pH 7.0, 60 mM EDTA, 0.1% (w/v) BSA, 0.02% sodium azide, 400 mM potassium fluoride) and 2.5 ul of a solution of europium cryptate-labelled anti-ADP antibody (HTRF Transscreener ADP kit) in HTRF Transscreener ADP detection buffer. The resulting mixture was incubated at 22° C. for 1 h to allow binding of the europium cryptate-labelled anti-ADP antibody to the ADP formed by the enzyme reaction and the d2-labelled ADP.
- Aminoacylation Assay Compounds were solubilized in DMSO. Serial 2-fold dilutions covering two concentration ranges, 10 mM to 19.5 μM and 100 μM to 195 nM, were prepared. 0.6 μl/well of the diluted compound solutions (50× the final assay concentration) were added to white 384-well polystyrene assay plates (Thermo Fisher Scientific/Matrix Technology Corp., Hudson, NH). Uninhibited control wells (MAX)received 2 μl of 30% (v/v) DMSO. Baseline wells (MIN) received 2 μl of a solution containing 30% (v/v)DMSO and 15 mM L-phenylalanine (Sigma-Aldrich). Assays were performed in a buffer consisting of 50 mM Tris-HCl (pH 8.0), 50 mM NH4Cl, 10 mM MgCl2, 2 mM DTT, 0.005% Tween 20 (Surfact-Amps-20, Thermo Fisher Scientific/Pierce Protein Research products, Suwanee, GA), and 0.1 mM EDTA-NaOH (pH 8.0). Compounds were preincubated with 15 μl/well of either 2 nM E. coli PheRS, 2 nM H. influenzae PheRS, or 0.8 nM P. aeruginosa PheRS in buffer for 30 min at 2× final assay concentrations. The reactions were initiated with 15 μl/well of a 2× substrate solution in buffer containing 2 μM E. coli phenylalanine tRNA (Sigma-Aldrich), 100 μM ATP, and 2 μM [3H]Phe with a specific radioactivity of 6.3 Ci/mmol (PerkinElmer Life Sciences). The reactions were quenched after 30 min with 15 μl/well of a solution containing 4 mg/ml PVT/PEI/WGA type A SPA beads (PerkinElmer Life Sciences), 262 mM sodium citrate (pH 2.0), and 150 mM NaCl. The plates were sealed with transparent film (PerkinElmer Life Sciences). The beads were allowed to settle for a minimum of 2 h before scintillation counting with a Top-Count plate reader (PerkinElmer Life Sciences). [3H] counts (CPM) for aminoacylated tRNA were measured for 1 min/well.
- Binding of the M6P Analogs of the Invention with CI-M6PR 96-Well plates (Maxisorp Nunc) are incubated overnight at 4° C. with 200 μl of PMP (pentamannose 6-phosphate) at the concentration of 200 μg ml−1, in carbonate buffer (NaHCO3/Na2CO3 at 0.1M, pH 9.6). The following day, the solution containing the residual PMP is discarded and the wells are saturated, for 1 h at ambient temperature, with 360 μl of 1% gelatin (Type A from Porcine Skin) diluted in PBS (1.9 mM NaH2PO4, 8.1 mM Na2PO4 and 154 mM NaCl, pH 7.4). The wells are then rinsed five times with PBS to which 0.2% gelatin has been added. All the washes and also the dilutions are carried out in the solution of PBS to which 0.2% gelatin has been added. The M6P analogs to be tested at the various concentrations (from 10−2 to 10−7 M) are pre-incubated in the presence of pre-biotinylated CI-M6PR (M6PRb) (2.5 μg mol−1) for 20 min, then 200 μl of the mixture are incubated in the wells for 2 h, at ambient temperature. After three washes, the wells are incubated for 1 h with a streptavidin-peroxidase solution (250 μl per well; 3.10−8 M). After three more washes, 200 μl of a solution of OPD (o-phenylenediamine, 1 mg ml−1 in citrate buffer, pH 5.0, and 1 μl 30% H2O2.ml−1; Sigma Aldrich) are added. After incubation for 20 min in the dark at ambient temperature, the optical densities are measured at the wavelength of 450 nm.The affinities of the phosphonate disaccharide 18 and carboxylate disaccharide 24 of formula (I) of the invention were measured and compared with those of their respective monosaccharide homologs.The affinity denotes the binding capacity (in the case in point by means of a covalent bond) of the M6P analogs for CI-M6PR.
- Effect of Compound I on CSF1R Kinase Activity 1. ELISA Assay: The enzyme reaction substrate poly(Glu, Tyr)4:1 was diluted to 20 μg/mL with potassium-free PBS (10 mM sodium phosphate buffer, 150 mM NaCl, pH7.2-7.4). Plates were coated with the dilution, allowed to incubate at 37° C. for 12-16 h, washed with T-PBS (0.1% Tween-20 in potassium-free PBS), and dried for later use. An ATP solution (final concentration of 5 μM) diluted with a reaction buffer (50 mM HEPES pH 7.4, 50 mM MgCl2, 0.5 mM MnCl2, 0.2 mM Na3VO4, 1 mM DTT), a test compound or solvent control, and a recombinant CSF1R kinase were added sequentially to individual wells to initiate the reaction. After reaction at 37° C. for 1 hour, the plates were washed with T-PBS, and the antibody PY99 dilution was added. The plates were incubated on a shaker at 37° C. for 0.5 h, and then washed with T-PBS. A horseradish peroxidase-labeled goat anti-mouse secondary antibody dilution was added, and the plates were incubated at 37° C. for 0.5 h. After the plates were washed, an OPD developing solution (diluted with 0.1 M citric acid-sodium citrate buffer (pH=5.4) containing 0.03% H2O2) was added at 2 mg/mL and the plates were allowed to react in the dark at 25° C. for 1 to 10 minutes. Finally, 2 M H2SO4 was added to stop the reaction. The plates were read with an adjustable wavelength microplate reader at a wavelength of 490 nm. The inhibition rate was calculated as follows.InhibitionRate%=(1‐CompoundODvalue‐Enzyme‐freecontrolODvalue)SolventODvalue‐Enzyme‐freecontrolODvalue×100%The IC50 values were calculated by a four-parameter regression program in the software embedded in the microplate reader.
- In Vitro Enzymatic Cathepsin D (CatD) FRET Assay Recombinant CatD was expressed in CHO cells. The assay buffer for CatD was 0.05 M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The CatD enzyme (9 nM) was pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays was effectively started by the addition of different FRET substrates (20 nM for CatD) and the combination was incubated for one hour. The FRET assay was terminated with by addition of tris buffer, which raises the pH to neutrality, and the fluorescence was determined. The FRET substrate was a peptide with commercially available fluorophore and quencher, on opposite sides of the CatD cleavage site. The CatD substrate peptide sequence was based on sequence #1 of Table 1 from Gulnik et al., FEBS Lett. 413(2):379-384 (1997). Proteolytic cleavage of the FRET substrate released quenching of fluorescence (CatD excitation 500 nm and emission 580 nm).Alternatively, a CatD assay may also be run according to the procedure described in Yasuda et al., J. Biochem. 125(6): 1137-1143 (1999). In addition, the CatD and Cathepsin E assays are described in International Patent Application Publication No. WO2011069934.The in vitro CatD FRET assay data for each of the Examples is provided in Table 2, conducted by the first procedure described above. As shown by the high micromolar CatD data (very poorly active or inactive against CatD), the compounds disclosed herein possess the unexpected property of little to no ability to inhibit the activity of CatD. Thus, with this surprising selectivity profile, the compounds provided herein are believed to minimize, reduce or completely eliminate any risk of retinal atrophy and abnormal development of the eye and of the retinal pigmented epithelium as it relates to the normal function and activity of CatD.
- In Vitro FRET Assay In vitro FRET assay was performed to evaluate the ability of select compounds to inhibit IRE1, the results of which are summarized in Table 3. To perform the in vitro FRET assay, 1× complete assay buffer (CAB; 1M DTT, 50 mM sodium citrate pH 7.15, 1 mM magnesium acetate, 0.02% tween 20) was used to dilute SignalChem IRE1α protein to a final concentration of 2 nM. Selected compounds were serially diluted with DMSO in a non-binding black 384-well plate for a total of 15 ul in each well. 2 ul of the serially diluted compound or DMSO control were then added to new wells containing 98 ul of 1×CAB, for a total volume of 100 ul, 10 ul of which were then transferred to wells of a new plate. 5 ul of the diluted IRE1α was then added to each well. 5 ul of a 400 mM XBP1 RNA probe was then added to each well. Fluorescence was then read over 30 minutes in kinetic mode (485/515 nm).Two RNA probes were used, XBP1 wildtype (SEQ ID NO: 2) which is able to be spliced by active IRE1α or XBP1 mutant (SEQ ID NO: 3) which is unable to be spliced. Each probe contained a 5′ 6-FAM modification and a 3′ IOWA Black FQ modification.A second FRET assay was performed to assess ATP-mediated inhibition. In this case, compounds and IRE1α were prepared and combined as discussed above, with the addition of ATP up to 1 mM final concentration. This mixture was incubated at room temperature for 60 minutes and then 5 ul of 400 nM XBP1 wildtype or mutant RNA probe was added. Plates were then read over 30 minutes in kinetic mode (485/515 nm) and rate of fluorescence increase calculated for different compound dilutions to determine IC50.
- Inhibition of Protease Activity Assay Full-length cDNA of human MALT1 gene (GenBank accession No: AB026118.1) amplified by PCR was inserted in flame to a SalI site located downstream of GST gene in a pGEX6P3 vector (GE Healthcare Japan Corp.) to prepare a vector (hereinafter, referred to as a pGEX6P3-MALT1 vector). Subsequently, E. coli for protein expression (BL21-RIL-codon plus-DE3, Agilent Technologies, Inc.) was transformed with the pGEX6P3-MALT1 vector and then analyzed by ampicillin resistance screening and colony PCR to obtain an E. coli strain expressing recombinant GST fusion MALT1. Protein expression was induced with isopropyl-(3-thiogalactopyranoside. After the expression induction, E. coli precipitates were recovered by centrifugation from the E. coli culture solution, and the E. coli precipitates were homogenized and then centrifuged to obtain a supernatant. The supernatant was purified using GSTrap FF column (GE Healthcare Japan Corp.) to obtain recombinant GST fusion MALT1.B) Evaluation of Inhibition of Protease Activity of MALT1:To 89 μL of an enzyme solution (4.8 g/mL GST fusion MALT1, 50 mmol/L MES, 150 mmol/L NaCl, 10% sucrose, 0.1% CHAPS, 10 mmol/L dithiothreitol, and 1 mol/L tri-ammonium citrate) per specimen, 1 μL of a test compound (DMSO-diluted solution) of each concentration was added to prepare a mixed solution. The mixed solution was incubated at room temperature for 30 minutes, followed by the measurement of the fluorescence value of the mixed solution (fluorescence value of the first measurement) (Ex: 380 nm, Em: 460 nm; Envision (Perkin Elmer Inc.)). Next, 10 μL of 200 μmol/L substrate (Ac-LRSR-AMC, SM Biochemicals LLC) was added (final concentration: 20 μmol/L) to the mixed solution, and the mixture was reacted by incubation at 30° C. for 80 minutes, followed by the measurement of the fluorescence value of the reaction solution (fluorescence value of the second measurement) (Ex: 380 nm, Em: 460 nm; Envision (Perkin Elmer Inc.)).
- LAD2 Degranulation Assay (Protocol 1) LAD-2 cells were maintained at 37oC with 5% CO2 and grown in complete StemPro-34 media supplemented with 1% L-Glutamate, 1% penicillin/streptomycin, and 250 ng/mL human SCF. On the day of experiment, cells were collected by centrifugation and seeded in 384-well assay plates, at 4,000 cells per well in 10 ^l freshly made assay buffer (10 mM HEPES, 130 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5.6 mM glucose, 0.1% BSA, pH = 7.40). For dose-response assays, compounds solubilized in 100% DMSO at 30 mM were serially diluted (1:3) first in 100% DMSO, and 2 intermediate dilutions with assay buffer were used to establish a final top concentration of 3 ^M (in assay plate). Final DMSO concentration was kept constant at 0.1% across plates for all assays. Cortistatin-14 at 30 mM stock concentration was solubilized in HBSS + 0.1% BSA, stored at -20°C in 5 ^L aliquots and discarded after one freeze-thaw cycle. Final concentration of agonist used in both dose-response and single point assays was 1 ^M, prepared in assay buffer. Antagonist equilibrium was established with 30 minute preincubation, followed by no wash, direct addition, and stimulation of agonist for 30 minutes at 37oC. For detection of ^-hexosaminidase activity, assay plates were centrifuged and 5 ^l of cell-free supernatant from each assay well was transferred to a black-walled black-bottomed 384-well plate. Substrate solution (1 mM 4-Methylumbelliferyl N-acetyl-β-D-glucosaminide in citrate buffer, pH=4.5, 15 ^l) was added to each well and plates were incubated for 1 hour at 37oC. The plate was read on an EnVision XCite plate reader for umbelliferone fluorescence intensity (λex=355 nm, λem=460 nm). Background-subtracted data was analyzed using either GraphPad Prism or a custom Python script to calculate IC50 using either Log[Antagonist] vs. response (4 parameter) nonlinear regression (dose-response assays).
- LC-MS Based In Vitro Assay hAC protein preparation (10 μg) was preincubated with inhibitors (final DMSO concentration 1%) in assay buffer (100 mM sodium phosphate, 0.1% Nonidet P-40, 150 mM NaCl, 3 mM DTT, 100 mM sodium citrate, pH 4.5) for 30 min at 37° C. Reactions were started by the addition of 50 μM N-lauroyl ceramide (Nu-Chek Prep, Elysian, Minn.) and carried on for 30 min at 37° C. Reactions were stopped by addition of a mixture of chloroform/methanol (2:1) containing 1 nmol 11-lauroleic acid (NuChek Prep). The organic phases were collected, dried under nitrogen and analyzed by UPLC/MS (Acquity, Waters). In the negative-ion mode monitoring the reaction product (lauric acid, m/z=199) using 11-lauroleic acid as internal standard.Lipids were eluted on an Acquity UPLC BEH C18 column (50×2.1 mmID, particle size 1.7 μm), column flow at 0.5 mL/min for 1.5 min with a gradient of acetonitrile and water, both containing 0.25% acetic acid and 5 mM ammonium acetate (70% to 100% acetonitrile in 0.5 min, 100% acetonitrile for 0.5 min, 70% acetonitrile for 0.4 min). The column temperature was 40° C. Electrospray ionization (ESI) was in the negative mode, capillary voltage was 1 kV and cone voltage was 50 V. N2 was used as drying gas at a flow rate of 500 L/h and at a temperature of 400° C.The [M-H]− ion was monitored in the selected-ion monitoring mode (m/z values: lauric acid 199, 11-lauroleic acid 197.35). Calibration curves were generated with authentic lauric acid (Nu Check Prep). Inhibition of AC activity was calculated as reduction of lauric acid in the samples compared to vehicle controls. IC50 values were calculated by non-linear regression analysis of log [concentration]/inhibition curves using GraphPad Prism 5 (GraphPad Software Inc., CA-USA) applying a standard slope curve fitting.
- MALT1 Enzymatic Activity Inhibition Compound activity was determined using recombinant MALT1 protein (Creative Biomart, Cat #MALT1-28H) and Ac-LRSR-AMC Substrate in an in vitro enzymatic reaction. The enzymatic assay used to determine activity was a Fluorescence assay using a Microplate Reader ClarioStar Plus. The enzymatic reaction was carried out in 1× Assay Buffer (50 mM HEPES pH 7.2-7.4, 100 mM NaCl, 900 mM Sodium citrate, 10 mM DTT). The compounds were dispensed on a 384-well Diamond Well Plate (Axygen, Cat #P-384-120SQ-C—S) using the Biomek FX liquid handling system at 100× solutions of compounds in DMSO. 2.5×MALT1 mix (final concentration 1.5 ng/μl of MALT1) was prepared in 1× Assay Buffer and 8 μl of mixture per well were added into 384w white Reaction Plates with NBS (Corning, Cat #4513). 8 μl of 1× Assay Buffer were used for negative control. Plates were centrifuged for 1 min at 200 g. Next, the compounds were added to Reaction plates using Biomek station via following steps: 1 μl of 100× compounds (in DMSO) was mixed thoroughly with 19 μl of 1× Assay Buffer, then 4 μl of this mixture were added to Reaction plates with 8 μl of MALT1 mix. Plates were centrifuged for 1 min at 200 g and incubated for 10 minutes at rt. Next, 2.5× Ac-LRSR-AMC mix (final concentration 1 μM of Ac-LRSR-AMC) was prepared in 1× Assay Buffer and 8 μl of mixture per well were added to Reaction Plates using Biomek station. Plates were centrifuged for 1 min at 200 g, then the fluorescence was measured immediately using Microplate Reader. Plates were incubated for 30 minutes at 37° C., then for 30 minutes at rt. The fluorescence was measured after the whole incubation using Microplate Reader. The reaction signal was calculated as the subtraction of the first data set values from the second.
- Enzyme Kinase Activity Assay The enzyme reaction substrate Poly(Glu,Tyr) 4:1 was diluted with PBS without potassium ion (10 mM sodium phosphate buffer, 150 mM NaCl, pH7.2-7.4) to 20 μg/mL, 125 μL/well to coat the enzyme plate, and reacted at 37° C. for 12-16 hours. After the liquid was removed from the wells, the plate was washed three times with 200 μL/well of T-PBS (PBS containing 0.1% Tween-20) for 5 minutes each. The enzyme plate was dried in a 37° C. oven for 1-2 hours.50 μL of the ATP solution diluted with the reaction buffer (50 mM HEPES pH 7.4, 50 mM MgCl2, 0.5 mM MnCl2, 0.2 mM Na3VO4, 1 mM DTT) was added into each well at a 5 μM final concentration. The compounds were diluted to the appropriate concentration in DMSO, 1 μL/well or containing the corresponding concentrations of DMSO (negative control wells). The reaction was initiated by addition of each kinase domain recombinant protein diluted with 49 μL of reaction buffer. Two control wells without ATP were set in each experiment. The reaction mixtures were placed on a shaker (100 rpm) to react at 37° C. for 1 hour. The plates were washed with T-PBS for three times. 100 μL/well of the primary antibody PY99 dilution was added, and reacted on a shaker (100 rpm) at 37° C. for 0.5 hour. The plates were washed with T-PBS for three times. 100 μL/well of the secondary anti-horseradish peroxidase-labeled goat anti-mouse IgG dilution was added, and reacted on a shaker at 37° C. for 0.5 hour. The plates were washed with T-PBS for three times. 100 μL/well of 2 mg/mL OPD developing solution (diluted with 0.1M citric acid-sodium citrate buffer containing 0.03% H2O2 (pH=5.4)), and reacted in dark for 1-10 minutes at 25° C. (Ultrasound is needed in OPD dissolution, and the developing solution should be prepared on the site). The reaction was quenched with 50 μL/well of 2M H2SO4, and read out at 490 nm using a tunable microplate microplate reader SPECTRA MAX 190.
- Immumoenzymatic Assay Solutions of the tested compounds in DMEM medium {DMEM) 1x; Source: Cellgro; Catalogue: 10-013-CV} were prepared immediately before use. Eleven serial three fold dilutions with variation of concentrations from 20 nM to 0.2 pM were prepared. In 4 hours after seeding, serial dilutions of the compounds were added to the cells (100 ul to each well). Final concentration of tested compounds was varied from 10 nM to 0.1 pM, and DMSO4 0.5%. If it was necessary, higher concentrations of the disclosed azoles were investigated. Each dilution of the compound was tested on two identical wells. Then the cells were incubated for three days at 37° C./5% CO2 and fixed by addition of acetone/methanol (1:1) mixture in amount of 250 ul/well. In 1 min the cells were washed 3 times with PBS (Phosphate Buffered Saline) solution. Then the cells were blocked by addition of 10% fetal calf serum in PBS solution in amount of 150 ul/well for 1 h at room temperature. Then, the cells were incubated with mouse monoclonal antibodies to cor-antigen HCV, don C7-50 (Source: Affinity BioReagents; Catalogue: MA1-080) (100 ul/well, working dilution 1:500 in 10% fetal calf serum in PBS solution) for 2 h at 37° C. The cells were washed 6 times with PBS/0.05% Tween 20 solution, then, they were incubated for 1 h with goat anti-mouse immunoglobulin antibodies (conjugated with horseradish peroxidase, 100 ul/well, working dilution 1:2500 in 10% fetal calf serum in PBS solution). The cells were washed 6 times with PBS/0.05% Tween 20 solution, once with PBS solution, after that substrate (1 tablet of o-phenylenediamine (oPD)+12 ml citrate/phosphate buffer+5 ul 30% H2O2) in amount of 100 ul/well was added. The plates were kept for 30 min in the dark at room temperature. The reaction was arrested by the addition of 2N H2SO4 in amount of 100 ul/well, and optical density (wavelength 490 nm) was measured by means of multiscan plate reader Victor3 V 1420 (Perkin Elmer). IC50 values (azole concentration, lowering the level of virus RNA-replicon on 50%) for every tested azole were calculated with the help of XLfit 4 program.
- In Vitro Enzymatic Cathepsin D (Cat D) FRET (Fluorescence Resonance Energy Transfer) Assay Recombinant Cat D was expressed in CHO cells. The assay buffer for CatD is 0.05M citrate pH 3.5, 10% DMSO final, 5 mM CHAPS. The CatD enzyme (9 nM) is pre-incubated for one hour with inhibitors, typically in about 1 uL of DMSO according to a serial dilution, is added thereto. The assays are effectively started by the addition of different FRET substrates (20 nM for CatD) and the combination is incubated for one hour. The FRET assay is terminated with by addition of Tris buffer, which raises the pH to neutrality, and the fluorescence is determined. The FRET substrate is a peptide with commercially available fluorophore and quencher, on opposite sides of the BACE cleavage site. The CatD substrate peptide sequence is based on sequence #1 of Table 1 from Gulnik et al. FEBS Letters v413 p 379-384 1997. Proteolytic cleavage of the FRET substrate releases quenching of fluorescence (CatD excitation 500 nm and emission 580 nm).Alternatively, a CatD assay may also be run according to the procedure described in the article, Characterization of new fluorgenic substrates for the rapid and sensitive assay of cathepsin E and cathepsin D, J. Biochem., 125:1137, 1999. In addition, the CatD and cathepsin E assays are described in PCT publication WO2011069934. This WIPO publication describes BACE inhibitor compounds having an amide linker connecting two aromatic groups with extremely poor CatD and/or cathepsin E inhibitory activity (see Table 2).Where available, the in-vitro CatD FRET assay data for each of the Examples, conducted by the first procedure, is provided in Table 1. As shown by the high micromolar CatD data (very poorly active or inactive against CatD), the compounds of the present invention possess the unexpected property of little to no ability to inhibit the activity of CatD. Thus, with this surprising selectivity profile, the compounds of the present invention are believed to minimize, reduce or completely eliminate any risk of retinal atrophy and abnormal development of the eye and of the retinal pigmented epithelium as it relates to the normal function and activity of CatD.
- LAD2 Degranulation Assay (Protocol 2) LAD-2 cells were maintained at 37oC with 5% CO2 and grown in complete StemPro-34 media supplemented with 1% L-Glutamate, 1% penicillin/streptomycin, and 250 ng/mL human SCF. 18-24 hours prior to experiment, LAD-2 cells were collected and transferred to growth media without SCF. On the day of experiment, cells were collected by centrifugation and seeded in 384-well assay plates, at 4,000 cells per well in 17 ^l freshly made assay buffer (10 mM HEPES, 137 mM NaCl, 2.7 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5.6 mM glucose, 0.4 mM NaH2PO4, 0.04% BSA, pH = 7.40). For dose-response assays, compounds solubilized in 100% DMSO at 30 mM were serially diluted (1:3) first in 100% DMSO, and 2 intermediate dilutions with assay buffer were used to establish a final top concentration of 3 ^M (in assay plate). For single point assays, dilution of DMSO stocks to 250 nM (in assay plate) was performed in the same manner as described for dose-response assays. Final DMSO concentration was kept constant at 0.1% across plates for all assays. Cortistatin-14 at 30 mM stock concentration was solubilized in HBSS + 0.1% BSA, stored at -20°C in 5 ^L aliquots and discarded after one freeze-thaw cycle. Final concentration of agonist used in both dose-response and single point assays was 0.5 ^M, prepared in assay buffer. Antagonist equilibrium was established with 2 h preincubation, followed by no wash, direct addition, and stimulation of agonist for 30 minutes at 37oC. For detection of ^-hexosaminidase activity, assay plates were centrifuged and 5 ^L of cell-free supernatant from each assay well was transferred to a black-walled black-bottomed 384-well plate. Substrate solution (1 mM 4-Methylumbelliferyl N-acetyl-β-D-glucosaminide in citrate buffer, pH=4.5, 15 ^l) was added to each well and plates were incubated for 1 hour at 37oC. The plate was read on an EnVision XCite plate reader for umbelliferone fluorescence intensity (λex=355 nm, λem=460 nm). Background-subtracted data was analyzed using either GraphPad Prism or a custom Python script to calculate IC50 using either Log[Antagonist] vs. response (4 parameter) nonlinear regression (dose-response assays).
- Malachite Green ATPase Assay 1. Dilute test compounds to 500 uM in AR water (DMSO concentration will be 2.5%). Transfer 2.5 ul of these compounds directly from the daughter plate to the assay plate, giving a final assay concentration of 100 uM. To obtain 12 point IC50 values, perform serial dilutions 1:2 to produce a range of assay concentrations from 100 uM to 97.6 nM (2.5% DMSO), and transfer 2.5 uM of each concentration into the assay plate. Column 1 in the assay plate contains no compound, as a negative control. An additional row with no compound is also used as a background.2. Prepare ATP by diluting 100 mM stock to 925 uM with assay buffer, and aliquot 5 ul of diluted ATP to each well including controls (final assay concentration 370 uM).3. Add 5 ul of buffer to background row.4. Dilute enzyme preparation to 1.05 uM with assay buffer, and aliquot 5 uM into each compound well and to the negative control column.5. Collect the reagents to the bottom of the well, cover plate with plate seal and incubate overnight at 37deg C.6. First thing in the morning prepare the Malachite Green Reagent. Add 2 parts of Malachite Green Solution, 1 part of Polyvinyl Alcohol Solution, 1 part of Ammonium Molybdate Solution, and 2 parts of AR water. 7. Invert to mix, and leave for approximately 1 hour until the colour turns from brown to golden yellow.8. Add 40 uM of Malachite Green Reagent to each well, allow 5 mins for colour to develop.9. Add 5 ul of Sodium Citrate Reagent to each well (see comment 2)10. Re-cover with plate seal and shake on plate shaker for at least 15 mins.11. Measure Absorbance at 620 nM using a suitable plate reader (e.g. Victor, Perkin Elmer Life Sciences, Milton Keynes, UK). Under these conditions, the control absorbance is 0.9 to 1.4, and the background is 0.2-0.35 giving a signal to noise ratio of 12. The Z factor calculated from data obtained using these conditions is between 0.6
- Inhibition Assay In order to have an assay method more conducive to high-throughput screening than those published for measuring the NAE hydrolyzing activity of NAAA, we developed the fluorogenic PEA analog N-(4-methyl coumarin)palmitamide (PAMCA), which is hydrolyzed to fluorescent 7-amino-4-methyl coumarin (AMC) and palmitic acid. For three point concentration inhibition assays with hNAAA the following procedure is used. Purified activated NAAA (final concentration of 0.25 μg/mL) is incubated in assay buffer (100 mM citrate-phosphate buffer, pH 4.5, 3 mM DTT, 0.1% Triton X-100, 0.05% BSA, and 150 mM NaCl) made up to a total volume of 180 μL, followed by addition of the compound dissolved in 10 μL DMSO (along with DMSO neat for the control sample) with the final concentrations for each compound of 100, 10, and 1 μM, in triplicate on a 96 well plate. These samples are allowed to incubate for 15 min at room temperature and then 10 μL of a PAMCA stock solution in DMSO (final PAMCA concentration [5 μM]) was added. After 5 minutes of agitation on a shaking plate, the reaction is allowed to proceed at 37° C. for 120 minutes, with fluorescence readings taken every 10 minutes at a wavelength of 460 nm (using an excitation wavelength of 360 nm) on a Synergy HT Plate Reader using Gen5 software from Bio-Tek. The enzyme activity is calculated by converting the relative fluorescence units to AMC formed, using a standard curve of AMC. For compounds that inhibit hNAAA in range IC50 <1 μM full inhibition curves using eight different concentrations of inhibitor (8 point assay) are generated. The assay procedure used is the same as the three point assay. For ostensibly covalent compounds (as observed by a significant decrease in the slope of a plot of fluorescence vs. time in three point screen) samples are allowed to incubate for 2 hours at 37° C., instead of 15 minutes, before addition of 10 μL of a PAMCA stock solution in DMSO for a final PAMCA concentration of 5 μM. After 5 minutes of agitation on a shaking plate, the reaction is allowed to proceed at 37° C. for 120 minutes Inhibition constants are calculated using pro Fit software (Quantum Soft, Uetikon am See, Switzerland) and a Levenberg-Marquardt algorithm.
- Tyrosine Kinase FGFR1 Activity Assay 1. Enzyme reaction substrate μPoly(Glu,Tyr)4:1 was diluted with PBS without potassium ion (10 mM sodium phosphate buffer, 150 mM NaCl, pH7.2-7.4) to 201 μg/mL, an enzyme plate was coated at 125 μL/well, and incubated at 37° C. for 12-16 hours. The liquid from the well was discarded. The plate was washed for three times with T-PBS (0.1% Tween-20 in potassium-free PBS, 200 μL/well), 5 minutes for each time. The elisa plate was dried in 37° C. dryer for 1-2 hours.2. 49 μL of ATP solution diluted in reaction buffer (50 mM HEPES pH 7.4, 50 mM MgCl2, 0.5 mM MnCl2, 0.2 mM Na3VO4, 1 mM DTT) was added into each well, and 1 μL of the test compound was added to each well, and then 50 μL of FGFR1 kinase domain recombinant protein diluted in reaction buffer was added to initiate the reaction, and two control wells without ATP were required for each experiment. The reaction was performed on a Shaker (100 rpm) at 37° C. for 1 hour. The liquid from the well was discarded, and the plate was washed with T-PBS for three times.3. The antibody PY99 dilution (antibody diluted 1:500 in T-PBS with BSA 5 mg/mL) was added at 100 μL/well, and shaken for 0.5 hours on a 37° C. shaker. The liquid from the well was discarded, and the plate was washed with T-PBS for three times.4. The horseradish peroxidase-labeled goat anti-mouse second antibody dilution (antibody diluted 1:2000 in T-PBS with BSA 5 mg/mL) was added at 100 μL/well, and shaken for 0.5 hours on a 37° C. shaker. The liquid from the well was discarded, and the plate was washed with T-PBS for three times.5. 2 mg/mL OPD coloration solution (diluted with 0.1 M citric acid-sodium citrate buffer containing 0.03% H2O2 (pH=5.4)) was added at 100 μL/well, and reacted for 1-10 minutes at 25° C. in darkness.6. The reaction was quenched with 50 μL/well of 2M H2SO4, and the plate was read using a tunable microplate microplate reader VERSAmax at 490 nm.
- Enzyme Assay The enzyme reaction substrate Poly(Glu, Tyr)4:1 was diluted with potassium-free PBS (10 mM sodium phosphate buffer, 150 mM NaCl, pH 7.2-7.4) to 20 μg/ml and microwell plate was coated with 125 ml/well mixture. The reaction was carried out at 37° C. for 12-16 h. Then the liquid was discarded and the microwell plate was washed with 200 ml/well T-PBS (PBS containing 0.1% Tween-20) three times, 5 minutes each. The microwell plate was dried for 1-2 hours at 37° C. oven. Each well was added with reaction buffer (50 mM HEPES, pH 7.4, 50 mM MgCl2, 5 mM MnCl2, 0.2 mM Na3VO4, 1 mM DTT) diluted ATP solution (50 mL) whose final concentration is 5 μM. Drug was diluted with 1% DMSO to a suitable concentration. 10 μl/well of drug was added and then 40 μl reaction buffer diluted VEGFR-2 tyrosine kinase protein was added. The microwell plate was placed into a shaker (100 rpm) and the reaction was carried out at 37° C. for 1 h. The microwell plate was washed with T-PBS three times. Three enzyme-free control wells and corresponding concentration of DMSO control wells were required for each experiment. 100 ml of primary antibody PY99 (p-Tyr (PY99), Cell Signaling Technology, diluted with T-PBS containing 5 mg/ml BSA, 1:1000 dilution) was added to each well and the plate was placed into a shaker to react for 0.5 h at 37° C. The plate was washed with T-PBS three times. 100 ml of secondary antibody horseradish peroxidase-labeled goat anti-mouse IgG (diluted with T-PBS containing 5 mg/ml BSA, 1:2000 dilution) was added to each well and the plate was placed into a shaker to react for 0.5 h at 37° C. The plate was washed with T-PBS three times. 100 ml of 2 mg/ml of OPD developing solution (diluted with 0.1 M citric acid-sodium citrate buffer containing 0.03% of H2O2 (pH=5.4)) was added to each well and the reaction was carried out at 25° C. in the dark for 1-10 minutes. OPD was dissolved under ultrasound and developing solution was freshly prepared. 50 ml of 2 M H2SO4 was added to each well to quench the reaction and OD value was measured by wavelength tunable microplate reader SPECTRA MAX 190. Wavelength was 490 nm.