Target (3)
Compound (225)
Article Title (89)
Assay (190)
Marutani, E; Sakaguchi, M; Chen, W; Sasakura, K; Liu, J; Xian, M; Hanaoka, K; Nagano, T; Ichinose, F Cytoprotective effects of hydrogen sulfide-releasing Medchemcomm 5: 1577 -1583 (2014) Silverman, RB; Xue, F Intramolecular hydrogen-bonded nitric oxide synthase inhibitors US Patent US8927730 (2015) Singh, D; Silakari, O Sodium hydrogen exchanger inhibitory activity of benzotriazole derivatives. Eur J Med Chem 126: 183 -189 (2017) Göring, S; Taymans, JM; Baekelandt, V; Schmidt, B Indolinone based LRRK2 kinase inhibitors with a key hydrogen bond. Bioorg Med Chem Lett 24: 4630 -7 (2014) Ross, PD; Rekharsky, MV Thermodynamics of hydrogen bond and hydrophobic interactions in cyclodextrin complexes. Biophys J 71: 2144 -54 (1996) Park, H; McEachon, JD; Pollock, JA Synthesis and characterization of hydrogen peroxide activated estrogen receptor beta ligands. Bioorg Med Chem 27: 2075 -2082 (2019) Donkor, IO; Zheng, X; Han, J; Lacy, C; Miller, DD Significance of hydrogen bonding at the S(1)' subsite of calpain I. Bioorg Med Chem Lett 11: 1753 -5 (2001) Schumann, NC; Bruning, J; Marshall, AC; Abell, AD The role of N-terminal heterocycles in hydrogen bonding to α-chymotrypsin. Bioorg Med Chem Lett 29: 396 -399 (2019) Mazurov, AA; Kombo, DC; Akireddy, S; Murthy, S; Hauser, TA; Jordan, KG; Gatto, GJ; Yohannes, D Novel nicotinic acetylcholine receptor agonists containing carbonyl moiety as a hydrogen bond acceptor. Bioorg Med Chem Lett 23: 3927 -34 (2013) Liu, KK; Huang, X; Bagrodia, S; Chen, JH; Greasley, S; Cheng, H; Sun, S; Knighton, D; Rodgers, C; Rafidi, K; Zou, A; Xiao, J; Yan, S Quinazolines with intra-molecular hydrogen bonding scaffold (iMHBS) as PI3K/mTOR dual inhibitors. Bioorg Med Chem Lett 21: 1270 -4 (2011) Sciabola, S; Goetz, GH; Bai, G; Rogers, BN; Gray, DL; Duplantier, A; Fonseca, KR; Vanase-Frawley, MA; Kablaoui, NM Systematic N-methylation of oxytocin: Impact on pharmacology and intramolecular hydrogen bonding network. Bioorg Med Chem 24: 3513 -20 (2016) Yang, CY; Phillips, JG; Stuckey, JA; Bai, L; Sun, H; Delproposto, J; Brown, WC; Chinnaswamy, K Buried Hydrogen Bond Interactions Contribute to the High Potency of Complement Factor D Inhibitors. ACS Med Chem Lett 7: 1092 -1096 (2016) Böhm, M; Klebe, G Development of new hydrogen-bond descriptors and their application to comparative molecular field analyses. J Med Chem 45: 1585 -97 (2002) Mao, F; Chen, J; Zhou, Q; Luo, Z; Huang, L; Li, X Novel tacrine-ebselen hybrids with improved cholinesterase inhibitory, hydrogen peroxide and peroxynitrite scavenging activity. Bioorg Med Chem Lett 23: 6737 -42 (2013) Géraldy, M; Morgen, M; Sehr, P; Steimbach, RR; Moi, D; Ridinger, J; Oehme, I; Witt, O; Malz, M; Nogueira, MS; Koch, O; Gunkel, N; Miller, AK Selective Inhibition of Histone Deacetylase 10: Hydrogen Bonding to the Gatekeeper Residue is Implicated. J Med Chem 62: 4426 -4443 (2019) Soares, J; Keppler, BR; Wang, X; Lee, KH; Jarstfer, MB ortho-Quinone tanshinones directly inhibit telomerase through an oxidative mechanism mediated by hydrogen peroxide. Bioorg Med Chem Lett 21: 7474 -8 (2011) Oguma, T; Uehara, S; Nakahara, K; Okuyama, Y; Fuchino, K; Suzuki, N; Kan, Y; Kanegawa, N; Ogata, Y; Kusakabe, KI A Quantum Mechanics-Based Method to Predict Intramolecular Hydrogen Bond Formation Reflecting P-glycoprotein Recognition. ACS Med Chem Lett 14: 223 -228 (2023) Ahmad, S; Doweyko, LM; Dugar, S; Grazier, N; Ngu, K; Wu, SC; Yost, KJ; Chen, BC; Gougoutas, JZ; DiMarco, JD; Lan, SJ; Gavin, BJ; Chen, AY; Dorso, CR; Serafino, R; Kirby, M; Atwal, KS Arylcyclopropanecarboxyl guanidines as novel, potent, and selective inhibitors of the sodium hydrogen exchanger isoform-1. J Med Chem 44: 3302 -10 (2001) Zheng, YG; Zhang, WQ; Meng, L; Wu, XQ; Zhang, L; An, L; Li, CL; Gao, CY; Xu, L; Liu, Y Design, synthesis and biological evaluation of 4-aniline quinazoline derivatives conjugated with hydrogen sulfide (H Eur J Med Chem 202: (2020) Katragadda, M; Magotti, P; Sfyroera, G; Lambris, JD Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J Med Chem 49: 4616 -22 (2006) Labby, KJ; Xue, F; Kraus, JM; Ji, H; Mataka, J; Li, H; Martásek, P; Roman, LJ; Poulos, TL; Silverman, RB Intramolecular hydrogen bonding: a potential strategy for more bioavailable inhibitors of neuronal nitric oxide synthase. Bioorg Med Chem 20: 2435 -43 (2012) Watterson, SH; Dhar, TG; Ballentine, SK; Shen, Z; Barrish, JC; Cheney, D; Fleener, CA; Rouleau, KA; Townsend, R; Hollenbaugh, DL; Iwanowicz, EJ Novel indole-based inhibitors of IMPDH: introduction of hydrogen bond acceptors at indole C-3. Bioorg Med Chem Lett 13: 1273 -6 (2003) Gonzalez, AZ; Li, Z; Beck, HP; Canon, J; Chen, A; Chow, D; Duquette, J; Eksterowicz, J; Fox, BM; Fu, J; Huang, X; Houze, J; Jin, L; Li, Y; Ling, Y; Lo, MC; Long, AM; McGee, LR; McIntosh, J; Oliner, JD; Osgood, T; Rew, Y; Saiki, AY; Shaffer, P; Wortman, S; Yakowec, P; Yan, X; Ye, Q; Yu, D; Zhao, X; Zhou, J; Olson, SH; Sun, D; Medina, JC Novel inhibitors of the MDM2-p53 interaction featuring hydrogen bond acceptors as carboxylic acid isosteres. J Med Chem 57: 2963 -88 (2014) Wiedeman, PE; Fesik, SW; Petros, AM; Nettesheim, DG; Mollison, KW; Lane, BC; Or, YS; Luly, JR Retention of immunosuppressant activity in an ascomycin analogue lacking a hydrogen-bonding interaction with FKBP12. J Med Chem 42: 4456 -61 (1999) Foloppe, N; Fisher, LM; Howes, R; Kierstan, P; Potter, A; Robertson, AG; Surgenor, AE Structure-based design of novel Chk1 inhibitors: insights into hydrogen bonding and protein-ligand affinity. J Med Chem 48: 4332 -45 (2005) Moss, RJ; Petrie, CR; Meyer, RB; Nord, LD; Willis, RC; Smith, RA; Larson, SB; Kini, GD; Robins, RK Synthesis, intramolecular hydrogen bonding, and biochemical studies of clitocine, a naturally occurring exocyclic amino nucleoside. J Med Chem 31: 786 -90 (1988) Tardia, P; Stefanachi, A; Niso, M; Stolfa, DA; Mangiatordi, GF; Alberga, D; Nicolotti, O; Lattanzi, G; Carotti, A; Leonetti, F; Perrone, R; Berardi, F; Azzariti, A; Colabufo, NA; Cellamare, S Trimethoxybenzanilide-based P-glycoprotein modulators: an interesting case of lipophilicity tuning by intramolecular hydrogen bonding. J Med Chem 57: 6403 -18 (2014) Ettorre, A; D'Andrea, P; Mauro, S; Porcelloni, M; Rossi, C; Altamura, M; Catalioto, RM; Giuliani, S; Maggi, CA; Fattori, D hNK2 receptor antagonists. The use of intramolecular hydrogen bonding to increase solubility and membrane permeability. Bioorg Med Chem Lett 21: 1807 -9 (2011) Dawson, TK; Dziedzic, P; Robertson, MJ; Cisneros, JA; Krimmer, SG; Newton, AS; Tirado-Rives, J; Jorgensen, WL Adding a Hydrogen Bond May Not Help: Naphthyridinone vs Quinoline Inhibitors of Macrophage Migration Inhibitory Factor. ACS Med Chem Lett 8: 1287 -1291 (2017) Katz, BA; Elrod, K; Verner, E; Mackman, RL; Luong, C; Shrader, WD; Sendzik, M; Spencer, JR; Sprengeler, PA; Kolesnikov, A; Tai, VW; Hui, HC; Breitenbucher, JG; Allen, D; Janc, JW Elaborate manifold of short hydrogen bond arrays mediating binding of active site-directed serine protease inhibitors. J Mol Biol 329: 93 -120 (2003) Klumb, LA; Chu, V; Stayton, PS Energetic roles of hydrogen bonds at the ureido oxygen binding pocket in the streptavidin-biotin complex. Biochemistry 37: 7657 -63 (1998) Harada, T; Nakagawa, Y; Akamatsu, M; Miyagawa, H Evaluation of hydrogen bonds of ecdysteroids in the ligand-receptor interactions using a protein modeling system. Bioorg Med Chem 17: 5868 -73 (2009) Bao, Q; Zhang, L; Wang, N; Gabet, B; Yang, W; Gao, X; You, Q; Jiang, Z Hydrogen Peroxide Inducible JAK3 Covalent Inhibitor: Prodrug for the Treatment of RA with Enhanced Safety Profile. ACS Med Chem Lett 11: 2182 -2189 (2020) Hodge, C; Pierce, J A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of scytalone dehydratase Bioorg Med Chem Lett 3: 1605 -1608 (1993) Vaz, RJ; Gao, Z; Pribish, J; Chen, X; Levell, J; Davis, L; Albert, E; Brollo, M; Ugolini, A; Cramer, DM; Cairns, J; Sides, K; Liu, F; Kwong, J; Kang, J; Rebello, S; Elliot, M; Lim, H; Chellaraj, V; Singleton, RW; Li, Y Design of bivalent ligands using hydrogen bond linkers: synthesis and evaluation of inhibitors for human beta-tryptase. Bioorg Med Chem Lett 14: 6053 -6 (2004) Kuo, PY; Shie, TL; Chen, YS; Lai, JT; Yang, DY Enzyme inhibition potency enhancement by active site metal chelating and hydrogen bonding induced conformation-restricted cyclopropanecarbonyl derivatives. Bioorg Med Chem Lett 16: 6024 -7 (2006) Rusere, LN; Lockbaum, GJ; Lee, SK; Henes, M; Kosovrasti, K; Spielvogel, E; Nalivaika, EA; Swanstrom, R; Yilmaz, NK; Schiffer, CA; Ali, A HIV-1 Protease Inhibitors Incorporating Stereochemically Defined P2' Ligands To Optimize Hydrogen Bonding in the Substrate Envelope. J Med Chem 62: 8062 -8079 (2019) Mahanti, M; Pal, KB; Kumar, R; Schulze, M; Leffler, H; Logan, DT; Nilsson, UJ Ligand Sulfur Oxidation State Progressively Alters Galectin-3-Ligand Complex Conformations To Induce Affinity-Influencing Hydrogen Bonds. J Med Chem 66: 14716 -14723 (2023) Rosenberg, SH; Dellaria, JF; Kempf, DJ; Hutchins, CW; Woods, KW; Maki, RG; de Lara, E; Spina, KP; Stein, HH; Cohen, J Potent, low molecular weight renin inhibitors containing a C-terminal heterocycle: hydrogen bonding at the active site. J Med Chem 33: 1582 -90 (1990) Zhang, W; Zhang, D; Stashko, MA; DeRyckere, D; Hunter, D; Kireev, D; Miley, MJ; Cummings, C; Lee, M; Norris-Drouin, J; Stewart, WM; Sather, S; Zhou, Y; Kirkpatrick, G; Machius, M; Janzen, WP; Earp, HS; Graham, DK; Frye, SV; Wang, X Pseudo-cyclization through intramolecular hydrogen bond enables discovery of pyridine substituted pyrimidines as new Mer kinase inhibitors. J Med Chem 56: 9683 -92 (2014) Cowan, JA; Ohyama, T; Wang, D; Natarajan, K Recognition of a cognate RNA aptamer by neomycin B: quantitative evaluation of hydrogen bonding and electrostatic interactions. Nucleic Acids Res 28: 2935 -42 (2000) Ashwood, VA; Field, MJ; Horwell, DC; Julien-Larose, C; Lewthwaite, RA; McCleary, S; Pritchard, MC; Raphy, J; Singh, L Utilization of an intramolecular hydrogen bond to increase the CNS penetration of an NK(1) receptor antagonist. J Med Chem 44: 2276 -85 (2001) Gijsen, HJ; Berthelot, D; Zaja, M; Brône, B; Geuens, I; Mercken, M Analogues of morphanthridine and the tear gas dibenz[b,f][1,4]oxazepine (CR) as extremely potent activators of the human transient receptor potential ankyrin 1 (TRPA1) channel. J Med Chem 53: 7011 -20 (2010) Katz, BA; Elrod, K; Luong, C; Rice, MJ; Mackman, RL; Sprengeler, PA; Spencer, J; Hataye, J; Janc, J; Link, J; Litvak, J; Rai, R; Rice, K; Sideris, S; Verner, E; Young, W A novel serine protease inhibition motif involving a multi-centered short hydrogen bonding network at the active site. J Mol Biol 307: 1451 -86 (2001) Patterson, JR; Terrell, LR; Donatelli, CA; Holt, DA; Jolivette, LJ; Rivero, RA; Roethke, TJ; Shu, A; Stoy, P; Ye, G; Youngman, M; Lawhorn, BG Design and Optimization of an Acyclic Amine Series of TRPV4 Antagonists by Electronic Modulation of Hydrogen Bond Interactions. J Med Chem 63: 14867 -14884 (2020) Development of UTX-143, a selective sodium-hydrogen exchange subtype 5 inhibitor, using amiloride as a lead compound. Dichiara, M; Artacho-Cordón, A; Turnaturi, R; Santos-Caballero, M; González-Cano, R; Pasquinucci, L; Barbaraci, C; Rodríguez-Gómez, I; Gómez-Guzmán, M; Marrazzo, A; Cobos, EJ; Amata, E Dual Sigma-1 receptor antagonists and hydrogen sulfide-releasing compounds for pain treatment: Design, synthesis, and pharmacological evaluation. Eur J Med Chem 230: (2022) Napoletano, M; Norcini, G; Pellacini, F; Marchini, F; Morazzoni, G; Fattori, R; Ferlenga, P; Pradella, L Phthalazine PDE4 inhibitors. Part 3: the synthesis and in vitro evaluation of derivatives with a hydrogen bond acceptor. Bioorg Med Chem Lett 12: 5 -8 (2002) Zheng, B; D'Andrea, SV; Sun, LQ; Wang, AX; Chen, Y; Hrnciar, P; Friborg, J; Falk, P; Hernandez, D; Yu, F; Sheaffer, AK; Knipe, JO; Mosure, K; Rajamani, R; Good, AC; Kish, K; Tredup, J; Klei, HE; Paruchuri, M; Ng, A; Gao, Q; Rampulla, RA; Mathur, A; Meanwell, NA; McPhee, F; Scola, PM Potent Inhibitors of Hepatitis C Virus NS3 Protease: Employment of a Difluoromethyl Group as a Hydrogen-Bond Donor. ACS Med Chem Lett 9: 143 -148 (2018) Ahmad, S; Ngu, K; Combs, DW; Wu, SC; Weinstein, DS; Liu, W; Chen, BC; Chandrasena, G; Dorso, CR; Kirby, M; Atwal, KS Aminoimidazoles as bioisosteres of acylguanidines: novel, potent, selective and orally bioavailable inhibitors of the sodium hydrogen exchanger isoform-1. Bioorg Med Chem Lett 14: 177 -80 (2003) El-Gamil, DS; ElHady, AK; Chen, PJ; Hwang, TL; Abadi, AH; Abdel-Halim, M; Engel, M Development of novel conformationally restricted selective Clk1/4 inhibitors through creating an intramolecular hydrogen bond involving an imide linker. Eur J Med Chem 238: (2022) Shore, DGM; Sweeney, ZK; Beresford, A; Chan, BK; Chen, H; Drummond, J; Gill, A; Kleinheinz, T; Liu, X; Medhurst, AD; McIver, EG; Moffat, JG; Zhu, H; Estrada, AA Discovery of potent azaindazole leucine-rich repeat kinase 2 (LRRK2) inhibitors possessing a key intramolecular hydrogen bond - Part 2. Bioorg Med Chem Lett 29: 674 -680 (2019) Guzman-Perez, A; Wester, RT; Allen, MC; Brown, JA; Buchholz, AR; Cook, ER; Day, WW; Hamanaka, ES; Kennedy, SP; Knight, DR; Kowalczyk, PJ; Marala, RB; Mularski, CJ; Novomisle, WA; Ruggeri, RB; Tracey, WR; Hill, RJ Discovery of zoniporide: a potent and selective sodium-hydrogen exchanger type 1 (NHE-1) inhibitor with high aqueous solubility. Bioorg Med Chem Lett 11: 803 -7 (2001) Mellon, C; Aspiotis, R; Black, CW; Bayly, CI; Grimm, EL; Giroux, A; Han, Y; Isabel, E; McKay, DJ; Nicholson, DW; Rasper, DM; Roy, S; Tam, J; Thornberry, NA; Vaillancourt, JP; Xanthoudakis, S; Zamboni, R Lipophilic versus hydrogen-bonding effect in P3 on potency and selectivity of valine aspartyl ketones as caspase 3 inhibitors. Bioorg Med Chem Lett 15: 3886 -90 (2005) Gemma, S; Camodeca, C; Brindisi, M; Brogi, S; Kukreja, G; Kunjir, S; Gabellieri, E; Lucantoni, L; Habluetzel, A; Taramelli, D; Basilico, N; Gualdani, R; Tadini-Buoninsegni, F; Bartolommei, G; Moncelli, MR; Martin, RE; Summers, RL; Lamponi, S; Savini, L; Fiorini, I; Valoti, M; Novellino, E; Campiani, G; Butini, S Mimicking the intramolecular hydrogen bond: synthesis, biological evaluation, and molecular modeling of benzoxazines and quinazolines as potential antimalarial agents. J Med Chem 55: 10387 -404 (2012) Lange, JH; van der Neut, MA; den Hartog, AP; Wals, HC; Hoogendoorn, J; van Stuivenberg, HH; van Vliet, BJ; Kruse, CG Synthesis, SAR and intramolecular hydrogen bonding pattern of 1,3,5-trisubstituted 4,5-dihydropyrazoles as potent cannabinoid CB(1) receptor antagonists. Bioorg Med Chem Lett 20: 1752 -7 (2010) Pierce, AC; ter Haar, E; Binch, HM; Kay, DP; Patel, SR; Li, P CH...O and CH...N hydrogen bonds in ligand design: a novel quinazolin-4-ylthiazol-2-ylamine protein kinase inhibitor. J Med Chem 48: 1278 -81 (2005) Liu, C; Li, H; Xu, F; Jiang, X; Ma, H; Seeram, NP Cannabidiol Protects Human Skin Keratinocytes from Hydrogen-Peroxide-Induced Oxidative Stress via Modulation of the Caspase-1-IL-1β Axis. J Nat Prod 84: 1563 -1572 (2021) Hong, L; Fast, W Inhibition of human dimethylarginine dimethylaminohydrolase-1 by S-nitroso-L-homocysteine and hydrogen peroxide. Analysis, quantification, and implications for hyperhomocysteinemia. J Biol Chem 282: 34684 -92 (2007) Venables, BL; Sin, N; Wang, AX; Sun, LQ; Tu, Y; Hernandez, D; Sheaffer, A; Lee, M; Dunaj, C; Zhai, G; Barry, D; Friborg, J; Yu, F; Knipe, J; Sandquist, J; Falk, P; Parker, D; Good, AC; Rajamani, R; McPhee, F; Meanwell, NA; Scola, PM P3-P4 ureas and reverse carbamates as potent HCV NS3 protease inhibitors: Effective transposition of the P4 hydrogen bond donor. Bioorg Med Chem Lett 28: 1853 -1859 (2018) Ma, X; Lv, X; Qiu, N; Yang, B; He, Q; Hu, Y Discovery of novel quinoline-based mTOR inhibitors via introducing intra-molecular hydrogen bonding scaffold (iMHBS): The design, synthesis and biological evaluation. Bioorg Med Chem 23: 7585 -96 (2015) Tropak, MB; Kornhaber, GJ; Rigat, BA; Maegawa, GH; Buttner, JD; Blanchard, JE; Murphy, C; Tuske, SJ; Coales, SJ; Hamuro, Y; Brown, ED; Mahuran, DJ Identification of pharmacological chaperones for Gaucher disease and characterization of their effects on beta-glucocerebrosidase by hydrogen/deuterium exchange mass spectrometry. Chembiochem 9: 2650 -62 (2008) Peng, YH; Ueng, SH; Tseng, CT; Hung, MS; Song, JS; Wu, JS; Liao, FY; Fan, YS; Wu, MH; Hsiao, WC; Hsueh, CC; Lin, SY; Cheng, CY; Tu, CH; Lee, LC; Cheng, MF; Shia, KS; Shih, C; Wu, SY Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives with IDO1. J Med Chem 59: 282 -93 (2016) Bonardi, A; Micheli, L; Di Cesare Mannelli, L; Ghelardini, C; Gratteri, P; Nocentini, A; Supuran, CT Development of Hydrogen Sulfide-Releasing Carbonic Anhydrases IX- and XII-Selective Inhibitors with Enhanced Antihyperalgesic Action in a Rat Model of Arthritis. J Med Chem 65: 13143 -13157 (2022) Aso, K; Kobayashi, K; Mochizuki, M; Kanzaki, N; Sako, Y; Yano, T Discovery of pyrrolo[2,3-d]pyrimidin-4-ones as corticotropin-releasing factor 1 receptor antagonists with a carbonyl-based hydrogen bonding acceptor. Bioorg Med Chem Lett 21: 2365 -71 (2011) Uchida, T; Dojun, N; Sekine, Y; Ishimori, K Heme Proximal Hydrogen Bonding between His170 and Asp132 Plays an Essential Role in the Heme Degradation Reaction of HutZ from Vibrio cholerae. Biochemistry 56: 2723 -2734 (2017) Lee, D; Lee, S; Choi, J; Song, YK; Kim, MJ; Shin, DS; Bae, MA; Kim, YC; Park, CJ; Lee, KR; Choi, JH; Seo, J Interplay among Conformation, Intramolecular Hydrogen Bonds, and Chameleonicity in the Membrane Permeability and Cyclophilin A Binding of Macrocyclic Peptide Cyclosporin O Derivatives. J Med Chem 64: 8272 -8286 (2021) Rai, R; Katzenellenbogen, JA Effect of conformational mobility and hydrogen-bonding interactions on the selectivity of some guanidinoaryl-substituted mechanism-based inhibitors of trypsin-like serine proteases. J Med Chem 35: 4297 -305 (1992) Muley, L; Baum, B; Smolinski, M; Freindorf, M; Heine, A; Klebe, G; Hangauer, DG Enhancement of hydrophobic interactions and hydrogen bond strength by cooperativity: synthesis, modeling, and molecular dynamics simulations of a congeneric series of thrombin inhibitors. J Med Chem 53: 2126 -35 (2010) Oh, J; Quan, KT; Lee, JS; Park, I; Kim, CS; Ferreira, D; Thuong, PT; Kim, YH; Na, M NMR-Based Investigation of Hydrogen Bonding in a Dihydroanthracen-1(4 H)one from Rubia philippinensis and Its Soluble Epoxide Hydrolase Inhibitory Potential. J Nat Prod 81: 2429 -2435 (2018) Nakagawa, Y; Irie, K; Nakamura, Y; Ohigashi, H The amide hydrogen of (-)-indolactam-V and benzolactam-V8's plays a critical role in protein kinase C binding and tumor-promoting activities. Bioorg Med Chem Lett 11: 723 -8 (2001) Nasief, NN; Said, AM; Hangauer, D Modulating hydrogen-bond basicity within the context of protein-ligand binding: A case study with thrombin inhibitors that reveals a dominating role for desolvation. Eur J Med Chem 125: 975 -991 (2017) Abbate, F; Supuran, CT; Scozzafava, A; Orioli, P; Stubbs, MT; Klebe, G Nonaromatic sulfonamide group as an ideal anchor for potent human carbonic anhydrase inhibitors: role of hydrogen-bonding networks in ligand binding and drug design. J Med Chem 45: 3583 -7 (2002) Thaher, BA; Koch, P; Schattel, V; Laufer, S Role of the hydrogen bonding heteroatom-Lys53 interaction between the p38alpha mitogen-activated protein (MAP) kinase and pyridinyl-substituted 5-membered heterocyclic ring inhibitors. J Med Chem 52: 2613 -7 (2009) Verner, E; Katz, BA; Spencer, JR; Allen, D; Hataye, J; Hruzewicz, W; Hui, HC; Kolesnikov, A; Li, Y; Luong, C; Martelli, A; Radika, K; Rai, R; She, M; Shrader, W; Sprengeler, PA; Trapp, S; Wang, J; Young, WB; Mackman, RL Development of serine protease inhibitors displaying a multicentered short (<2.3 A) hydrogen bond binding mode: inhibitors of urokinase-type plasminogen activator and factor Xa. J Med Chem 44: 2753 -71 (2001) Moon, HR; Lee, HJ; Kim, KR; Lee, KM; Lee, SK; Kim, HO; Chun, MW; Jeong, LS Synthesis of 5'-substituted fluoro-neplanocin A analogues: importance of a hydrogen bonding donor at 5'-position for the inhibitory activity of S-adenosylhomocysteine hydrolase. Bioorg Med Chem Lett 14: 5641 -4 (2004) Ozawa, T; Tsuji, E; Ozawa, M; Handa, C; Mukaiyama, H; Nishimura, T; Kobayashi, S; Okazaki, K The importance of CH/pi hydrogen bonds in rational drug design: An ab initio fragment molecular orbital study to leukocyte-specific protein tyrosine (LCK) kinase. Bioorg Med Chem 16: 10311 -8 (2008) Kamaura, M; Kubo, O; Sugimoto, H; Noguchi, N; Miyashita, H; Abe, S; Matsuda, K; Tsujihata, Y; Odani, T; Iwasaki, S; Murata, T; Sato, K Discovery of a novel series of indolinylpyrimidine-based GPR119 agonists: Elimination of ether-a-go-go-related gene liability using a hydrogen bond acceptor-focused approach. Bioorg Med Chem 34: (2021) Wang, T; Lee, HJ; Tosh, DK; Kim, HO; Pal, S; Choi, S; Lee, Y; Moon, HR; Zhao, LX; Lee, KM; Jeong, LS Design, synthesis, and molecular modeling studies of 5'-deoxy-5'-ureidoadenosine: 5'-ureido group as multiple hydrogen bonding donor in the active site of S-adenosylhomocysteine hydrolase. Bioorg Med Chem Lett 17: 4456 -9 (2007) Fu, W; Zhang, M; Liao, J; Tang, Q; Lei, Y; Gong, Z; Shan, L; Duan, M; Chai, X; Pang, J; Tang, C; Wang, X; Xu, X; Li, D; Sheng, R; Hou, T Discovery of a Novel Androgen Receptor Antagonist Manifesting Evidence to Disrupt the Dimerization of the Ligand-Binding Domain via Attenuating the Hydrogen-Bonding Network Between the Two Monomers. J Med Chem 64: 17221 -17238 (2021) Sakamoto, T; Koga, Y; Hikota, M; Matsuki, K; Murakami, M; Kikkawa, K; Fujishige, K; Kotera, J; Omori, K; Morimoto, H; Yamada, K Design and synthesis of novel 5-(3,4,5-trimethoxybenzoyl)-4-aminopyrimidine derivatives as potent and selective phosphodiesterase 5 inhibitors: scaffold hopping using a pseudo-ring by intramolecular hydrogen bond formation. Bioorg Med Chem Lett 24: 5175 -80 (2014) Eibl, C; Tomassoli, I; Munoz, L; Stokes, C; Papke, RL; Gündisch, D The 3,7-diazabicyclo[3.3.1]nonane scaffold for subtype selective nicotinic acetylcholine receptor (nAChR) ligands. Part 1: the influence of different hydrogen bond acceptor systems on alkyl and (hetero)aryl substituents. Bioorg Med Chem 21: 7283 -308 (2013) Grunewald, GL; Dahanukar, VH; Caldwell, TM; Criscione, KR Examination of the role of the acidic hydrogen in imparting selectivity of 7-(aminosulfonyl)-1,2,3,4-tetrahydroisoquinoline (SK&F 29661) toward inhibition of phenylethanolamine N-methyltransferase vs the alpha 2-adrenoceptor. J Med Chem 40: 3997 -4005 (1998) Alonso, JA; Andrés, M; Bravo, M; Buil, MA; Calbet, M; Castro, J; Eastwood, PR; Esteve, C; Ferrer, M; Forns, P; Gómez, E; González, J; Lozoya, E; Mir, M; Moreno, I; Petit, S; Roberts, RS; Sevilla, S; Vidal, B; Vidal, L; Vilaseca, P Structure-activity relationships (SAR) and structure-kinetic relationships (SKR) of bicyclic heteroaromatic acetic acids as potent CRTh2 antagonists III: the role of a hydrogen-bond acceptor in long receptor residence times. Bioorg Med Chem Lett 24: 5127 -33 (2014) Bohacek, RS; McMartin, C Definition and display of steric, hydrophobic, and hydrogen-bonding properties of ligand binding sites in proteins using Lee and Richards accessible surface: validation of a high-resolution graphical tool for drug design. J Med Chem 35: 1671 -84 (1992) Huber, JD; Bentzien, J; Boyer, SJ; Burke, J; De Lombaert, S; Eickmeier, C; Guo, X; Haist, JV; Hickey, ER; Kaplita, P; Karmazyn, M; Kemper, R; Kennedy, CA; Kirrane, T; Madwed, JB; Mainolfi, E; Nagaraja, N; Soleymanzadeh, F; Swinamer, A; Eldrup, AB Identification of a potent sodium hydrogen exchanger isoform 1 (NHE1) inhibitor with a suitable profile for chronic dosing and demonstrated cardioprotective effects in a preclinical model of myocardial infarction in the rat. J Med Chem 55: 7114 -40 (2012) Kyriakis, E; Karra, AG; Papaioannou, O; Solovou, T; Skamnaki, VT; Liggri, PGV; Zographos, SE; Szennyes, E; Bokor, É; Kun, S; Psarra, AG; Somsák, L; Leonidas, DD The architecture of hydrogen and sulfur σ-hole interactions explain differences in the inhibitory potency of C-β-d-glucopyranosyl thiazoles, imidazoles and an N-β-d glucopyranosyl tetrazole for human liver glycogen phosphorylase and offer new insights to structure-based design. Bioorg Med Chem 28: (2020) Kotsikorou, E; Navas, F; Roche, MJ; Gilliam, AF; Thomas, BF; Seltzman, HH; Kumar, P; Song, ZH; Hurst, DP; Lynch, DL; Reggio, PH The importance of hydrogen bonding and aromatic stacking to the affinity and efficacy of cannabinoid receptor CB2 antagonist, 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide (SR144528). J Med Chem 56: 6593 -612 (2013) Zhao, J; Mao, Q; Lin, F; Zhang, B; Sun, M; Zhang, T; Wang, S Intramolecular hydrogen bond interruption and scaffold hopping of TMC-5 led to 2-(4-alkoxy-3-cyanophenyl)pyrimidine-4/5-carboxylic acids and 6-(4-alkoxy-3-cyanophenyl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-ones as potent pyrimidine-based xanthine oxidase inhibitors. Eur J Med Chem 229: (2022)
ChEMBL_796755 (CHEMBL1938070) Inhibition of human OSC expressed in Saccharomyces cerevisiae by gas chromatography ChEMBL_1555051 (CHEMBL3767629) Inhibition of catalase (unknown origin) using hydrogen peroxide as substrate ChEMBL_141046 (CHEMBL746894) Screened in AP1 cells expressing human NHE-1 for sodium hydrogen exchange activity ChEMBL_306146 (CHEMBL831381) Inhibitory concentration against human sodium/hydrogen exchanger (NHE-1) in PS120 variant cells ChEMBL_1901234 (CHEMBL4403456) Inhibition of rat spleen microsome HO-1 preincubated for 10 mins followed by beta-NADPH addition and measured after 15 mins by gas chromatography ChEMBL_1901235 (CHEMBL4403457) Inhibition of rat brain microsome HO-2 preincubated for 10 mins followed by beta-NADPH addition and measured after 15 mins by gas chromatography ChEMBL_141048 (CHEMBL872728) Compound is screened in AP1 cells expressing human NHE-2 for sodium hydrogen exchange activity ChEMBL_141050 (CHEMBL748618) Compound is screened in AP1 cells expressing human NHE-3 for sodium hydrogen exchange activity ChEMBL_141051 (CHEMBL748619) Compound is screened in AP1 cells expressing human NHE-5 for sodium hydrogen exchange activity ChEMBL_988011 (CHEMBL2438669) Inhibition of HO-2 in Sprague-Dawley rat brain microsomal fractions assessed as carbon monoxide formation from methemalbumin after 10 mins by gas chromatography analysis ChEMBL_988012 (CHEMBL2438670) Inhibition of HO-1 in Sprague-Dawley rat spleen microsomal fractions assessed as carbon monoxide formation from methemalbumin after 10 mins by gas chromatography analysis ChEMBL_1901236 (CHEMBL4403458) Inhibition of Sprague-Dawley rat spleen microsome HO-1 preincubated for 10 mins followed by beta-NADPH addition and measured after 15 mins by gas chromatography ChEMBL_1901237 (CHEMBL4403459) Inhibition of Sprague-Dawley rat brain microsome HO-2 preincubated for 10 mins followed by beta-NADPH addition and measured after 15 mins by gas chromatography ChEMBL_1886417 (CHEMBL4388094) Inhibition of ALR2 in rat sciatic nerve assessed as reduction in sorbitol accumulation incubated for 3 hrs in presence of 28 mM glucose by gas chromatographic analysis ChEMBL_202731 (CHEMBL809059) Sodium/hydrogen exchanger antiport activity was measured using [22]Na+ uptake inhibition assay in acidified rabbit erythrocytes ChEMBL_1287262 (CHEMBL3110842) Inhibition of human recombinant His-tagged LSD1 (171 to 836) assessed as hydrogen peroxide formation after 5 mins ChEMBL_582869 (CHEMBL1051107) Binding affinity to ecdysone receptor ligand binding domain in Drosophila melanogaster assessed as number of hydrogen bonds formed ChEMBL_607020 (CHEMBL1074648) Inhibition of PARP1 in human HeLa cells assessed as inhibition of hydrogen peroxide-induced poly(ADP-ribosyl)ation ChEMBL_1886412 (CHEMBL4388089) Inhibition of ALR2 in rat erythrocytes assessed as reduction in sorbitol accumulation incubated for 3 hrs in presence of 28 mM glucose by gas chromatographic analysis relative to control ChEMBL_1886422 (CHEMBL4388099) Inhibition of ALR2 in rat lens assessed as reduction in sorbitol accumulation incubated for 3 hrs in presence of 28 mM glucose by gas chromatographic analysis relative to control ChEMBL_908733 (CHEMBL3066160) Inhibition of Arabidopsis thaliana recombinant ACS5 expressed in Escherichia coli (BL21) assessed as oxidation of ACC to ethylene preincubated for 30 min measured after 10 min by gas chromatography ChEMBL_2152147 (CHEMBL5036694) Inhibition of human recombinant HAO1 assessed as fluorescent intensity using hydrogen peroxide as substrate by HRP interference detection assay ChEMBL_1631038 (CHEMBL3873744) Inhibition of Stat1 (unknown origin) expressed in LPS/INF-gamma-stimulated human MONO-MAC-6 cells assessed as reduction in GAS dependent transcription activity measured after 4 hrs by luciferase reporter gene assay ChEMBL_629407 (CHEMBL1105760) Inhibition of human recombinant MAO-A assessed as inhibition of production of hydrogen peroxide after 15 mins by Amplex Red fluorimetric method ChEMBL_629410 (CHEMBL1105763) Inhibition of human recombinant MAO-B assessed as inhibition of production of hydrogen peroxide after 15 mins by the Amplex Red fluorimetric method ChEMBL_1994916 (CHEMBL4628811) Binding affinity to full length human CYP46A1 expressed in Escherichia coli DH5alpha as cholesterol-24 hydroxylation using cholesterol as substrate incubated for 30 mins in presence of NADPH cytochrome P450 oxidoreductase by gas chromatography-mass spectrometry ChEMBL_2446254 Inhibition of human leukocyte MPO chlorinating activity using hydrogen peroxide as substrate preincubated for 15 mins followed by substrate addition and measured after 8 mins ChEMBL_663024 (CHEMBL1250927) Inhibition of human recombinant MAOA expressed in baculovirus infected BTI insect cells assessed as hydrogen peroxide production after 15 mins by Amplex red assay ChEMBL_663025 (CHEMBL1250928) Inhibition of human recombinant MAOB expressed in baculovirus infected BTI insect cells assessed as hydrogen peroxide production after 15 mins by Amplex red assay ChEMBL_743261 (CHEMBL1769015) Inhibition of human recombinant MAO-B expressed in baculovirus infected BTI-TN-5B1-4 insect sells assessed as hydrogen peroxide production by fluorimetric method ChEMBL_743262 (CHEMBL1769016) Inhibition of human recombinant MAO-A expressed in baculovirus infected BTI-TN-5B1-4 insect sells assessed as hydrogen peroxide production by fluorimetric method ChEMBL_1473802 (CHEMBL3418527) Inhibition of human recombinant MAO-A expressed in Sf9 cells using 5-hydroxytryptamine substrate assessed as hydrogen peroxide production after 1 hr by fluorescence assay ChEMBL_1473827 (CHEMBL3418726) Inhibition of human recombinant MAO-B expressed in Sf9 cells using 5-phenylacetaldehyde substrate assessed as hydrogen peroxide production after 1 hr by fluorescence assay ChEMBL_2122235 (CHEMBL4831382) Inhibition of Mycobacterium tuberculosis recombinant DprE1 expressed in Escherichia coli assessed as formation of resorufin using FPR as substrate by Amplex Red hydrogen/peroxidase coupled assay ChEMBL_2165791 (CHEMBL5050652) Inhibition of Mycobacterium tuberculosis H37Rv recombinant DprE1 expressed in Escherichia coli assessed as formation of resorufin using FPR as substrate by Amplex Red hydrogen/peroxidase coupled assay ChEMBL_661995 (CHEMBL1251643) Inhibition of human recombinant MAOA expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as hydrogen peroxide production from p-tyramine by amplex red assay ChEMBL_661996 (CHEMBL1251644) Inhibition of human recombinant MAOB expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as hydrogen peroxide production from p-tyramine by amplex red assay Inhibition Assay The inhibition of the activity of xanthine oxidase (XO) is tested through a coupled enzymatic reaction of xanthine oxidase, horseradish peroxidase (HRP) and a substrate thereof. First, xanthine oxidase oxidizes hypoxanthine to produce xanthine and hydrogen peroxide, and further oxidizes xanthine to produce uric acid and hydrogen peroxide. Then, hydrogen peroxide reacts with 10-acetyl-3,7-dihydroxyphenoxazine (Ampliflu Red) under catalytic action of horseradish peroxidase so as to produce resorufin, a compound with strong fluorescence. The fluorescence intensity of resorufin is determined by using a fluorescence microplate, which is in direct proportion to the activity of xanthine oxidase. ChEMBL_2055417 (CHEMBL4710418) Inhibition of Mycobacterium tuberculosis DprE1 expressed in Escherichia coli BL21 (DE3) cells assessed as formation of resorufin using FPR as substrate by Amplex Red hydrogen/peroxidase coupled assay ChEMBL_2355736 Inhibition of human NOX1 transfected in CHO cells assessed as PMA-induced hydrogen peroxide pre-incubated for 10 and measured after 15 mins by Amplex-red based fluorescence assay ChEMBL_2520737 Inhibition of human neutrophil elastase using MeOSuc-AAPV-MCA as substrate measured at 60 mins interval for up to several hrs in presence of hydrogen peroxide by fluorescence assay ChEMBL_657398 (CHEMBL1246374) Inhibition of human recombinant MAOA expressed in baculovirus infected insect BTI-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine by amplex red assay ChEMBL_657399 (CHEMBL1246375) Inhibition of human recombinant MAOB expressed in baculovirus infected insect BTI-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine by amplex red assay ChEMBL_2355737 Inhibition of human NOX2 transfected in PLB-985 cells assessed as PMA-induced hydrogen peroxide pre-incubated for 10 and measured after 15 mins by Amplex-red based fluorescence assay ChEMBL_2355741 Inhibition of human DUOX1 transfected in HEK cells assessed as PMA-induced hydrogen peroxide production preincubated for 15 mins and measured after 10 mins by Amplex-red based fluorescence assay ChEMBL_2355742 Inhibition of human DUOX2 transfected in HEK cells assessed as PMA-induced hydrogen peroxide production preincubated for 15 mins and measured after 10 mins by Amplex-red based fluorescence assay ChEMBL_2152146 (CHEMBL5036693) Inhibition of human recombinant HAO1 assessed as detecting hydrogen peroxide using glycolic acid as substrate preincubated for 30 min followed by substrate addition measured after 15 mins by microplate reader ChEMBL_2355740 Inhibition of human NOX5 transfected in HEK cells assessed as PMA-induced hydrogen peroxide production pre-incubated for 10 mins and measured after 15 mins by Amplex-res based fluorescence assay ChEMBL_786455 (CHEMBL1920950) Inhibition of human recombinant MAOA expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as hydrogen peroxide production from p-tyramine after 15 mins by amplex red assay ChEMBL_786456 (CHEMBL1920951) Inhibition of human recombinant MAOB expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as hydrogen peroxide production from p-tyramine after 15 mins by amplex red assay Biological Assay All assays were performed in room temperature with purified recombinantly expressed human SSAO. The enzyme activity was measured with benzylamine as substrate and utilized the production of hydrogen peroxide for detection. ChEMBL_2355769 Inhibition of NOX2 (unknown origin) transfected in HEK293 cells assessed as inhibition of hydrogen peroxide production pe-incubated for 15 mins and measured after 10 mins by Amplex Red based fluorescence analysis ChEMBL_769477 (CHEMBL1831417) Inhibition of human recombinant MAO-A expressed in baculovirus infected BTI-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine after 15 mins by microplate fluorescence assay ChEMBL_769479 (CHEMBL1833115) Inhibition of human recombinant MAO-B expressed in baculovirus infected BTI-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine after 15 mins by microplate fluorescence assay ChEMBL_2355738 Inhibition of human NOX3 transfected in HEK293-T-REx cells assessed as PMA-induced hydrogen peroxide production pre-incubated for 10 mins and measured after 15 mins by Amplex-res based fluorescence assay ChEMBL_2355739 Inhibition of human NOX4 transfected in HEK293-T-REx cells assessed as PMA-induced hydrogen peroxide production pre-incubated for 10 mins and measured after 15 mins by Amplex-res based fluorescence assay ChEMBL_795153 (CHEMBL1936625) Inhibition of human recombinant MAO-A expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as inhibition of hydrogen peroxide production from p-tryptamine after 15 mins by fluorimetric method ChEMBL_795208 (CHEMBL1936776) Inhibition of human recombinant MAO-B expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as inhibition of hydrogen peroxide production from p-tryptamine after 15 mins by fluorimetric method ChEMBL_772924 (CHEMBL1838812) Inhibition of human recombinant MAO-A expressed in insect BT1-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine up to 15 mins by amplex red-based fluorometric assay ChEMBL_772926 (CHEMBL1838814) Inhibition of human recombinant MAO-B expressed in insect BT1-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine up to 15 mins by amplex red-based fluorometric assay ChEMBL_946070 (CHEMBL2342563) Inhibition of recombinant human MAO-B expressed in baculovirus infected BT1-TN-5B1-4 cells assessed as inhibition of production of hydrogen peroxide from p-tyramine after 15 mins by Amplex Red assay ChEMBL_946072 (CHEMBL2342565) Inhibition of recombinant human MAO-A expressed in baculovirus infected BT1-TN-5B1-4 cells assessed as inhibition of production of hydrogen peroxide from p-tyramine after 15 mins by Amplex Red assay ChEMBL_1515820 (CHEMBL3614582) Inhibition of recombinant human DAAO expressed in HEK cells using D-serine as substrate assessed as formation of alpha-keto acid, ammonia, hydrogen peroxidase after 20 mins by horseradish peroxidase/o-phenylenediamine-based assay ChEMBL_2355732 Inhibition of human NOX2 expressed in HEK cells assessed as inhibition of ionomycin-induced hydrogen peroxide production pre-incubated for 15 mins followed by ionomycin stimulation and measured after 10 mins by Fluorescence polarization assay ChEMBL_2355733 Inhibition of human NOX1 expressed in HEK cells assessed as inhibition of ionomycin-induced hydrogen peroxide production pre-incubated for 15 mins followed by ionomycin stimulation and measured after 10 mins by Fluorescence polarization assay In vitro D-Amino Acid Oxidase Assay D-amino acid oxidase (DAAO) was assayed in a 96-well plate format. D-serine was oxidatively deaminated by porcine D-amino acid oxidase in the presence of molecular oxygen and flavin adenosine dinucleotide (FAD) to yield the corresponding alpha-keto acid, ammonia and hydrogen peroxide. The resulting hydrogen peroxide was quantified using horseradish peroxidase and o-phenylenediamine, which displays a defined yellow absorbance at 411 nm when it becomes oxidized. ChEMBL_2355728 Inhibition of human NOX1 expressed in CHO cells assessed as inhibition of PMA-induced hydrogen peroxide production pretreated for 15 mins followed by PMA stimulation and measured after 10 mins by Amplex Red based fluorescence analysis ChEMBL_2355729 Inhibition of NOX2 in human PLB-985 cells assessed as inhibition of PMA-induced hydrogen peroxide production pretreated for 15 mins followed by PMA stimulation and measured after 10 mins by Amplex Red based fluorescence analysis ChEMBL_2355731 Inhibition of human NOX5 expressed in HEK293 cells assessed as inhibition of ionomycin-induced hydrogen peroxide production pretreated for 15 mins followed by ionomycin stimulation and measured after 10 mins by Amplex Red based fluorescence analysis ChEMBL_2355770 Inhibition of NOX1 (unknown origin) expressed in HT 29 cells assessed as inhibition of PMA-induced hydrogen peroxide production pre-incubated for 15 mins and followed by PMA stimulation and measured after 10 mins by Amplex Red based analysis ChEMBL_1292875 (CHEMBL3122614) Inhibition of human recombinant MAO-B expressed in baculovirus infected insect cells assessed as hydrogen peroxide production using p-tyramine as substrate incubated for 30 mins prior to substrate addition measured for 45 mins by Amplex Red monoamine oxidase assay ChEMBL_1292876 (CHEMBL3122615) Inhibition of human recombinant MAO-A expressed in baculovirus infected insect cells assessed as hydrogen peroxide production using p-tyramine as substrate incubated for 30 mins prior to substrate addition measured for 45 mins by Amplex Red monoamine oxidase assay ChEMBL_2355730 Inhibition of human NOX4 expressed in HEK293T cells assessed as inhibition of teracycline-induced hydrogen peroxide production pretreated for 18 hrs with tetracycline and treated with compound for 15 mins and measured at 10 mins post-compound treatment by Amplex Red based fluorescence analysis Enzyme Activity Assay The formation of ADP from ATP was quantified using a coupled enzyme assay (DiscoverX) in with a fluorescent resorufin dye is generated from the interaction of ADP with hydrogen peroxide and 10-acetyl-3,7-dihydroxy-phenoxazine (excitation and emission wavelenghts of 540 and 590 nm). Absorbance Microorganism-Based Dose Response Followup to Identify Inhibitors of Streptokinase Expression Keywords: Group A streptococcus, GAS, streptokinase, expression, virulence, inhibition, dose response, EC50 Assay Overview: The goal of this assay is to identify compounds that specifically reduce streptokinase expression without inhibiting cell growth. Active compounds from the primary screen were tested in 6-point, 3-fold dilution doses with starting concentration at 15 uM as follows. GAS UMAA2166 at OD600 equal to 0.015 were plated onto 384-well plates (Corning 3570) and incubated with test compounds in Todd-Hewitt Broth medium supplemented with 100 ug/ml of streptomycin for 6 hours before cells were pelleted. Supernatant of cells was removed and tested for SK activity by measuring activation of plasminogen in an absorbance-based assay as described in the Protocol section. Cell pellets were assayed for viability by BacTiterGlo reagent (Promega G8233). Normalized SK activity values were generated by dividing values of SK activity by the values of viability reading, and used Absorbance Microorganism-Based Dose Response Followup to Identify Inhibitors of Streptokinase Expression. Keywords: Group A streptococcus, GAS, streptokinase, expression, virulence, inhibition, dose response, EC50 Assay Overview: The goal of this assay is to identify compounds that specifically reduce streptokinase expression without inhibiting cell growth. Active compounds from the primary screen were tested in 6-point, 3-fold dilution doses with starting concentration at 15 uM as follows. GAS UMAA2166 at OD600 equal to 0.015 were plated onto 384-well plates (Corning 3570) and incubated with test compounds in Todd-Hewitt Broth medium supplemented with 100 ug/ml of streptomycin for 6 hours before cells were pelleted. Supernatant of cells was removed and tested for SK activity by measuring activation of plasminogen in an absorbance-based assay as described in the Protocol section. Cell pellets were assayed for viability by BacTiterGlo reagent (Promega G8233). Normalized SK activity values were generated by dividing values of SK activity by the values of viability reading, and used Absorbance Microorganism-Based Dose Response HTS to Identify Inhibitors of Streptokinase Expression Keywords: Group A streptococcus, GAS, streptokinase, expression, virulence, inhibition, dose response, EC50 Assay Overview: The goal of this assay is to identify compounds that specifically reduce streptokinase expression without inhibiting cell growth. Active compounds from the primary screen were tested in 6-point, 3-fold dilution doses with starting concentration at 15 uM as follows. GAS UMAA2166 at OD600 equal to 0.015 were plated onto 384-well plates (Corning 3570) and incubated with test compounds in Todd-Hewitt Broth medium supplemented with 100 ug/ml of streptomycin for 6 hours before cells were pelleted. Supernatant of cells was removed and tested for SK activity by measuring activation of plasminogen in an absorbance-based assay as described in the Protocol section. Cell pellets were assayed for viability by BacTiterGlo reagent (Promega G8233). Normalized SK activity values were generated by dividing values of SK activity by the values of viability reading, and used 17β-HSD13 Rapid-Fire Mass Spectrometry Assay Recombinant human 17β-HSD13 was expressed and purified from sf9 cells at Charles River Labs (Saffron Walden, UK). Leukotriene B4 (Catalog #71160-24-2) and 12-oxoleukotriene B4 (Catalog #20140) were purchased from Cayman Chemicals (Ann Arbor, MI). NAD+ (Catalog #N8285), BSA (Catalog #A7030), DMSO (Catalog #D2650), and Tween-20 (Catalog #11332465001) were purchased from Sigma (St. Louis, MO). Formic acid (Catalog #28905) was from ThermoFisher Scientific and 384 deep well PP microplates (Catalog #784261) were from Greiner Bio-One. In a typical IC50 assay performed in a 384w PP microplate, test compounds (0-100 μM) were incubated with HSD17B13 (80 nM), LTB4 (10 μM), and NAD+ (0.5 mM) in 10 μL assay buffer (20 mM Tris (pH 7.5), BSA (0.005%), and Tween-20 (0.01%)) at RT for 3 h. The assays were quenched by adding 20 μL of 0.15% aqueous formic acid and the plates were frozen at −80° C. RF/MS analysis was performed at PureHoney Technologies (Billerica, MA) on a RapidFire RF300 system (Agilent Technologies, Inc.) coupled to an API 4000 triple quadrupole mass spectrometer (Sciex) equipped with Agilent RapidFire cartridge type A (C4). The mobile phase was 0.09% formic acid and 0.01% trifluoracetic acid in water (Buffer A) and 0.09% formic acid and 0.01% trifluoracetic acid in 80% aqueous acetonitrile (Buffer B). The RapidFire method conditions were the following: 250 ms aspirate, 3000 ms load/desalt, 4000 ms elute, and 500 ms re-equilibrate. RF-MS/MS was performed in negative polarity (−4500 V), the source temperature was 650° C., and gas 1 and gas 2 settings for nitrogen were set to 50. The curtain gas and collision gas were also nitrogen and were set to 20 and 12, respectively. Leukotriene B4 (335.3) and 12-oxoLeukotriene B4 (333.3) SRM transitions were optimized with Discovery Quant software and extracted ion counts for these analytes were determined. Growth Inhibition Assay The [3H]-hypoxanthine growth inhibition assay (Desjardins et al., 1979 Antimicrobial Agents Chemother 16: 710-718) was used to evaluate the in vitro antimalarial activity of the compounds. Briefly, synchronised parasite cultures (>90% rings, 6 to 8 h post invasion) in [3H]-RPMI-LPLF complete medium with 1% parasitaemia and 2% haematocrit were exposed to the compounds at ten two-fold concentrations. Chloroquine was used as a reference drug. Uninfected RBCs at 2% haematocrit were used as background controls. Two drug exposure periods were evaluated (48 h and 96 h). For the 48 h exposure period, the plates were incubated in the gas mixture at 37° C. for approximately 20 h (about 24 h post-invasion). To each well, 0.2 uCi of tritiated hypoxanthine (GE Healthcare, Amersham) solution in [3H]RPMI-1640-LPLF was added. The plates were incubated for a further 24 h at 37° C. in the gas Mixture and then frozen at 20° C. For the 96 h exposure period, the plates were incubated. HSD17B13 rapid-fire mass spectrometry assay (RF/MS assay) Recombinant human HSD17B13 was expressed and purified from sf9 cells at Charles River Labs (Saffron Walden, UK). Leukotriene B4 (Catalog #71160-24-2) and 12-oxoleukotriene B4 (Catalog #20140) were purchased from Cayman Chemicals (Ann Arbor, Michigan.). NAD+ (Catalog #N8285), BSA (Catalog #A7030), DMSO (Catalog #D2650), and Tween-20 (Catalog #11332465001) were purchased from Sigma (St. Louis, MO). Formic acid (Catalog #28905) was from ThermoFisher Scientific and 384 deep well PP microplates (Catalog #784261) were from Greiner Bio-One. In a typical IC50 assay performed in a 384w PP microplate, test compounds (0-100 μM) were incubated with HSD17B13 (80 nM), LTB4 (10 μM), and NAD+ (0.5 mM) in 10 μL assay buffer (20 mM Tris (pH 7.5), BSA (0.005%), and Tween-20 (0.01%)) at RT for 3 h. The assays were quenched by adding 20 μL of 0.15% aqueous formic acid and the plates were frozen at −80° C. RF/MS analysis was performed at PureHoney Technologies (Billerica, MA) on a RapidFire RF300 system (Agilent Technologies, Inc.) coupled to an API 4000 triple quadrupole mass spectrometer (Sciex) equipped with Agilent RapidFire cartridge type A (C4). The mobile phase was 0.09% formic acid and 0.01% trifluoracetic acid in water (Buffer A) and 0.09% formic acid and 0.01% trifluoracetic acid in 80% aqueous acetonitrile (Buffer B). The RapidFire method conditions were the following: 250 ms aspirate, 3000 ms load/desalt, 4000 ms elute, and 500 ms re-equilibrate. RF-MS/MS was performed in negative polarity (−4500 V), the source temperature was 650° C., and gas 1 and gas 2 settings for nitrogen were set to 50. The curtain gas and collision gas were also nitrogen and were set to 20 and 12, respectively. Leukotriene B4 (335.3) and 12-oxoLeukotriene B4 (333.3) SRM transitions were optimized with Discovery Quant software and extracted ion counts for these analytes were determined. Data Analysis. HSD17B13 enzyme activity was measured as percent conversion of extracted ion counts and normalized to high and low controls to determine percent residual activity at various concentrations of test compounds. Data were fitted to normalized activity (variable slope) versus concentration fit in GraphPad Prism 7 to determine IC50. All experiments were run in duplicates. Biochemical ATX Assay (Assay A) 5 nM recombinant ATX (Cayman Chemicals) was supplemented to 50 mM Tris buffer (pH 8.0) containing 3 mM KCl, 1 mM CaCl2, 1 mM MgCl2 0.14 mM NaCl, and 0.1% bovine serum albumin. Test compounds were dissolved in DMSO and tested in the range of 0.1 nM to 10 μM. The enzymatic reaction (22.5 μL) was started by addition of 2.5 μL 10 μM 18:1 LPC (Avanti Lipids, Alabaster, Ala., USA). After 2-h incubation at room temperature, the reaction was stopped by addition of 20 μL water containing 500 nM 20:4 LPA as internal standard and 100 μL 1-butanol for extracting LPA. Subsequently, the plates were centrifuged at 4000 rpm, 4° C., for 2 min. The resultant upper butanol phase was directly used for injection at a RapidFire system (Agilent).The RapidFire autosampler was coupled to a binary pump (Agilent 1290) and a Triple Quad 6500 (ABSciex, Toronto, Canada). This system was equipped with a 10-μL loop, 5 μL Waters Atlantis HILIC cartridge (Waters, Elstree, UK), 90% acetonitrile containing 10 mM ammonium acetate as eluent A and 40% acetonitrile containing 10 mM ammoniumacetate as eluent B. For details see (Bretschneider et al., SLAS Discovery, 2017). 1 The MS was operated in negative mode with a source temperature of 550° C., curtain gas=35, gas 1=65, and gas 2=80. The following transitions and MS parameters (DP: declustering potential and CE: collision energy) for the respective LPAs were determined: 18:1 LPA at 435.2/152.8, DP=−40, CE=−28 and 20:4 LPA at 457.2/152.8, DP=−100, CE=−27). Myeloperoxidase Assay Assays were performed at 22 °C with 2 nM MPO and 10 μM hydrogen peroxide (H2O2) in 20 mM NaH2PO4 buffer, pH 6.5 containing 140 mM NaCl, 10 mM taurine, and 1 mM L-tyrosine. Inhibitor compounds were preincubated with MPO for 15 min prior to the addition of H2O2, and the accumulation of taurine chloramine was determined after 1 min. NAAA Assay Recombinant NAAA or native rat lung NAAA was incubated at 37 °C. for 30 min in 0.2 ml of sodium hydrogen phosphate buffer (50 mM, pH 5.0) containing 0.1% Triton X-100, 3 mM dithiothreitol (DTT) and 50 mM heptadecenoylethanolamide as substrate. The reaction was terminated by the addition of 0.2 ml cold methanol containing 1 mmol of heptadecanoic acid (HDA, NuChek Prep, Elysian, Minn.). Samples were analyzed by LC/MS (liquid chromatography/mass spectrometry). Heptadecanoic acid was eluted on an XDB Eclipse C18 column isocratically at 2.2 ml/min for 1 min with a solvent mixture of 95% methanol and 5% water, both containing 0.25% acetic acid and 5 mM ammonium acetate. The column temperature was 50 °C. ESI was in the negative mode, capillary voltage was 4 kV, and fragmentor voltage was 100 V. N2 was used as drying gas at a flow rate of 13 liters/min and a temperature of 350 °C. Nebulizer pressure was set at 60 psi. CRBN/DDB1 Protein Activity Microplate reader (BMG PHERAstar FSX), ECHO (LABCYTE Echo 665), microplate thermostatic oscillator (Hangzhou Ruicheng Instrument Co., Ltd.), disodium hydrogen phosphate (Sigma-Aldrich (Shanghai) Trading Co., Ltd.), sodium dihydrogen phosphate (Sigma-Aldrich (Shanghai) Trading Co., Ltd.), bovine serum albumin (Sigma-Aldrich (Shanghai) Trading Co., Ltd.), Anti-6His-Tb crypate Gold (Cisbio Bioassays Company CISBIO), CRBN/DDB1 protein (HitGen Inc.), 384-well plate (Grenier Bio-one (Shanghai) Co., Ltd.). In Vitro Assay A reliable 96-well plate D-amino acid oxidase (DAAO) assay was developed based on previously published reports (J. Biol. Chem. 277: 27782 (2002)). Briefly, D-serine (5 mM) was oxidatively deaminated by human recombinant D-amino acid oxidase in the presence of molecular oxygen and flavin adenosine dinucleotide (FAD, 1 μM), to yield the corresponding α-keto acid, ammonia and hydrogen peroxide. The resulting hydrogen peroxide was quantified using horseradish peroxidase (0.01 mg/mL) and o-phenylenediamine (180 μg/mL), which displays a defined yellow absorbance at 411 nm when it becomes oxidized. All reactions were carried out for 20 min at room temperature in a 100-μL volume in Tris buffer (50 mM, pH 8.5). Additionally, stock solutions and serial dilutions of potential DAO inhibitors were made in 10:90 DMSO:buffer with a final assay DMSO concentration of 1%. Enzyme Assay The functional activity of compounds inhibiting the DAAO enzyme was determined by utilizing the co-product of the catalysis of D-Serine, H2O2 which can be quantitatively measured using the Amplex Red (Invitrogen) detection. Amplex Red reagent is a colorless substrate that reacts with hydrogen peroxide (H2O2) with a 1:1 stoichiometry in the presence of hydrogen peroxide to produce highly fluorescent resorufin (excitation/emission maxima=570/585 nm). The changes in fluorescence were monitored by a fluorescence plate reader, Envision (Perkin Elmer) and increases in DAAO activity were readily detected upon addition of D-Serine and suppression of this response observed with the application of test compounds.Human DAAO enzyme was supplied by the Takeda Pharmaceutical Company (Osaka) and each batch was tested and used at concentrations giving comparable levels of activity. The Km of D-Serine was measured for each enzyme batch to maintain consistency; this Km was used in subsequent assays. Inhibition Assay NSC-87877 ranked among top 10% (175th) of the compounds with the best GLIDE scores for the docking to the human Shp2 PTP domain in our virtual screening of 2368 3D structures derived from the NCI Diversity Set. Computer docking of NSC-87877 (FIG. 2) suggested that the B-ring sulfonic acid group forms hydrogen bond with the backbone NH group of Arg-465. Arg-465 is a conserved residue in the PTP signature motif (motif 9) VHCSXGXGR[T/S]G located at the base of the PTP catalytic cleft (Andersen et al., 2001). The A-ring sulfonic acid forms hydrogen bonds with the side-chain NH3 group of Lys-280 and the side-chain NH2 group of Asn-281. Lys-280/Asn-281 are non-conserved PTP residues located adjacent to the phosphotyrosine recognition loop (motif 1) (Andersen et al., 2001). The interaction between aromatic rings of the compound and the protein contributes to the binding through hydrophobic stabilization. PAR Assay Cellular activity of PARP-1 inhibitors was assessed by measuring the inhibition of the hydrogen peroxide induced PAR formation in HeLa cells (ECACC). Cellular PAR levels were measured by immunocytochemistry, and quantified using an ArrayScan vTi instrument (Cellomics Thermo Scientific). Studies were performed as follows: 6000 cells/well were seeded in 96 well plates (Perkin Elmer) in MEM/10% FCS and incubated for 24 hours at 37 C., 5% carbon dioxide. Test compounds were then added at the required concentration for 30 minutes. DNA damage was then induced adding hydrogen peroxide at the concentration of 0.1 mM for 15 minutes. Concentration curves were prepared in MEM/10% FCS from compound stocks in DMSO, and final DMSO concentration was 0.002% (v/v). Duplicate wells for each concentration point were prepared with a typical highest compound concentration of 20 uM and serial dilution 1:3. Plates were dried and fixed adding cold methanol-acetone (70:30) solution for 15 minutes at room temperature. Fluorescence Binding Assay The fluorescence buffer was degassed and aerated with pure nitrogen gas to remove dissolved oxygen. The assay was carried out on a FluoroMax-2 fluorometer. The excitation and emission wavelengths were 284 nm and 341 nm, respectively. The inhibitor solution was titrated into EGFR kinase solution, and the emission fluorescence intensity was read after addition of inhibitor, and the average of five measurements was recorded. A blank assay was performed in exactly the same manner except that the buffer without inhibitor was used for the titration. Dissociation constants (Kd) were determined by nonlinear fitting of the fluorescence data using a modified static quenching model. Enzymatic Activity Assay The inhibition of purified recombinant human GAC by varying concentrations of inhibitors is assessed via a dual-coupled enzymatic assay. The glutamate produced by the glutaminase reaction is used by glutamate oxidase to produce α-ketoglutarate, ammonia, and hydrogen peroxide, with this hydrogen peroxide subsequently being used by horseradish peroxidase to produce resorufin in the presence of Amplex UltraRed. The assay buffer consisted of 50 mM Hepes (pH 7.4), 0.25 mM EDTA and 0.1 mM Triton X-100. GAC was incubated with potassium phosphate (10 minutes at room temperature) prior to incubation with inhibitor (10 minutes at room temperature). The final reaction conditions were as follows: 2 nM GAC, 50 mM potassium phosphate, 100 mU/mL glutamate oxidase (Sigma), 1 mM glutamine (Sigma), 100 mU/mL horseradish peroxidase (Sigma), 75 μM Amplex UltraRed (Life Technologies), and 1% (v/v) DMSO. The production of resorufin was monitored on a Perkin Elmer Envision plate reader (excitation 530 nm, emission 590 nm) either in a kinetics or endpoint mode (at 20 minutes). IC50 values were calculated using a four-parameter logistic curve fit. Biological Assays of the SSAO Enzyme Inhibitors Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≤2%.Hydrogen Peroxide Detection:In a horseradish peroxidase (HRP) coupled reaction, hydrogen peroxide oxidation of 10-acetyl-3,7-dihydroxyphenoxazine produced resorufin, which is a highly fluorescent compound (Zhout and Panchuk-Voloshina. Analytical Biochemistry 253 (1997) 169-174; Amplex Red Hydrogen Peroxide/peroxidase Assay kit, Invitrogen A22188). Enzyme and compounds in 50 mM sodium phosphate, pH 7.4 were set to pre-incubate in flat-bottomed microtiter plates for approximately 15 min before initiating the reaction by addition of a mixture of HRP, benzylamine and Amplex reagent. Benzylamine concentration was fixed at a concentration corresponding to the Michaelis constant, determined using standard procedures. Fluorescence intensity was then measured at several time points during 1-2 h, exciting at 544 nm and reading the emission at 590 nm. For the human SSAO assay final concentrations of the reagents in the assay wells were: SSAO enzyme 1 ug/mL, benzylamine 100 uM, Amplex reagent 20 uM, HRP 0.1 U/mL and varying concentrations of test compound. The inhibition was measured as % decrease of the signal compared to a control without inhibitor (only diluted DMSO). The background signal from a sample containing no SSAO enzyme was subtracted from all data points. Data was fitted to a four parameter logistic model and IC50 values were calculated using the GraphPad Prism 4 or XLfit 4 programs. Inhibition of DAO (Diamine Oxidase) Test purpose: The following method is used to determine the selective inhibitory activity of the compound of the present invention on DAO.Test Materials:Human recombinant DAO (Recombinant Human ABP-1/DAO) was purchased from R&D, Cat. No. 8298-AO;Amplex® Red Hydrogen PeroxidePeroxidase Assay Kit was purchased from Invitrogen, Cat. No. A22188;1,4-Diaminobutane dihydrochloride was purchased from Aladdin, Cat. No. D106194-25G.Test Method:The test compound was dissolved in DMSO and diluted 5 times to a total of 6 concentrations. In a 384-well plate, 24 μL of human recombinant DAO (1 μg/mL) was added into each well. 1 μL of test compounds at different concentrations were added to each well containing human recombinant DAO, and the plate was incubated at 37° C. for 30 min. After incubating for 30 min, 25 μL of Amplex® Red Hydrogen Peroxide Peroxidase Assay Kit (containing 100 μM Amplex® Red and 0.2 U/ml HRP) containing 1 M 1,4-butanediamine dihydrochloride was added into the corresponding wells, and the plate was incubated in the dark at 37° C. for 30 min. After 30 min, PHERAstar FSX microplate reader of BMG LABTECH was used to read the fluorescence value (RFU) under excitation at 540 nm and emission at 580 nm. Biological Assay of the SSAO Enzyme Inhibitors All assays were performed in room temperature with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 2006, 46, 321-331). The enzyme activity was measured with benzylamine as substrate and utilized the production of hydrogen peroxide for detection. In a horseradish peroxidise (HRP) coupled reaction, hydrogen peroxide oxidation of 10-acetyl-3,7-dihydroxyphenoxazine produced resorufin, which is a highly fluorescent compound (Zhout and Panchuk-Voloshina. Analytical Biochemistry 1997, 253, 169-174; Amplex Red Hydrogen Peroxide/peroxidise Assay kit, Invitrogen A22188).Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer (50 mM sodium phosphate, pH 7.4) yielded a final DMSO concentration ≤2%. Enzyme and compounds were set to pre-incubate in flat-bottomed microtiter plates for approximately 60 minutes before initiating the reaction by addition of a mixture of HRP, benzylamine and Amplex reagent. Fluorescence intensity was then measured at several time points (15 minutes, 20 minutes and 30 minutes) exciting at 544 nm and reading the emission at 590 nm). Final concentrations of the reagents in the assay wells were: SSAO enzyme 2 μg/ml, benzylamine 100 μM, Amplex reagent 20 μM, HRP 0.1 U/mL and varying concentrations of test compound. The inhibition was measured as % decrease of the signal compared to a control without inhibitor (only diluted DMSO). The background signal from a sample containing no SSAO enzyme was subtracted from all data points. Data was fitted to a four parameter logistic model and IC50 values were calculated using the GraphPad Prism 4 or XLfit 4 programs. MAO Enzyme Inhibition Assay The effects of the test compounds on hMAO isoform enzymatic activity were evaluated by measuring their effects on the production of hydrogen peroxide (and therefore, of resorufin) from p-tyramine, using the Amplex Red MAO assay kit (Molecular Probes, Eugene, Oregon, USA) and microsomal MAO isoforms prepared from insect cells infected with recombinant baculovirus containing cDNA inserts for human MAO-A or MAO-B. The production of H2O2 was quantified in a multidetection microplate fluorescence reader (FLX800, Bio-Tek Instruments, Inc., Winooski, VT) based on the fluorescence generated (excitation, 545 nm; emission, 590 nm) over a 15 min period, in which the fluorescence increased linearly. Nox4 Assay Cells: HEK (CJ Nox4) stably expressing Nox4 was purchased from Redoxis AB (Lund). The adherent cells were cultivated in RPMI 1640 with L-Glutamine were supplemented with FBS (10%), penicillin (10 U/ml) streptomycin (100 μg/ml) and neomycin (200 μg/ml) at 37° C. in air with 5% CO2.Hydrogen peroxide produced by Nox4 was measured (fluorescence emission: 590 and excitation: 544) using Amplex red (Molecular Probes) in Fluorescan Ascent plate reader Type 374. Cells were collected from cultures by Trypsin mediated detachment of adherent cells. Cells were seeded in 96-well plates at a density of 50 000 cells in 200 l assay volume. Inhibitors were added for 30 min (37° C.) and then reagents was added to give a final concentration of Amplex Red 35 mM and 0.17 U/ml horseradish peroxidase. Nox4 activity was measured up to 100 min with readings every minute. Inhibition curves of different Nox4 inhibitors were evaluated at 50% inhibition (IC50) in comparison to cell control without inhibitor present. Y-axes: turnover of hydrogen peroxide; x-axes: concentration of inhibitor. Inhibitors were diluted in a compound plate in DMSO (100%) then transferred to Hanks buffer solution and in assay plate DMSO were 2% in all the wells.Compounds (Nox inhibitors) were diluted at 3× working concentration and titrated from 200 μM to 0.003 μM in 11 steps. In Vitro HO Activity Assay HO activity in rat spleen (HO-1) and brain (HO-2) microsomal fractions was determined by the quantitation of CO formed from the degradation of methemalbumin. Incubations for HO activity analysis were done under conditions for which the rate of CO formation was linear with respect to time and microsomal protein concentration. Briefly, reaction mixtures consisting of 100 mM phosphate buffer (pH 7.4), 50 uM methemalbumin, and 1 mg/mL protein were pre-incubated with the inhibitors at final concentrations ranging from 0.1 to 100 uM for 10 min at 37 deg C. Reactions were initiated by adding NADPH at a final concentration of 1 mM and incubations were performed for an additional 15 min at 37 deg C. Reactions were stopped by instantly freezing the reaction mixture on dry ice, and CO formation was monitored by gas chromatography. Fluorescence Based Assay The fluorescence based assay to monitor the activity of human nSMase2 in the presence or absence of potential inhibitors has been described recently. Figuera-Losada, et al. Lysate of cells expressing recombinant nSMase2 is used to catalyze the hydrolysis of sphingomyelin (SM) to ceramide and phosphorylcholine. Phosphorylcholine undergoes dephosphorylation in a reaction catalyzed by alkaline phosphatase (4 U/mL) to produce choline which in turn is oxidized by choline oxidase (0.1 U/mL) to betaine and hydrogen peroxide (H2O2). Hydrogen peroxide is made to react with Amplex red (50 μM) in the presence of horseradish peroxidase (HRP, 1 U/mL) to generate the fluorescent molecule resorufin. Generation of fluorescence is monitored by measuring relative florescence units (RFU) with excitation at 530 nm and emission at 590 nm. Extent of fluorescence is directly proportional to the extent of SM hydrolysis. Substrate stock solution is prepared in 2% Triton X-100 and sonicated for 1 min. Reactions are carried out for 1 h at 37° C. in 100 mM Tris-HCl pH 7.4, 10 mM MgCl2, 0.2% Triton X-100. This assay has been optimized in 384-well format (50 μL total volume per well) under conditions where nSMase2-catalyzed hydrolysis of SM is linear with respect to nSMase2 concentration, time of incubation and SM concentration (FIGS. 3A-3C). The assay has high reliability (Z′=0.8-0.9). It is used for compound screening, IC50 determinations and mode of inhibition studies Enzymatic Activity Assay A protocol for determination of the carbonic anhydrase enzymatic activity at rt using the pH indicator method is described below:1 μL inhibitor (50 mM stock solution in DMSO) is diluted to a final test concentration ranging from 100 μM down to 1 nM (or 1 μL water in controls) and incubated for 2 min with 0.5 to 2 EU human Carboanhydrase I (180 U/mg) in 400 μL water and 200 μL phenol red indicator solution (20 mg/L). An enzymactic unit (EU) is defined as an amount which doubles the non catalyzied rate. The hydration reaction is initiated by adding 100 μL 0.5M bicarbonate buffer (0.3M Na2CO3; 0.2M NaHCO3) and subsequent dumping of CO2 through a needle (0.7×30 mm; 22 G×1.25) into the assay solution at a rate of 10 mL gas/minute. The time to colour change (pH 7.2) is determined with a microchronometer or stop watch. Inhibition Assay Assay buffers consist of 20 mM citric acid, 60 mM disodium hydrogen orthophosphate, 1 mM EDTA, 0.1% CHAPS, 4 mM DTT, pH 5.8 for legumain, 50 mM dihydrogen sodium orthophosphate, 1 mM EDTA, 5 mM DTT, pH 6.25 for cathepsin B and cathepsin Land 100 mM Tris, 0.1% CHAPS, 10% sucrose, 10 mM DTT, pH 7.4 for caspase-3. Concentrations of substrates during the measurement were 10 nM (legumain, cathepsin Land caspase-3) and 50 nM (cathepsin B) and concentration of enzymes were 100 nM for cathepsin Land caspase-3, 270 nM for legumain and 360 nM for cathepsin B. Each enzyme was incubated with inhibitor concentrations ranging from 1 nM to 1 mM in the presence of the substrates. ROS Inhibiting Drug A high-throughput screen was performed to find molecules that inhibit ROS production by neutrophils. Extracted human neutrophils were purified and kept in culture. The cells were then exposed to various drugs and ROS production was monitored over time. Compounds that also scavenged hydrogen peroxide (H2O2) and/or lowered neutrophil ATP levels (reflecting toxicity) were removed. The top hits from the screen were selected for further analysis.160 basal hits were tested for their ability to inhibit neutrophil ROS production. 64 molecules were able to inhibit ROS production in the presence of PMA activation. 67 molecules were able to inhibit ROS production in the presence of N-Formylmethionine-leucyl-phenylalanine (fMLP). Of those 47 molecules were able to inhibit ROS production by both stimulation methods. TarO Biochemical Enzymatic Assay The TarO biochemical enzymatic assay is a liquid chromatography-mass spectroscopy (LC-MS) based end point assay that measures C55-P-P-GlcNAc (LIPID III) production. The TarO biochemical enzymatic assay was performed in a 384-well microtiter plate (Labcyte) with a reaction volume of 20 μl. The reaction mix contained 0.1 μgs/μl of TarO membrane preparation derived from MRSA COL (lysostaphin/lysozyme treated, centrifuged at 40K rpm, and re-suspended in 50 mM Tris pH 7.5, 10 mM MgCl2), 1500 μM UDP-GlcNAc, x 75 μM C55-P substrates in 83 mM Tris pH 8.0, 6.7 mM MgCl2, 6 mM CHAPS, and 8.3% DMSO buffer. The enzyme reactions were quenched by extraction in 40 μl of 1-pentanol containing 0.04 μM 15C C55-PP-GlcNAc, which was used as an internal standard. A 10 μl volume of the quenched reaction mixture (pH≈3) from each well was injected onto a reversed-phase column (C4, 5 m, 2.1×50 mm, Thermo Scientific Biobacis-4) and eluted using a NH4Ac/H2O/MeOH gradient (solvent A: 10 mM NH4Ac in water, pH 5.6; solvent B: NH4Ac (1 M)-Isopropanol (1:90, v/v, pH 5.6). The HPLC conditions were as follows: 15% solvent B for 15 seconds followed by a gradient to 90% solvent B in 90 seconds; then solvent B was kept at 95% for 10 seconds followed by a gradient to 8% solvent B in 0.1 minute. The column was then equilibrated at 15% B for 1 minute before the next injection. The flow rate was kept constant at 600 μl/minute. Mass spectrometric detection was carried out in the negative-ion mode using selected reaction monitoring (SRM). Typical mass spectrometric conditions were as follows: heated capillary temperature, 210° C.; spray voltage, 2500 V; desolvation gas (N2), 40 l/h; auxiliary gas (N2), 35 l/h. Selected ion current (SIC) chromatograms of C55-PP-GlcNAc and internal standard 15C C55-PP-GlcNAc were plotted and integrated using LCQuan incorporated in Xcalibur software (ThermoFinnigan). The linearity of C55-PP-GlcNAc concentration versus mass spectrometric signal (AC55-PP-GlcNAc/A15C-C55-PP-GlcNAc) was determined with purified C55-PP-GlcNAc. The IC50 values were calculated using the nonlinear regression analysis (sigmoidal dose response fit allowing for a variable slope) of percent inhibition data with minimum and maximum values set to 0 and 100 percent. TarO Biochemical Enzymatic Assay The TarO biochemical enzymatic assay is a liquid chromatography-mass spectroscopy (LC-MS) based end point assay that measures C55-P-P-GlcNAc (LIPID III) production. The TarO biochemical enzymatic assay was performed in a 384-well microtiter plate (Labcyte) with a reaction volume of 20 μl. The reaction mix contained 0.1 μgs/μl of TarO membrane preparation derived from MRSA COL (lysostaphin/lysozyme treated, centrifuged at 40K rpm, and re-suspended in 50 mM Tris pH 7.5, 10 mM MgCl2), 1500 μM UDP-GlcNAc, ×75 μM C55-P substrates in 83 mM Tris pH 8.0, 6.7 mM MgCl2, 6 mM CHAPS, and 8.3% DMSO buffer. The enzyme reactions were quenched by extraction in 40 μl of 1-pentanol containing 0.04 μM 15C C55-PP-GlcNAc, which was used as an internal standard. A 10 μl volume of the quenched reaction mixture (pH≈3) from each well was injected onto a reversed-phase column (C4, 5 μm, 2.1×50 mm, Thermo Scientific Biobacis-4) and eluted using a NH4Ac/H2O/MeOH gradient (solvent A: 10 mM NH4Ac in water, pH 5.6; solvent B: NH4Ac (1 M)-Isopropanol (1:90, v/v, pH 5.6). The HPLC conditions were as follows: 15% solvent B for 15 seconds followed by a gradient to 90% solvent B in 90 seconds; then solvent B was kept at 95% for 10 seconds followed by a gradient to 8% solvent B in 0.1 minute. The column was then equilibrated at 15% B for 1 minute before the next injection. The flow rate was kept constant at 600 μl/minute. Mass spectrometric detection was carried out in the negative-ion mode using selected reaction monitoring (SRM). Typical mass spectrometric conditions were as follows: heated capillary temperature, 210° C.; spray voltage, 2500 V; desolvation gas (N2), 40 l/h; auxiliary gas (N2), 35 l/h. Selected ion current (SIC) chromatograms of C55-PP-GlcNAc and internal standard 15C C55-PP-GlcNAc were plotted and integrated using LCQuan incorporated in Xcalibur software (ThermoFinnigan). The linearity of C55-PP-GlcNAc concentration versus mass spectrometric signal (AC55-PP-GlcNAc/A15C-C55-PP-GlcNAc) was determined with purified C55-PP-GlcNAc. The IC50 values were calculated using the nonlinear regression analysis (sigmoidal dose response fit allowing for a variable slope) of percent inhibition data with minimum and maximum values set to 0 and 100 percent. Biochemical Assay Affinity evaluation of the tested compounds and their selectivity with respect to the different PARP isoforms of interest was assessed in a displacement assay. The identification of compounds capable of binding several PARP proteins is carried out through a screening method including the steps of a) providing a reaction mixture containing: the PARP protein isoform under investigation, a compound of formula (IP): wherein R11 is hydrogen atom or a methyl group, B is (CH2)n NH group wherein n is 2 to 6; m is 0 or 1 and X- is a counterion, andserial dilutions of the test compound; b) comparing the polarization signal generated in the absence of the test compound with the one generated in the presence of different concentrations of the test compound, andc) evaluating the ability of the test compound to displace the compound of formula (IP) as defined above indicated from a decreased fluorescence polarization level. Biochemical Assay Affinity evaluation of the tested compounds and their selectivity with respect to the different PARP isoforms of interest was assessed in a displacement assay.The identification of compounds capable of binding several PARP proteins is carried out through a screening method including the steps ofa) providing a reaction mixture containing:the PARP protein isoform under investigation,a compound of formula (IP): wherein R10 is hydrogen or methyl, B is (CH2)nNH group, n is 2 to 6; m is 0 or 1 and X− is a counterion, and serial dilutions of the test compound;b) comparing the polarization signal generated in the absence of the test compound with the one generated in the presence of different concentrations of the test compound, andc) evaluating the ability of the test compound to displace the compound of formula (IP) as defined above indicated from a decreased fluorescence polarization level. The polarization signal can be measured, e.g., by a plate reader such as the Saphire2 (Tecan). Biological Assay All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≤2%. Biological Assay All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≦2%. Biological Assay SSAO: All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≤2%. Biological Assay SSAO: All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration≤2%. Biological Assays All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≤2%. Dundee MALDI-TOF Mass Spectrometry Assay (ICs) USP30 (25 ng/μl) tested against K48-linked diubiquitin (5.6 μM). USP30 was diluted in a buffer containing 40 mM Tris, 0.01% BSA, 1 mM DTT and K48 in 40 mM Tris, 0.01% BSA. The compounds were pre-incubated with the USP30 for 5 mins at room temp before the K48 dimer addition. The assay mixture was then incubated for 45 mins at room temp. The assay was stopped by the addition of TFA to a final concentration of 2% (v/v). Acidified samples of the DUB assays were mixed with 0.5 mM 15N-ubiquitin and then with one part of 2% (v/v) TFA and one part of 2.5 DHAP matrix solution (7.6 mg of 2.5 DHAP in 375 ml ethanol and 125 ml of an aqueous 12 mg ml 1 diammonium hydrogen citrate). Then 250 nl of these solutions were spotted onto an MTP AnchorChip 1,536 TF and this is analysed on the Bruker rapifleX MALDI-TOF. Inhibition Assay The DAAO inhibitory activity was measured by assaying the amount of hydrogen peroxide (H2O2) produced by reacting DAAO protein with flavin adenine dinucleotide (FAD) and D-alanine. The amount of H2O2 was determined by measuring the fluorescence generated on conversion of Amplex red (manufactured by Invitrogen Co.) into resorufin by the reaction of H2O2 with horseradish peroxidase (HRP). 4 uL of 4% dimethyl sulfoxide (DMSO) buffer (50 mM sodium phosphate (pH 7.5), 0.02% CHAPS) solution of the test compound was added to 384-well black, low volume plate, a mixed solution (4 uL) of recombinant human DAAO protein (15 nM), which had been expressed in Escherichia coli and purified, and 18 uM FAD was added, and the mixture was incubated at room temperature for 15 min. After incubation, a mixed buffer (4 uL) of 2.25 mM D-alanine, 1.5 U/mL HRP and 150 uM Amplex red was added, the mixture was incubated at room temperature for 30 min. In vitro MKP-1 Phosphatase Dose Response Active/Probe Assessment Assay - Effects of Catalase The MKP-1 dose response Active/Probe assessment-Catalase assay has been developed to evaluate the effects of adding 100 U/mL of Catalase on the MKP-1 inhibition of actives identified in the MH-76391 In vitro MKP-1 HTS assay AID #374, and subsequently confirmed in the HTS dose response confirmation assay AID #551. Protein tyrosine phosphatases have an active site cysteine that is very susceptible to inactivation by oxidation. In addition, a number of compounds such as quinone-like compounds are capable of generating reactive oxygen species via redox cycling in the presence of DTT. Adding Catalase to inactivate hydrogen peroxide (H2O2)does not affect the activity of MKP-1 in the assay but can reverse the inhibition of some inhibitors or significantly increase their IC50 values.The MKP-1 Phosphatase Dose Response Active/Probe Assessment Assay - Effects of Catalase has been Developed and Run at the University of Pittsburgh Molecular Screening Center (PMLSC) part of the Molecular Library S Choline Release Assasy The purpose of this assay is to detect autotaxin inhibition using a choline release assay.Test compound (10 mM stocks in 100% DMSO) is serially diluted in 100% DMSO resulting in 10 concentrations of 100× inhibitor in half area 96 well plates (Corning 3992). Each of these 10 wells in 100% DMSO is diluted 1:33.33 in assay buffer in round bottom 96 well plates (Fisher 12565502) resulting in 3× concentrations in well containing 3% DMSO. The assay buffer is 50 mM Tris pH8.0, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 0.01% TRITON X-100 (Sigma T9284) and 0.01% fatty acid free bovine serum albumin (Sigma A8806). A 20 μl aliquot of each 3× test compound is then added to black flat bottom 96 well plates (Corning 3991) in singlicate. A 20 μl aliquot per well of 3× recombinant human autotaxin, (Echelon, E-4000) (full length human autotaxin with a C-terminal His tag transfected into 293E cells and purified via nickel chelate and size exclusion chromatography) is then added to every well except for the no enzyme control wells. A 20 μl aliquot per well of assay buffer is added to the no enzyme control wells. A 20 μl aliquot of a 3× cocktail containing choline oxidase (Sigma C5896), horseradish peroxidase (Sigma P8125), amplex ultrared (Invitrogen A36006) and the autotaxin substrate lysophosphatidylcholine (LPC) 16:0 (Avanti Polar Lipids 855675P) is added to each well while avoiding exposure to light. The final concentrations in the well of choline oxidase, horseradish peroxidase, amplex ultrared and LPC 16:0 are 0.4 units/ml, 4 units/ml, 40 μM and 30 μM respectively. The plate is then sealed with aluminum foil seals and incubated at 37° C. for 1 hour in a Labline Imperial III incubator. During this incubation, LPC is cleaved by autotaxin resulting in Lysophosphatidic Acid (LPA) 16:0 and choline. The choline that is released is oxidized by choline oxidase resulting in betaine and hydrogen peroxide. The hydrogen peroxide reacts with the horseradish peroxide and amplex ultrared to form the fluorescent molecule resorufin. In Vitro DAAO Enzyme Assay The functional activity of compounds inhibiting the DAAO enzyme was determined by utilizing the co-product of the catalysis of D-Serine, H2O2 which can be quantitatively measured using the Amplex (trade mark) Red (Invitrogen) detection. Amplex Red reagent is a colorless substrate that reacts with hydrogen peroxide (H2O2) with a 1:1 stoichiometry in the presence of hydrogen peroxide to produce highly fluorescent resorufin (excitation/emission maxima=570/585 nm). The changes in fluorescence were monitored by a fluorescence plate reader, Envision (Perkin Elmer) and increases in DAAO activity were readily detected upon addition of D-Serine and suppression of this response observed with the application of test compounds.Human DAAO enzyme was supplied by the Takeda Pharmaceutical Company (Osaka) and each batch was tested and used at concentrations giving comparable levels of activity. The Km of D-Serine was measured for each enzyme batch to maintain consistency; this Km was used in subsequent assays.On the day of the assay compounds were serially diluted in DMSO before being diluted 1:20 with assay buffer (20 mM Tris ph 7.4). A 5 μl portion of assay buffer was added to the wells of a 384 clear base black-walled plate (Corning), 5 μl of diluted compound was then added via automated plate to plate transfer using the Bravo liquid handler (Agilent technologies) followed by 5 μl of human DAAO enzyme and then 5 μl D-Serine 50 mM was added to all but the negative control wells (final concentration of 10 mM). Finally 5 μl Amplex red reagent (Invitrogen) was added to all wells as per manufacturer's protocol. The plate was incubated for 60 minutes in the dark at 25° C. and the fluorescence in each well was measured in the Envision plate reader.The IC50 values for compounds were determined from ten point half log scale dose-response studies and represent the concentration of compound required to prevent 50% inhibition of DAAO activity in the presence of 10 mM D-Serine. Concentration response curves were generated using the average of duplicate wells for each data point and analyzed using non-linear regression and four parameter curve fit. In Vitro DAAO Enzyme Assay The functional activity of compounds inhibiting the DAAO enzyme was determined by utilizing the co-product of the catalysis of D-Serine, H2O2 which can be quantitatively measured using the Amplex Red (Invitrogen) detection. Amplex Red reagent is a colorless substrate that reacts with hydrogen peroxide (H2O2) with a 1:1 stoichiometry in the presence of hydrogen peroxide to produce highly fluorescent resorufin (excitationemission maxima=570/585 nm). The changes in fluorescence were monitored by a fluorescence plate reader, Envision (Perkin Elmer) and increases in DAAO activity were readily detected upon addition of D-Serine and suppression of this response observed with the application of test compounds.Human DAAO Enzyme was supplied by the Takeda Pharmaceutical Company (Osaka) and each batch was tested and used at concentrations giving comparable levels of activity. The Km of D-Serine was measured for each enzyme batch to maintain consistency; this Km was used in subsequent assays.On the day of the assay compounds were serially diluted in DMSO before being diluted 1:20 with assay buffer (20 mM Tris ph 7.4). A 5 μl portion of assay buffer was added to the wells of a 384 clear base black walled plate (Corning), 5 μl of diluted compound was then added via automated plate to plate transfer using the Bravo liquid handler (Agilent technologies) followed by 5 μl of human DAAO enzyme and then 5 μl D-Serine 50 mM was added to all but the negative control wells (final concentration of 10 mM). Finally 5 μl Amplex red reagent (Invitrogen) was added to all wells as per manufacturer's protocol. The plate was incubated for 60 minutes in the dark at 25° C. and the fluorescence in each well was measured in the Envision plate reader.The IC50 values for compounds were determined from ten point half log scale dose-response studies and represent the concentration of compound required to prevent 50% inhibition of DAAO activity in the presence of 10 mM D-Serine. Concentration response curves were generated using the average of duplicate wells for each data point and analyzed using nonlinear regression and four parameter curve fit. Inhibitory Activity Assay 1. Materials, Kits and EquipmentsSodium L-ascorbate (Cat: A4034-100G, SIGMA)4-(dimethylamino)benzaldehyde (Cat: 156477-25g, SIGMA)Trichloroacetic acid (Cat: T0699-100ML, SIGMA)L-Tryptophan (Cat: T8941-25G, SIGMA)Methylene blue (Cat: M9140-25G, SIGMA)Potassium dihydrogen phosphate (Cat: 10017618, Sinopharm Chemical Reagent)Disodium hydrogen phosphate (Cat: 20040618, Sinopharm Chemical Reagent)Constant temperature water tank (Cat: DK-8D, Shanghai Jinghong Experimental Equipment)Multifunctional microplate reader (Cat: M5, Molecular Devices)96-well reaction plate (Cat: 3590, costar)IDO1 protease (commercially available)Desktop Microplate Reader: SpectraMax M5 Microplate Reader (Molecular Devices)Test compounds: self-madePositive control agent: INCB024360 (commercially available)2. Reagent Preparation100 mM PBS:100 mM disodium hydrogen phosphate and 100 mM potassium dihydrogen phosphate mixed in a ratio of 3:5, pH 6.5IDO1 assay buffer:100 mM PBS containing 400 μM L-tryptophan, 20 mM ascorbate, 20 μM methylene blue and 1000 U/ml catalase, pH 6.530% trichloroacetic acidddH2 O solution of 30% trichloroacetic acidEhrlich reagent1% (w/v) diluted solution of 4-(dimethylamino) benzaldehyde compoundAll compounds were dissolved with DMSO. During the assay, each compound was diluted to a concentration as needed. The compound of each concentration was added to multi-wells, and the final concentration of DMSO was controlled at 1%.3. Test Methoda.) the reaction mixture was prepared by adding 50 nM IDO1 and the desired concentration of the test compound to 100 μL of IDO1 assay buffer. IDO1 and assay buffer need to be preheated to 37° C.b.) The mixture was reacted in a constant temperature water tank at 37° C. for 30 minutes.c.) 50 μL of 30% trichloroacetic acid was added.d.) The above mixture was reacted in a constant temperature water tank at 52° C. for 30 minutes.e.) The reaction mixture was centrifuged at 12000 g for 10 minutes at room temperature.f.) 100 μL of the obtained supernatant and 100 μL of Ehrlich reagent were mixed.g.) the absorbance at 480 nm was measured using an M5 microplate reader.4. Data AnalysisInhibition rate=(ODpostive−ODsample)/(ODpositive−ODnegative)*100%5. Results and Discussion In Vitro DGAT2 Assay For determination of IC50 values, the reactions were carried out in 384-well white Polyplates (Perkin Elmer) in a total volume of 20 μL. To 14 of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 20 minutes. To this mixture, 10 μL of hDGAT2 membrane fraction (0.01 mg/mL) diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock) was added. After this mixture was preincubated at room temperature for 2 hours, DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). cAMP Assays To optimize functional activity directed toward Gas coupling, a HEK293/CRE-Luc cell line developed by HDB stably expressing the GLP-1 Receptor was used. 200× concentration of compound working solutions were prepared (Agilent Technologies Bravo) with 1/2 log serial dilution in 384-well Echo LDV plate (Labcyte, Cat #LP-0200). 50 nL/well 200× concentration of compound working solutions were moved to 384-well white low volume plate (Greiner, Cat #784075) using Labcyte ECHO550. 1×105 cells/mL HEK293/GLP1R/CRE-LUC(HD Biosciences) cell suspensions prepared with assay buffer[DPBS containing 0.5 mM IBMX(Sigma, Cat #15879) and 0.1% BSA(GENVIEW, Cat #FA016-100g)], 10 uL cell suspensions were added to each well of previous generated assay plate which already contains 50n1 compound at 200× concentration using ThermoFisher Multidrop Combi(1000 cells/well). Seal the plate and incubate at 37° C. with 5% CO2 for 30 min.After incubation the cAMP assay signal was generated using cAMP dynamic 2 Kit (Cisbio). 5 μL cAMP-d2 working solution was added to each well, followed with 5 μL Anti-cAMP antibody-cryptate working solution added to each well using ThermoFisher Multidrop Combi. Incubate at room temperature for 1 hour protected from light. Read the fluorescence at 665 and 615 nm with Reader PerkinElmer EnVision.% Activity=100%×(mean RLU of test sample−mean RLU of vehicle control)/(mean RLU of MAX control−mean RLU of vehicle control)). Biological Activity Assay Assaying the inhibition of KDM1A can be determined in vitro, in cultured cells, and in animals. There are a variety of spectrophotometric methods to detect the results of demethylation of methylated lysines, viz., detecting the products of KDM1A demethylase oxidative activity on a peptide fragment of at least 18 amino acid representing the N-terminus of the histone H3 substrate that contains a monomethyl at the fourth lysine residue. Hydrogen peroxide, one product of the KDM1A demethylase reaction, reacts with horseradish peroxidase and dihydroxyphenoxazine (ADHP) to produce the fluorescent compound resorufin (excitation=530-560 nm:emission=590 nm). The KDM1A demethylase enzyme activity can obtained from mammalian cells or tissues expressing KDM1A from an endogenous or recombinant gene and purified or assayed from a whole cell extract. These methods can be used to determine the concentration of the disclosed compounds can inhibit fifty percent of the enzyme activity (IC50). In one aspect, the disclosed compounds exhibit inhibition fifty percent of the KDM1A enzyme activity at a concentration of less than 500 nM, less than 100 nM, less than 50 nM or less than 10 nM. Biological Assay Most known kinase inhibitors bind in the ATP-binding pocket of the active site19,20. These inhibitors exploit many of the same hydrophobic contacts as the purine ring of ATP and make at least one conserved hydrogen bond to the hinge region. Potent inhibitors also occupy at least one hydrophobic pocket adjacent to the ATP-binding site. These additional hydrophobic interactions increase both binding affinity and target selectivity of the inhibitor because there is substantial heterogeneity among different kinases in these regions. Examination of the TgCDPK1 sequence in the vicinity of the ATP-binding pocket (FIG. 2b ) shows that it contains a glycine residue at a position that has been termed the gatekeeper residue because it constrains access to the ATP-binding site21-23. The glycine at this position in TgCDPK1 (Gly128) is expected to create a much larger pocket off the ATP-binding site than is typically seen in protein kinases and comparison of the TgCDPK1 structure with other kinases shows that this is indeed the case. This difference in the active site architectures can be exploited for design of selective inhibitors against TgCDPK1. In vitro MKP-3 Phosphatase Dose Response Active/Probe Assessment Assay - Effects of Catalase The MKP-3 dose response Active/Probe assessment-Catalase assay has been developed to evaluate the effects of adding 100 U/mL of Catalase on the MKP-3 inhibition of actives identified in the MKP-3 HTS run at (Burham Institute) SDCCG center AID 425 and the MKP-1 HTS run at the PMLSC AID 374, and subsequently confirmed in the MKP-3 & MKP-1 HTS dose response confirmation assays AID's 553 & 551. Protein tyrosine phosphatases have an active site cysteine that is very susceptible to inactivation by oxidation. In addition, a number of compounds such as quinone-like compounds are capable of generating reactive oxygen species via redox cycling in the presence of DTT. Adding Catalase to inactivate hydrogen peroxide (H2O2)does not affect the activity of MKP-3 in the assay but can reverse the inhibition of some inhibitors or significantly increase their IC50 values. The MKP-3 Phosphatase Dose Response Active/Probe Assessment Assay - Effects of Catalase has been Developed and Run at the Univers HepaRG-CAR Cell-Based Assay for Quantitation of Glycolate Oxidase Inhibition A HepaRG human hepatic cell line was transfected for stable overexpression of the constitutive androstane receptor (i.e., HepaRG-CAR cells), as reported by van der Mark et al. (Drug Metab. Dispos., 2017, 45:56-67. Overexpression of CAR in these cells resulted in higher levels of glycolate oxidase (GOX) expression compared to the parental HepaRG cells. HepaRG-CAR cells were plated in a 12-wells plate and incubated for 4 weeks until fully differentiated.To measure cellular glycolate flux, the HepaRG-CAR cells were incubated in Williams medium supplemented with 10% fetal bovine serum (FBS), 5 μg/mL insulin, 50 μM hydrocortisone hemisuccinate, 2 mM glutamine, 5000 U/mL penicillin and 5 mg/mL streptomycin. Test compounds were added to the medium at 0, 0.3, 1, 3 or 10 μM and incubated for 30 minutes, after which 500 μM glycolate was also added. After incubation for 48 hours, 400 μL medium was taken from the culture plate and added to 60 μL 37% HCl.Internal standards (2,2-d2 glycolate, 1,2-13C2 oxalate and 13C2-glyoxylate) and hydroxylamine were added followed by another 30 minute incubation at 80° C. The acids were extracted using ethyl acetate with NaCl. The organic phase was dried under nitrogen and derivatized with N-tert-butyldimethylsilyl-N-methyl trifluoroacetamide (MTBSTFA) for 30 minutes at 80° C. The amounts of glycolate, glyoxylate and oxalate were determined by gas chromatography-mass spectrometry (GC-MS) analysis, using a 25 meter CP-Sil 5 CB low bleed column. A standard curve was used to calculate the concentrations of each acid in the culture medium. LOX Inhibition Assay Lysyl oxidase (LOX) is an extracellular copper dependent enzyme which oxidizes peptidyl lysine and hydroxylysine residues in collagen and lysine residues in elastin to produce peptidyl alpha-aminoadipic-delta-semialdehydes. This catalytic reaction can be irreversibly inhibited by β-aminopropionitrile (BAPN) that binds to the active site of LOX (Tang S. S., Trackman P C and Kagan H. M., Reaction of aortic lysyl oxidase with beta-aminoproprionitrile. J Biol Chem 1983; 258: 4331-4338). There are five LOX family members; these are LOX, LOXL1, LOXL2, LOXL3 and LOXL4. LOX and LOXL family members can be acquired as recombinant active proteins from commercial sources, or extracted from animal tissues like bovine aorta, tendons, pig skin; or prepared from cell cultures. The inhibitory effects of the compounds of the present invention were tested against the given LOX-LOXL preparation using a method based on the detection of hydrogen peroxide with an Amplex Red oxidation assay (Zhou et al. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal. Biochem. 1997; 253, 162-168). The assay was developed using either 384 or 96 well format. Briefly, in a standard black, clear bottom 384 well plate assay 25 μL of a dilution of any of the isoenzymes and orthologues in 1.2 M urea, 50 mM sodium borate buffer (pH 8.2) were added into each well in the presence of 1 μM mofegiline and 0.5 mM pargyline (to inhibit SSAO and MAO-B and MAO-A, respectively; not necessary if the enzyme is from a recombinant or purified form). Test compounds were dissolved in DMSO and tested in a Concentration Response Curve (CRC) with 11 data points, typically in the micromolar or nanomolar range after incubation with the enzyme for 30 min at 37° C. Twenty five μL of a reaction mixture containing twice the KM concentration of putrescine (Sigma Aldrich, e.g. 20 mM for LOX, or 10 mM for LOXL2 and LOXL3), 120 μM Amplex Red (Sigma Aldrich) and 1.5 U/mL horseradish peroxidase (Sigma Aldrich) prepared in 1.2 M urea, 50 mM sodium borate buffer (pH 8.2) were then added to the corresponding wells. The above volumes were doubled in the case of 96 wells plate. The fluorescence (RFU) was read every 2.5 min for 30 min at a range of temperatures from 37° C., excitation 565 nm and emission 590 (Optima; BMG labtech). The slope of the kinetics for each well was calculated using MARS data analysis software (BMG labtech) and this value was used to deduce the IC50 value (Dotmatics). In Vitro Assay For determination of IC50 values, the reactions were carried out in 384-well white Polyplates (Perkin Elmer) in a total volume of 20 μL. To 1 μL of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 15 minutes. hDGAT2 membrane fractions were diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock). 10 μL of this enzyme working solution was added to the plates and incubation continued for 2 hours at room temperature. DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). Plates were centrifuged at 2,000×g for 1 minute in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). In Vitro Assay For determination of IC50 values, the reactions were carried out in 384-well white polypropylene plates (Nunc) in a total volume of 20 μL. To 1 μL of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 15 minutes. hDGAT2 membrane fractions were diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical, dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock). 10 μL of this enzyme working solution was added to the plates and incubation continued for 2 hours at room temperature. DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). Plates were centrifuged at 2,000×g for 1 minute in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). DGAT2 activity was measured by quantifying the generated product [14C]tridecanoylglycerol in the upper organic phase. In Vitro DGAT2 Assay For determination of IC50 values, the reactions were carried out in 384-well white Polyplates (Perkin Elmer) in a total volume of 20 uL. To 1 uL of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 uL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 20 minutes. To this mixture, 10 uL of hDGAT2 membrane fraction (0.01 mg/mL) diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock) was added. After this mixture was preincubated at room temperature for 2 hours, DGAT2 reactions were initiated by the addition of 4 uL of substrates containing 30 uM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 uM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 uL of 1% H3PO4. After the addition of 45 uL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). Plates were centrifuged at 2,000×g for 1 min in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). DGAT2 activity was measured by quantifying the generated product [14C]tridecanoylglycerol in the upper organic phase. In Vitro DGAT2 Assay For determination of IC50 values, the reactions were carried out in 384-well white Polyplates (Perkin Elmer) in a total volume of 20 μL. To 1 μL of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 15 minutes. hDGAT2 membrane fractions were diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock). 10 μL of this enzyme working solution was added to the plates and incubation continued for 2 hours at room temperature. DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, CA). Plates were centrifuged at 2,000×g for 1 minute in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). DGAT2 activity was measured by quantifying the generated product [14C]tridecanoylglycerol in the upper organic phase. In Vitro DGAT2 Assay For determination of IC50 values, the reactions were carried out in 384-well white polypropylene plates (Nunc) in a total volume of 20 μL. To 1 μL of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 15 minutes. hDGAT2 membrane fractions were diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical, dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock). 10 μL of this enzyme working solution was added to the plates and incubation continued for 2 hours at room temperature. DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, CA). Plates were centrifuged at 2,000×g for 1 minute in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). DGAT2 activity was measured by quantifying the generated product [14C]tridecanoylglycerol in the upper organic phase. UPLC/MS Assay NAAA protein preparation (10 ug) was pre-incubated with various concentrations of test compound or vehicle control in 100 mM NaH2PO4, 100 mM Tri Sodium Citrate Dehydrate, 0.1% Triton-X 100, 3 mM DTT, pH 4.5 for 30 min at 37° C. Duplicate samples were then incubated with 50 uM C17:1 10-cis-heptadecenoylethanolamide (Avanti Polar Lipids, Alabaster, Ala. USA) at 37° C. for 30 minutes. The reaction was terminated by the addition of 0.2 mL of cold methanol containing 1 nmol of heptadecanoic acid (NuChek Prep, Elysian, Minn. USA) as internal standard. Samples were then analyzed by UPLC/MS. Heptadecenoic and heptadecanoic acids were eluted on an Acquity UPLC BEH C18 column (50 mm length, 2.1 mm i.d., 1.7 um pore size, Waters) isocratically at 0.5 mL/min for 1.5 min with a solvent mixture of 95% methanol and 5% water, both containing 0.25% Acetic Acid and 5 mM Ammonium Acetate. The column temperature was 40° C. Electrospray ionization was in the negative mode, capillary voltage was 0.5 kV, cone voltage was 25 kV, desolvation temperature was 500° C. N2 was used as drying gas at a flow rate of 1000 L/hour and a temperature of 500° C. The [M-H]- ion was monitored in the selected-ion monitoring mode (m/z values: heptadecenoic acid 267.37, heptadecanoic acid 269.37). Calibration curves were generated using commercial heptadecenoic acid (NuCheck Prep). Inhibition of NAAA activity was calculated as reduction of heptadecenoic acid in the samples compared to vehicle controls. IC50 values were calculated by non-linear regression analysis of log [concentration]/inhibition curves using GraphPad Prism 5 (GraphPad Software Inc., CA-USA) applying a standard slope curve fitting. Biochemical Assay In brief, this Rpn11 bioassay employs a fluorescent polarization readout based on the ability of the 26S proteasome to cleave the protein substrate including four tandem ubiquitin proteins fused to a peptide having a unique cysteine labeled with a fluorophore. Cleavage of this substrate by Rpn11 at the junction between the fourth ubiquitin and the peptide, releases the low molecular weight fluorescent peptide. Accordingly, inhibition of fluorescence correlates with inhibition of Rpn11. Inhibition is reported as the half maximal inhibitory concentration (IC50) for the candidate compound.The catalytic JAB1/MPN/Mov34 metalloenzyme (JAMM) motif of Rpn11 is found in 7 different human proteins including the Csn5 subunit of the COP9 signalosome, AMSH, AMSH-LP, the BRCC36 subunit of BRISC, MPND, and MYSM1. All of these enzymes cleave the isopeptide linkage that joins ubiquitin (or the ubiquitin-like protein Nedd8 in the case of Csn5) to a second molecule of ubiquitin or to a substrate. The conserved JAMM domain has the consensus sequence EXnHS/THX7SXXD, in which the histidine (His) and aspartic acid (Asp) residues bind the Zn2+ ion and the fourth coordination site is occupied by a water molecule that is engaged in hydrogen bonding with a conserved glutamic acid (Glu). The Zn2+ acts as a Lewis acid and increases the nucleophilic character of the bound water enough to allow hydrolytic cleavage of the isopeptide bond. In Vitro Measurements of porcine D-Amino Acid Oxidase (DAAO) Activities The pkDAAO (porcine kidney DAAO) activity was measured by using D-Proine as a substrate to produce hydrogen peroxide (H2O2). The produced H2O2 would be oxidized by peroxidase, and the produced free radicals would further react with 1,2-Phenylenediamine (OPD) reagent. The reaction product had an absorbance on 450 nm. The OD450 would be measured to represent the activity of pkDAAO. All compounds were dissolved in DMSO. Each compound was diluted with DMSO in 3 or 4-fold serial dilution to create a 9-point dose response curve. Each sample was added in triplicate, 10 μL/well, into 96-well assay microplate. Positive control wells were added with 10 μL of DMSO. The diluted compounds were incubated with pkDAAO in dark for 10 minutes and then reacted with D-Proline. The final reaction mixture was composed of 0.01 U/mL pkDAAO, 0.03% OPD, 25 U/mL HRP and 40 mM D-Proline in PBS. The reaction plates were then incubated in the dark at room temperature. The OD450 absorbance readout was detected at 0 and 20 minute by Molecular Device Spectra Max Plus reader. The percentage of inhibition values for each well were calculated with the following equation:The percentage of inhibition=(OD450sample, 20 min−OD450sample, 0 min)/(OD450DMSO, 20 min−OD450DMSO, 0 min)×100%The nonlinear curve fitting model in GraphPad Prism 5 was used to calculate IC50 value for each compound. Kinase Assay Kinase assays were performed in 96 well microtiter plates that were coated overnight with 30 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.2-7.4. The plates were incubated with 1% BSA and then washed four times with PBS prior to starting the reaction. Reactions were carried out in 120 μL reaction volumes containing 3.6 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 0.5 ng of purified protein. Following a ten minute incubation at 25 °C., the reactions were washed four times with PBS containing 0.05% Tween-20. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate was diluted 1:10000 in PBS-Tween-20 and added to the wells for 30 minutes. Following four washes with PBS-Tween-20, 100 μl of 0-phenylenediamine Dihydrochloride in Phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2 SO4 to each well and read using a microplate ELISA reader set at 492 nm. PDGFRbeta Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 36 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. PDGFRbeta Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 36 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. LC-MS Based In Vitro Assay hAC protein preparation (10 μg) was preincubated with inhibitors (final DMSO concentration 1%) in assay buffer (100 mM sodium phosphate, 0.1% Nonidet P-40, 150 mM NaCl, 3 mM DTT, 100 mM sodium citrate, pH 4.5) for 30 min at 37° C. Reactions were started by the addition of 50 μM N-lauroyl ceramide (Nu-Chek Prep, Elysian, Minn.) and carried on for 30 min at 37° C. Reactions were stopped by addition of a mixture of chloroform/methanol (2:1) containing 1 nmol 11-lauroleic acid (NuChek Prep). The organic phases were collected, dried under nitrogen and analyzed by UPLC/MS (Acquity, Waters). In the negative-ion mode monitoring the reaction product (lauric acid, m/z=199) using 11-lauroleic acid as internal standard.Lipids were eluted on an Acquity UPLC BEH C18 column (50×2.1 mmID, particle size 1.7 μm), column flow at 0.5 mL/min for 1.5 min with a gradient of acetonitrile and water, both containing 0.25% acetic acid and 5 mM ammonium acetate (70% to 100% acetonitrile in 0.5 min, 100% acetonitrile for 0.5 min, 70% acetonitrile for 0.4 min). The column temperature was 40° C. Electrospray ionization (ESI) was in the negative mode, capillary voltage was 1 kV and cone voltage was 50 V. N2 was used as drying gas at a flow rate of 500 L/h and at a temperature of 400° C.The [M-H]− ion was monitored in the selected-ion monitoring mode (m/z values: lauric acid 199, 11-lauroleic acid 197.35). Calibration curves were generated with authentic lauric acid (Nu Check Prep). Inhibition of AC activity was calculated as reduction of lauric acid in the samples compared to vehicle controls. IC50 values were calculated by non-linear regression analysis of log [concentration]/inhibition curves using GraphPad Prism 5 (GraphPad Software Inc., CA-USA) applying a standard slope curve fitting. Competitive Fluorescence Polarization (FP) Assay Binding affinities (Kd) with the JAK2 JH2 domain were evaluated using the previously developed FP assay and compound 1 as a control. An initial screening is conducted at 50 μM, and binding affinities (Kd's) are measured for those exhibiting >50% binding at 50 μM (Table 1). Replacement of the oxazole in 2 with 6-membered aromatic rings produced compounds with a binding range of 0.129 to 1.4 μM. Conversion of the carboxylate of compound 7 to a methyl ester (8) or just hydrogen (9) resulted in large loss of binding affinity, and the addition of a benzyloxy group gave compounds (11-14) with an affinity range of 0.033 to 0.075 μM. Selectivity measurements were conducted subsequently for the most potent JAK2 JH2 ligands. In a flat black bottom 96 well plate (Corning), 200 μL of FP buffer were added to column 1 (blank), 150 μL to column 2, and 140 μL to columns 3-12. 10 μL of 2.96 μM of JAK2-JH2 WT (3.52 μM for JAK2-JH2-VF, and 6.93 μM for JAK2-JH1), were added to columns 3-12, followed by the addition of 2 μL of DMSO to columns 1-3. 2 μL of inhibitor in DMSO at different concentrations were added from column 4 to 12. 50 μL of 24 nM of tracer were added to columns 2-12. Fluorescence polarization was measured at λexc=485±20 nm, λem=535±25 nm for 1 hour. Experiments were carried out by quadruplicates in three independent experiments. Inhibitory Activity of Human Recombinant SSAO/VAP-1 Test purpose: The following method is used to determine the inhibitory activity of the compounds in the examples of the present invention on human recombinant SSAO/VAP-1.Test Materials:Human recombinant SSAO/VAP-1 (VAP-1, human) was purchased from Sigma, Cat. No. SRP6241;Amplex® Red Monoamine Oxidase Assay Kit was purchased from Invitrogen, Cat. No. A12214;384-well plate was purchased from Corning, Cat. No. 6005174;Amplex® Red Hydrogen PeroxidePeroxidase Assay Kit was purchased from Invitrogen, Cat. No. A22188.Benzylamine hydrochloride was purchased from Sigma, Cat. No. B5136-25G;DMSO (Dimethyl Sulfoxide) was purchased from Sigma, Cat. No. D2650-100ML;Test Method:The test compound was dissolved in DMSO and diluted 4 times to a total of 10 concentrations. In a 384-well plate, 25 μL of human recombinant SSAO/VAP-1 (1.6 μg/mL) was added into each well. 100 nL of test compounds at different concentrations were added to each well containing human recombinant SSAO/VAP-1, and the plate was incubated at room temperature for 30 min. After incubating for 30 min, 25 μL of Amplex® Red Monoamine Oxidase Assay Kit (a reaction mixture containing 200 μM Amplex Red reagent, 1 U/mL HRP and 1 mM benzylamine hydrochloride) was added into the corresponding wells, and the plate was incubated at room temperature in the dark for 60 min. After 60 min, PerkinElmer's Envision was used to read the fluorescence value (RFU) under excitation at 530-560 nm and emission at 590 nm. PDGFRbeta Kinase Assay Biochemical PDGFRbeta kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 36 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 ul of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. PDGFRbeta Kinase Assay Biochemical PDGFRbeta kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per wellPBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 36 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 ul of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. Cell-based ELISA Assay The ELISA portion of the assay was performed in a black Maxisorp 96-well plate that was coated overnight at 4° C. with 100 uL/well of the cell lysate (1:10 dilution of the lysate with PBS containing protease inhibitors, phosphatase inhibitors, and PMSF). The following day the wells were washed 3 times with 300 uL/well of Wash buffer (Tris-buffered saline with 0.1% Tween 20). The wells were blocked with 100 ul/well Blocking buffer (Tris buffered saline w/0.05% Tween 20 and 2.5% Bovine serum albumin). Each well was then washed two times with 300 uL/well of wash buffer. The anti O-GlcNAc antibody RL-2 (Abeam, Cambridge, Mass.), diluted 1:1000 in blocking buffer, was added at 100 uL/well. The plate was sealed and incubated at 37° C. for 2 h with gentle shaking. The wells were then washed 3-times with 300 uL/well wash buffer. To detect the amount of RL-2 bound horse-radish peroxidase (HRP) conjugated goat anti-mouse secondary antibody (diluted 1:3000 in blocking buffer) was added at100 uL/well. The plate was incubated for 60 min at 37° C. with gentle shaking. Each well was then washed 3-times with 300 uL/well wash buffer. The detection reagent was added, 100 uL/well of Amplex Ultra RED reagent (prepared by adding 30 uL of 10 mM Amplex Ultra Red stock solution to 10 mL PBS with 18 uL 3% hydrogen peroxide, H2O2). The detection reaction was incubated for 15 minutes at room temperature and then read with excitation at 530 nm and emission at 590 nm. Cell-based ELISA The ELISA portion of the assay is performed in a black Maxisorp 96-well plate that is coated overnight at 4° C. with 100 uL/well of the cell lysate (1:10 dilution of the lysate with PBS containing protease inhibitors, phosphatase inhibitors, and PMSF). The following day the wells are washed 3 times with 300 uL/well of Wash buffer (Tris-buffered saline with 0.1% Tween 20). The wells are blocked with 100 uL/well Blocking buffer (Tris buffered saline w/0.05% Tween 20 and 2.5% Bovine serum albumin). Each well is then washed two times with 300 uL/well of wash buffer. The anti O-GlcNAc antibody RL-2 (Abcam, Cambridge, Mass.), diluted 1:1000 in blocking buffer, is added at 100 uL/well. The plate is sealed and incubated at 37° C. for 2 h with gentle shaking. The wells are then washed 3-times with 300 uL/well wash buffer. To detect the amount of RL-2 bound horse-radish peroxidase (HRP) conjugated goat anti-mouse secondary antibody (diluted 1:3000 in blocking buffer) is added at 100 uL/well. The plate is incubated for 60 min at 37° C. with gentle shaking. Each well is then washed 3-times with 300 uL/well wash buffer. The detection reagent is added, 100 uL/well of Amplex Ultra RED reagent (prepared by adding 30 ul of 10 mM Amplex Ultra Red stock solution to 10 mL PBS with 18 uL 3% hydrogen peroxide, H2O2). The detection reaction is incubated for 15 minutes at room temperature and then read with excitation at 530 nm and emission at 590 nm. Kinase Assay Biochemical KDR kinase assays were performed in 96 well microliter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mLs per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mLs per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mLs per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mLs per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microliter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. Kinase Assay PDGFRβ: Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. Kinase Assay VEGFR2: Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. PDGFRβ Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. PDGFRβ Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microliter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-β protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. PDGFRβ Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFRβ protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mis per well wash with PBS-Tween-20, 100 μl of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. PDGFRβ Kinase Assay Biochemical PDGFR3 kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30 °C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microliter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 M ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30 °C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mis per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30 μC., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mis per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 2.7 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 ul of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. Biochemical Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values. Biochemical Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 μl per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values. Biological Assay 1. Determination of Inhibitory Activity of Human Recombinant SSAO/VAP-1Test purpose: The following methods can be used to determine the inhibitory activity of the compounds described in the examples of the invention for human recombinant SSAO/VAP-1.Test Materials:Human recombinant SSAO/VAP-1 (VAP-1, human) purchased from Sigma, Cat. No. SRP6241;Amplex Red Monoamine Oxidase Assay Kit purchased from Invitrogen, Cat. No. A12214;384-Well plate purchased from Corning, Cat. No. 6005174;Amplex Red Hydrogen PeroxidePeroxidase Assay Kit purchased from Invitrogen, Cat. No. A22188.Benzylamine hydrochloride purchased from Sigma, Cat. No. B5136-25G;DMSO (Dimethyl Sulfoxide) purchased from Sigma, Cat. No. D2650-100ML;Test Method:The test compound was dissolved in DMSO and diluted to 10 concentrations in 4-fold serial dilution. In 384 well plates, 25 μL of human recombinant SSAO/VAP-1 (1.6 μg/ml) was added to each well. 100 nL of test compounds with different concentrations were added to each well containing human recombinant SSAO/VAP-1 and incubated at room temperature for 30 min. After 30 min incubation, 25 μl of Amplex red monoamine oxidase assay kit (containing a reaction mixture of 200 μm Amplex Red reagent, 1 U/ml HRP and 1 mm benzylamine hydrochloride) was added to the corresponding well, and incubated at room temperature and dark for 60 min. After 60 min, the relative fluorescence unit (RFU) was read by using PerkinElmer's Envision at 530-560 nm excitation and 590 nm emission. The curve was drawn by using the graph pad prism 5 software, and the IC50 value was calculated. The results were shown in table 1, wherein the compound listed in table 1 is the compound in the preparation examples with the same compound number: DAAO Inhibitory Activity Assay The DAAO inhibitory activity was measured by assaying the amount of hydrogen peroxide (H2O2) produced by reacting DAAO protein with flavin adenine dinucleotide (FAD) and D-alanine. The amount of H2O2 was determined by measuring the fluorescence generated on conversion of Amplex red (manufactured by Invitrogen Co.) into resorufin by the reaction of H2O2 with horseradish peroxidase (HRP). 4 μL of 4% dimethyl sulfoxide (DMSO) buffer (50 mM sodium phosphate (pH 7.5), 0.02% CHAPS) solution of the test compound was added to 384-well black, low volume plate, a mixed solution (4 μL) of recombinant human DAAO protein (15 nM), which had been expressed in Escherichia coli and purified, and 18 μM FAD was added, and the mixture was incubated at room temperature for 15 min. After incubation, a mixed buffer (4 μL) of 2.25 mM D-alanine, 1.5 U/mL HRP and 150 μM Amplex red was added, the mixture was incubated at room temperature for 30 min, and the fluorescence (excitation wavelength 530 nm, fluorescence wavelength 590 nm) was measured using an Envision plate reader (manufactured by Perkin Elmer Co.). To cross-check the artificial inhibition of Amplex red conversion or the HRP activity inhibition of the test compound, the fluorescence was also measured under the conditions of 30 μM H2O2 addition in the absence of DAAO. Taking the fluorescence value in the absence of the test compound as 100% and the fluorescence value in the absence of DAAO as 0%, the DAAO activity was regarded to have been inhibited when the fluorescence value decreased by 50% in the presence of the test compound, and the concentration of the test compound at that time was taken as the IC50 value (nM). Enzyme Assay The functional activity of compounds inhibiting the DAAO enzyme was determined by utilizing the co-product of the catalysis of D-Serine, H2O2, which can be quantitatively measured using the Amplex (trade mark) Red (Invitrogen) detection. Amplex Red reagent is a colorless substrate that reacts with hydrogen peroxide (H2O2) with a 1:1 stoichiometry to produce highly fluorescent resorufin (excitation/emission maxima=570/585 nm). The changes in fluorescence were monitored by a fluorescence plate reader, Envision (Perkin Elmer) and increases in DAAO activity were readily detected upon addition of D-Serine and suppression of this response observed with the application of test compounds.Human DAAO enzyme was supplied by the Takeda Pharmaceutical Company (Osaka) and each batch was tested and used at concentrations giving comparable levels of activity. The Km of D-Serine was measured for each enzyme batch to maintain consistency; this Km was used in subsequent assays.On the day of the assay compounds were serially diluted in DMSO before being diluted 1:20 with assay buffer (20 mM Tris ph 7.4). A 5 μl portion of assay buffer was added to the wells of a 384 clear base black-walled plate (Corning), 5 μl of diluted compound was then added via automated plate-to-plate transfer using the Bravo liquid handler (Agilent technologies) followed by 5 μl of human DAAO enzyme, and then 5 μl D-Serine 50 mM was added to all but the negative control wells (final concentration of 10 mM). Finally 5 μl Amplex red reagent (Invitrogen) was added to all wells as per manufacturer's protocol. The plate was incubated for 60 minutes in the dark at 25° C. and the fluorescence in each well was measured in the Envision plate reader. Inhibitory Activity on Human ACC1 and the ACC2 Recombinant human ACC1 and recombinant human ACC2, which were prepared by the method mentioned above, were preincubated with assay buffer solution (50 mM HEPES-KOH (pH 7.4), 10 mM magnesium chloride, 6-10 mM potassium citrate, 4 mM reduced form of glutathione, 1.5 mg/ml bovine serum albumin) for one hour. Then, 0.2 μL of each this invention compound solution (in DMSO) were dispensed to 384-well microplate, 5 μL of the preincubated enzyme solution and 5 μL of substrate solution (50 mM HEPES-KOH (pH 7.4), 1 mM ATP, 0.8 mM acetyl CoA and 25-50 mM potassium bicarbonate) were added to microplate. After centrifugation and shaking, the reaction mixtures were incubated in a humidified box at room temperature for 1 to 3 hours. After the incubation, the enzyme reactions were stopped by the addition of EDTA. Then, after the samples were cocrystallized with CHCA (α-cyano-4-hydroxy cinnamic acid) matrices on MALDI target plate, by using the matrix assist laser deionization time-of-flight mass spectrometer (MALDI-TOF MS), samples were measured in reflector negative mode. Deprotonated ions of acetyl CoA (AcCoA) of substrate and malonyl CoA (MalCoA) of the reaction product were detected, then, the conversion rates of acetyl CoA to malonyl CoA was calculated by the intensity of [MalCoA-H]−/(Intensity of [MalCoA-H]−+Intensity of [AcCoA-H]−) using each signal strength. The 50% inhibitory concentration (IC50) was calculated from the inhibition rate of the enzymatic reaction at each concentration of the compounds. In addition, potassium citrate concentrations in assay buffer solution, potassium hydrogen carbonate concentrations in substrate solution and incubation time were adjusted by each lot of enzyme. Kinase Assay PDGFRβ: Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 300° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values. Kinase Assay The cytoplasmic domain of the human VEGF receptor (VEGFR-2) was expressed as a Histidine-tagged fusion protein following infection of insect cells using an His engineered baculovirus. His-VEGFR-2 was purified to homogeneity, as determined by SDS-PAGE, using nickel resin chromatography. Kinase assays were performed in 96 well microtiter plates that were coated overnight with 30 .mu g of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.2-7.4. The plates were incubated with 1% BSA and then washed four times with PBS prior to starting the reaction. Reactions were carried out in 120 .mu L reaction volumes containing 3.6 .mu M ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl.sub.2, 0.1 mM MnCl.sub.2 and 0.2 mM Na.sub.3 VO.sub.4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 0.5 ng of purified protein. Following a ten minute incubation at 25.degree. C., the reactions were washed four times with PBS containing 0.05% Tween-20. 100 .mu l of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate was diluted 1:10000 in PBS-Tween-20 and added to the wells for 30 minutes. Following four washes with PBS-Tween-20, 100 .mu l of O-phenylenediamine Dihydrochloride in Phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 .mu l of 2.5N H.sub.2 SO.sub.4 to each well and read using a microplate ELISA reader set at 492 nm. Kinase Assay VEGFR2: Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values. PDGFRβ Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values. PDGFRbeta Kinase Assay Biochemical PDGFRβ kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS +0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 36 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain PDGFR-b protein (Millipore). Following a 60 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values. The Measurement of Inhibitory Activity on Human ACC1 and the ACC2 Recombinant human ACC1 and recombinant human ACC2, which were prepared by the method mentioned above, were preincubated with assay buffer solution (50 mM HEPES-KOH (pH 7.4), 10 mM magnesium chloride, 6-10 mM potassium citrate, 4 mM reduced form of glutathione, 1.5 mg/ml bovine serum albumin) for one hour. Then, 0.2 μL of each this invention compound solution (in DMSO) were dispensed to 384-well microplate, 5 μL of the preincubated enzyme solution and 5 μL of substrate solution (50 mM HEPES-KOH (pH 7.4), 1 mM ATP, 0.8 mM acetyl CoA and 25-50 mM potassium bicarbonate) were added to microplate. After centrifugation and shaking, the reaction mixtures were incubated in a humidified box at room temperature for 1 to 3 hours. After the incubation, the enzyme reactions were stopped by the addition of EDTA. Then, after the samples were cocrystallized with CHCA (α-cyano-4-hydroxy cinnamic acid) matrices on MALDI target plate, by using the matrix assist laser deionization time-of-flight mass spectrometer (MALDI-TOF MS), samples were measured in reflector negative mode. Deprotonated ions of acetyl CoA (AcCoA) of substrate and malonyl CoA (MalCoA) of the reaction product were detected, then, the conversion rates of acetyl CoA to malonyl CoA was calculated by the intensity of [MalCoA-H]−/(Intensity of [MalCoA-H].+Intensity of [AcCoA-H] ) using each signal strength. The 50% inhibitory concentration (IC50) was calculated from the inhibition rate of the enzymatic reaction at each concentration of the compounds. In addition, potassium citrate concentrations in assay buffer solution, potassium hydrogen carbonate concentrations in substrate solution and incubation time were adjusted by each lot of enzyme. VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS +0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of O-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values. VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 μg/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 μL reaction volumes containing 2.7 μM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 μl of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 μl of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 μl of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values. VEGFR2 Kinase Assay Biochemical KDR kinase assays were performed in 96 well microtiter plates that were coated overnight with 75 ug/well of poly-Glu-Tyr (4:1) in 10 mM Phosphate Buffered Saline (PBS), pH 7.4. The coated plates were washed with 2 mls per well PBS+0.05% Tween-20 (PBS-T), blocked by incubation with PBS containing 1% BSA, then washed with 2 mls per well PBS-T prior to starting the reaction. Reactions were carried out in 100 uL reaction volumes containing 2.7 uM ATP in kinase buffer (50 mM Hepes buffer pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2 and 0.2 mM Na3VO4). Test compounds were reconstituted in 100% DMSO and added to the reaction to give a final DMSO concentration of 5%. Reactions were initiated by the addition 20 ul per well of kinase buffer containing 200-300 ng purified cytoplasmic domain KDR protein (BPS Bioscience, San Diego, Calif.). Following a 15 minute incubation at 30° C., the reactions were washed 2 mls per well PBS-T. 100 ul of a monoclonal anti-phosphotyrosine antibody-peroxidase conjugate diluted 1:10,000 in PBS-T was added to the wells for 30 minutes. Following a 2 mls per well wash with PBS-Tween-20, 100 ul of 0-Phenylenediamine Dihydrochloride in phosphate-citrate buffer, containing urea hydrogen peroxide, was added to the wells for 7-10 minutes as a colorimetric substrate for the peroxidase. The reaction was terminated by the addition of 100 ul of 2.5N H2SO4 to each well and read using a microplate ELISA reader set at 492 nm. IC50 values for compound inhibition were calculated directly from graphs of optical density (arbitrary units) versus compound concentration following subtraction of blank values. IL4I1 Enzymatic Assay nterleukin 4 inducible protein 1 (IL4I1) is an L-amino oxidase that catalyzes the oxidation of aromatic residues (Phe, Trp and Tyr): L-amino acid+H2O+O2→2-oxo acid+NH3+H2O2. Equal molar of H2O2 and the corresponding alpha-ketoacid are produced when IL4I1 and substrate are added. In this assay, the hydrogen peroxide generated by IL4I1 are then detected through a coupled reaction with Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine) and Horse Peroxidase (HRP) to produce Resorufin product that could be detected in the form of fluorescence signals. The assessment of the inhibitory effect of small molecules (EC50) on IL4I1 is measured by the effectiveness of the compounds to inhibit the production of H2O2.Using this assay, the potency (EC50) of each compound was determined from a ten-point (1:3 serial dilution; top compound concentration of 10000 nM) titration curve using the following outlined procedure. To each well of a black flat-bottom Greiner (Cat #781076) 384 well-plate, 25 nL of compound (0.1% DMSO in final assay volume of 25 μL) was dispensed, followed by the addition of 12.5 μL of 1× assay buffer (50 mM Hepes 7.0 and 0.005% Tween20 (Sigma, Cat #P8341; low peroxide grade)) containing 2 nM of recombinant IL4I1 (R&D Systems, Cat #5684-AO-020). Plates were placed in an ambient temperature humidified chamber for a four-hour pre-incubation with compound. Subsequently, each reaction was initiated by the addition of 12.5 μL 1× assay buffer containing 2 mM of each aromatic amino acids (Phe/Tyr/Trp), 0.1 mM Amplex Red and 2 U/mL of HRP. The final reaction in each well of 25 μL consists of 1 nM of IL4I1, 1 mM of each residues (Phe, Tyr and Trp), 0.05 mM Amplex Red and 1 U/mL of HRP. It should be noted that the concentrations of Amplex Red and HRP used here are in excess such that the conversion of H2O2 to Resorufin product occurs instantaneously and non-rate limiting. PAR Assay Studies were performed as follows: 6000 cells/well were seeded in 96 well plates (Perkin Elmer) in MEM/10% FCS and incubated for 24 hs at 37° C., 5% carbon dioxide. Test compounds were then added at the required concentration for 30'. DNA damage was then induced adding hydrogen peroxide at the concentration of 0.1 mM for 15 min. Concentration curves were prepared in MEM/10% FCS from compound stocks in dimethylsulfoxide (DMSO), and final DMSO concentration was 0.002% (v/v). Duplicate wells for each concentration point were prepared with a typical highest compound concentration of 20 μM and serial dilution 1:3. Plates were dried and fixed adding a cold methanol-acetone (70:30) solution for 15 min at RT; fixing solution was aspired and wells were air dried for 5 min and then dehydrated in PBS. Non-specific binding sites were blocked by incubating wells for 30 min in PBS containing 5% (w/v) FBS 0.05% Tween20. Wells were then incubated for 1 h at RT in PBS containing anti PAR mouse monoclonal antibody (Anti-PAR, Mouse mAb 10H, Tulip Cat No 1020) diluted 1:200 in blocking solution. After 3 washes in PBS, wells were incubated in PBS (w/v) 5% FBS 0.05% Tween20 containing 2 μg/mL Cy2-conjugated Goat anti mouse secondary antibody (Amersham Pharmacia Biotech cat. No PA 42002) (Absorption maximum 489 nm, fluorescence maximum 506 nm) and 1 μg/mL DAPI (Absorption maximum 359 nm, fluorescence maximum 461 nm) (4',6-diamidino-2-phenylindole dilactate) (Sigma cat. No D9564), a high sensitivity dye for nucleic acid staining. After washing further 3 times in PBS, cellular PAR immunoreactivity was assessed using the ArrayScan vTi instrument, with a Zeiss 10×0.5 N.A. objective, and applying the Cytotoxicity.V3 algorithm (Cellomics/Thermo Fisher) with a XF100 filter. At least 10 fields, corresponding to at least 900 cells, were read for each well. In Vitro DGAT2 Assay For determination of IC50 values, the reactions were carried out in 384-well white Polyplates (Perkin Elmer) in a total volume of 20 μL. To 1 μL of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 15 minutes. hDGAT2 membrane fractions were diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock). 10 μL of this enzyme working solution was added to the plates and incubation continued for 2 hours at room temperature. DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). Plates were centrifuged at 2,000×g for 1 minute in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). DGAT2 activity was measured by quantifying the generated product [14C]tridecanoylglycerol in the upper organic phase.Background activity obtained using 50 μM of (R)-1-(2-((S)-1-(4-Chloro-1H-pyrazol-1-yl)ethyl)-3H-imidazo[4,5-b]pyridin-5-yl)piperidin-3-yl)(pyrrolidin-1-yl)methanone (WO 2013150416, Example 196-A) for complete inhibition of DGAT2 was subtracted from all reactions. Inhibitors were tested at eleven different concentrations to generate IC50 values for each compound. The eleven inhibitor concentrations employed typically included 50, 15.8, 5, 1.58, 0.50, 0.16, 0.05, 0.016, 0.005, 0.0016, and 0.0005 μM. The data were plotted as percentage of inhibition versus inhibitor concentration and fit to the equation, y=100/[1+(x/IC50)z], where IC50 is the inhibitor concentration at 50% inhibition and z is the Hill slope (the slope of the curve at its inflection point). In Vitro DGAT2 Assay and Determination of IC50 Values for DGAT2 Inhibitors For determination of IC50 values, the reactions were carried out in 384-well white polypropylene plates (Nunc) in a total volume of 20 μL. To 1 μL of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 15 minutes. hDGAT2 membrane fractions were diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock). 10 μL of this enzyme working solution was added to the plates and incubation continued for 2 hours at room temperature. DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). Plates were centrifuged at 2,000×g for 1 minute in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). DGAT2 activity was measured by quantifying the generated product [14C]tridecanoylglycerol in the upper organic phase.Background activity obtained using 50 μM of ((R)-1-(2-((S)-1-(4-chloro-1H-pyrazol-1-yl)ethyl)-3H-imidazo[4,5-b]pyridin-5-yl)piperidin-3-yl)(pyrrolidin-1-yl)methanone (WO 2013150416, Example 196-A) for complete inhibition of DGAT2 was subtracted from all reactions. Inhibitors were tested at eleven different concentrations to generate IC50 values for each compound. The eleven inhibitor concentrations employed typically included 50, 15.8, 5, 1.58, 0.50, 0.16, 0.05, 0.016, 0.005, 0.0016, and 0.0005 μM. The data were plotted as percentage of inhibition versus inhibitor concentration and fit to the equation, y=100/[1+(x/IC50)z], where IC50 is the inhibitor concentration at 50% inhibition and z is the Hill slope (the slope of the curve at its inflection point). Assays for Epoxide Hydrolase Activity Any of a number of standard assays for determining epoxide hydrolase activity can be used to determine inhibition of sEH. For example, suitable assays are described in Gill, et al., Anal Biochem 131:273-282 (1983); and Borhan, et al., Analytical Biochemistry 231:188-200 (1995)). Suitable in vitro assays are described in Zeldin et al., J Biol. Chem. 268:6402-6407 (1993). Suitable in vivo assays are described in Zeldin et al., Arch Biochem Biophys 330:87-96 (1996). Assays for epoxide hydrolase using both putative natural substrates and surrogate substrates have been reviewed (see, Hammock, et al. In: Methods in Enzymology, Volume III, Steroids and Isoprenoids, Part B, (Law, J. H. and H. C. Rilling, eds. 1985), Academic Press, Orlando, Fla., pp. 303-311 and Wixtrom et al., In: Biochemical Pharmacology and Toxicology, Vol. 1: Methodological Aspects of Drug Metabolizing Enzymes, (Zakim, D. and D. A. Vessey, eds. 1985), John Wiley & Sons, Inc., New York, pp. 1-93. Several spectral based assays exist based on the reactivity or tendency of the resulting diol product to hydrogen bond (see, e.g., Wixtrom, supra, and Hammock. Anal. Biochem. 174:291-299 (1985) and Dietze, et al. Anal. Biochem. 216:176-187 (1994)).The enzyme also can be detected based on the binding of specific ligands to the catalytic site which either immobilize the enzyme or label it with a probe such as dansyl, fluoracein, luciferase, green fluorescent protein or other reagent. The enzyme can be assayed by its hydration of EETs, its hydrolysis of an epoxide to give a colored product as described by Dietze et al., 1994, supra, or its hydrolysis of a radioactive surrogate substrate (Borhan et al., 1995, supra). The enzyme also can be detected based on the generation of fluorescent products following the hydrolysis of the epoxide. Numerous methods of epoxide hydrolase detection have been described (see, e.g., Wixtrom, supra).The assays are normally carried out with a recombinant enzyme following affinity purification. They can be carried out in crude tissue homogenates, cell culture or even in vivo, as known in the art and described in the references cited above. Method for Testing Activities of Compounds at Enzyme Level Principles of activity test at enzyme level: firstly, dihydroorotate (DHO) is oxidatively dehydrogenated by DHODH to form orotic acid (Orotate, OA), accompanied by reduction of Flavin mononucleotide (FMN) into reduced flavin mononucleotide (FMNH2) by accepting 2H+ and 2e−, then coenzyme Q (CoQ) as hydrogen acceptor accepting electron and proton from FMNH2 is reduced into reduced coenzyme Q (CoQH2), reduced coenzyme Q transfers electrons to a chromogenic substrate, diclofenac sodium salt (DCIP), and finally DCIP was reduced. DCIP shows maximum absorption at 600 nm, while the reduced DCIP does not show absorption at 600 nm. The degree to which the substrate DHO is oxidized can be judged based on the degree of decrease in absorbance. The degree of oxidation of the substrate DHO per unit time is the initial rate of the enzymatic reaction. Upon addition of a inhibitor, the initial rate of the enzymatic reaction is reduced.The purified DHODH was diluted to 10 nM with a test buffer (50 mM HEPES, pH 8.0, 150 mM KCl, 0.1% Triton X-100), and coenzyme Q and DCIP were added to a final concentration of 100 μM and 120 μM, respectively, mixed well, added into a 96-well plate and incubated for 5 min at room temperature. Substrate DHO was added to start the reaction with a DHO final concentration of 500 μM. The absorbance was read at 600 nm using a BioTek microplate reader and read every 30 s for 6 min. For inhibitor activity test, different concentrations of inhibitors were added into the above reaction system, the initial rate of enzymatic reaction when no inhibitor was added was V0, and the initial rate of enzymatic reaction after an inhibitor was added was Vi, and the inhibition rate of a compound was calculated by the formula (1−Vi/V0)×100%. For calculating IC50 of a compound, Origin 8.0 was used to test the inhibition rate at least 8 concentrations. In the experimental procedure, Brequinar (purchased from Sigma-Aldrich Reagent) was used as a positive control and at least three parallels were set in each experiment. In Vitro DGAT2 Assay For determination of IC50 values, the reactions were carried out in 384-well white Polyplates (Perkin Elmer) in a total volume of 20 μL. To 14 of compounds dissolved in 100% DMSO and spotted at the bottom of each well, 5 μL of 0.04% bovine serum albumin (BSA) (fatty acid free, Sigma Aldrich) was added and the mixture was incubated at room temperature for 20 minutes. To this mixture, 10 μL of hDGAT2 membrane fraction (0.01 mg/mL) diluted in 100 mM Hepes-NaOH, pH 7.4, 20 mM MgCl2 containing 200 nM methyl arachidonyl fluorophosphonate (Cayman Chemical; dried from ethyl acetate stock solution under argon gas and dissolved in DMSO as 5 mM stock) was added. After this mixture was preincubated at room temperature for 2 hours, DGAT2 reactions were initiated by the addition of 4 μL of substrates containing 30 μM [1-14C]decanoyl-CoA (custom-synthesized by Perkin Elmer, 50 mCi/mmol) and 125 μM 1,2-didecanoyl-sn-glycerol (Avanti Polar Lipids) dissolved in 12.5% acetone. The reaction mixtures were incubated at room temperature for 40 min and the reactions were stopped by addition of 5 μL of 1% H3PO4. After the addition of 45 μL MicroScint-E (Perkin-Elmer), plates were sealed with Top Seal-A covers (Perkin-Elmer) and phase partitioning of substrates and products was achieved using a HT-91100 microplate orbital shaker (Big Bear Automation, Santa Clara, Calif.). Plates were centrifuged at 2,000×g for 1 min in an Allegra 6R Centrifuge (Beckman Coulter) and then were sealed again with fresh covers before reading in a 1450 Microbeta Wallac Trilux Scintillation Counter (Perkin Elmer). DGAT2 activity was measured by quantifying the generated product [14C]tridecanoylglycerol in the upper organic phase. Background activity obtained using 50 μM of (1R, 2R)-2-({3′-Fluoro-4′-[(6-fluoro-1, 3-benzothiazol-2-yl)amino]-1,1′-biphenyl-4-yl}carbonyl)cyclopentanecarboxylic acid (US 20040224997, Example 26) or (R)-1-(2-((S)-1-(4-Chloro-1H-pyrazol-1-yl)ethyl)-3H-imidazo[4,5-b]pyridin-5-yl)piperidin-3-yl)(pyrrolidin-1-yl)methanone (WO 2013150416, Example 196-A) for complete inhibition of DGAT2 was subtracted from all reactions. Inhibitors were tested at eleven different concentrations to generate IC50 values for each compound. The eleven inhibitor concentrations employed typically included 50, 15.8, 5, 1.58, 0.50, 0.16, 0.05, 0.016, 0.005, 0.0016, and 0.0005 μM. The data were plotted as percentage of inhibition versus inhibitor concentration and fit to the equation, y=100/[1+(x/IC50)z], where IC50 is the inhibitor concentration at 50% inhibition and z is the Hill slope (the slope of the curve at its inflection point). Table 3 below provides the IC50 values of the Examples for inhibition of DGAT2 in accordance with the above-described assay. Results are reported as geometric mean IC50 values. Biochemical Assay Preferably, for the screening method above cited, both the PARP protein and the 5H-phenanthridin-6-one-derived probe of formula (IP) are pre-mixed, or the PARP protein and the test compound are pre-mixed. In a further preferred screening method, the PARP proteins are PARP-1, PARP-2 and PARP-3. The term PARP protein encompasses full-length native proteins as well as fragments thereof. More preferably, R11 is hydrogen or methyl, m is 0 or 1; when m is 1, n is 3 or 6, X′″ is trifluoroacetate. The 5H-phenanthridin-6-one-derived probe (IP) was selected for its capability of binding to the PARP proteins, both encompassing full-length native proteins and fragments thereof.The polarization signal can be measured, e.g., by a plate reader such as the Saphire2 (Tecan). Data analysis was performed, e.g., by using the Dynafit software. Displacement data were also fitted, e.g., by using Excel spreadsheet (Microsoft Inc. Seattle, USA) to a four parameter logistic model (4PL), or Hill-Slope model. The assay was used to test compounds of the present invention. The displacement ability of the test compounds of formula (I) is in correlation with the compounds affinity for the NAD pocket of the enzyme. Specific probes of formula (IP) used in the assay are: P1. 9-Dimethylamino-11,11-dimethyl-1-(3-{methyl-[(6-oxo-5,6-dihydro-phenanthridin-2-ylcarbamoyl)-methyl]-carbamoyl}-propyl-2,3,4,11-tetrahydro-naphtho[2,3-g]quinolinium trifluoroacetate; P2. 9-Dimethylamino-11,11-dimethyl-1-[3-(3-{[(6-oxo-5,6-dihydro-phenanthridin-2-ylcarbamoyl)-methyl]-amino}-propylcarbamoyl)-propyl])-2,3,4,11-tetrahydro-naphtho[2,3-g]quinolinium trifluoroacetate; P3. 9-Dimethylamino-11,11-dimethyl-1-[3-(6-{[(6-oxo-5,6-dihydro-phenanthridin-2-ylcarbamoyl)-methyl]-amino}-hexylcarbamoyl)-propyl]-2,3,4,11-tetrahydro-naphtho[2,3-g]quinolinium trifluoroacetate.A compound of formula (IP) as defined above can be prepared as described in WO 2010/133647.The assay is based on the use of a probe of formula (IP) that binds to the NAD binding pocket and takes advantage of the significant change in the polarization signal observed upon binding of the probe to PARP-1, -2 and -3. The ability of the probe of formula (IP) to bind full-length PARP-1, -2 and -3 has been previously reported (WO 2010/133647). The assay has been validated as described in WO 2010/133647. Biological Assay The compounds of the invention can be tested for their ability to inhibit LSD1. The ability of the compounds of the invention to inhibit LSD1 can be tested as follows. Human recombinant LSD1 protein was purchased from BPS Bioscience Inc (catalog reference number 50100: human recombinant LSD1, GenBank accession no. NM_015013, amino acids 158-end with N-terminal GST tag, MW: 103 kDa). In order to monitor LSD1 enzymatic activity and/or its inhibition rate by our inhibitor(s) of interest, di-methylated H3-K4 peptide (Anaspec) was chosen as a substrate. The demethylase activity was estimated, under aerobic conditions, by measuring the release of H2O2 produced during the catalytic process, using the Amplex Red hydrogen peroxide/peroxidase assay kit (Invitrogen).Briefly, a fixed amount of LSD1 was incubated on ice for 15 minutes, in the absence and/or in the presence of at least eight 3-fold serial dilutions of the respective inhibitor (e.g., from 0 to 75 uM, depending on the inhibitor strength). Tranylcypromine (Biomol International) was used as a control for inhibition. Within the experiment, each concentration of inhibitor was tested in duplicate. After leaving the enzyme interacting with the inhibitor, KM of di-methylated H3-K4 peptide was added to each reaction and the experiment was left for 30 minutes at 37° C. in the dark. The enzymatic reactions were set up in a 50 mM sodium phosphate, pH 7.4 buffer. At the end of the incubation, Amplex Red reagent and horseradish peroxidase (HPR) solution were added to the reaction according to the recommendations provided by the supplier (Invitrogen), and left to incubate for 5 extra minutes at room temperature in the dark. A 1 uM H2O2 solution was used as a control of the kit efficiency. The conversion of the Amplex Red reagent to resorufin due to the presence of H2O2 in the assay, was monitored by fluorescence (excitation at 540 nm, emission at 590 nm) using a microplate reader (Infinite 200, Tecan). Arbitrary units were used to measure level of H2O2 produced in the absence and/or in the presence of inhibitor. The maximum demethylase activity of LSD1 was obtained in the absence of inhibitor and corrected for background fluorescence in the absence of LSD1. The IC50 value of each inhibitor was calculated with GraphPad Prism Software. Biological Assay The compounds of the invention can be tested for their ability to inhibit LSD1. The ability of the compounds of the invention to inhibit LSD1 can be tested as follows. Human recombinant LSD1 protein was purchased from BPS Bioscience Inc (catalog reference number 50100: human recombinant LSD1, GenBank accession no. NM_015013, amino acids 158-end with N-terminal GST tag, MW: 103 kDa). In order to monitor LSD1 enzymatic activity and/or its inhibition rate by our inhibitor(s) of interest, di-methylated H3-K4 peptide (Anaspec) was chosen as a substrate. The demethylase activity was estimated, under aerobic conditions, by measuring the release of H2O2 produced during the catalytic process, using the Amplex Red hydrogen peroxide/peroxidase assay kit (Invitrogen).Briefly, a fixed amount of LSD1 was incubated on ice for 15 minutes, in the absence and/or in the presence of at least eight 3-fold serial dilutions of the respective test compound (e.g., from 0 to 75 uM, depending on the inhibitor strength). Tranylcypromine (Biomol International) was used as a control for inhibition. Within the experiment, each concentration of inhibitor was tested in duplicate. After leaving the enzyme interacting with the inhibitor, KM of di-methylated H3-K4 peptide was added to each reaction and the experiment was left for 30 minutes at 37° C. In the dark. The enzymatic reactions were set up in a 50 mM sodium phosphate, pH 7.4 buffer. At the end of the incubation, Amplex Red reagent and horseradish peroxidase (HPR) solution were added to the reaction according to the recommendations provided by the supplier (Invitrogen), and left to incubate for 5 extra minutes at room temperature in the dark. A 1 uM H2O2 solution was used as a control of the kit efficiency. The conversion of the Amplex Red reagent to resorufin due to the presence of H2O2 in the assay, was monitored by fluorescence (excitation at 540 nm, emission at 590 nm) using a microplate reader (Infinite 200, Tecan). Arbitrary units were used to measure level of H2O2 produced in the absence and/or in the presence of inhibitor. The maximum demethylase activity of LSD1 was obtained in the absence of inhibitor and corrected for background fluorescence in the absence of LSD1. The IC50 value of each inhibitor was calculated with GraphPad Prism Software. Inhibition of LSD1 The compounds of the invention can be tested for their ability to inhibit LSD1. The ability of the compounds of the invention to inhibit LSD1 can be tested as follows. Human recombinant LSD1 protein was purchased from BPS Bioscience Inc (catalog reference number 50100: human recombinant LSD1, GenBank accession no. NM_015013, amino acids 158-end with N-terminal GST tag, MW: 103 kDa). In order to monitor LSD1 enzymatic activity and/or its inhibition rate by our inhibitor(s) of interest, di-methylated H3-K4 peptide (Anaspec) was chosen as a substrate. The demethylase activity was estimated, under aerobic conditions, by measuring the release of H2O2 produced during the catalytic process, using the Amplex Red hydrogen peroxide/peroxidase assay kit (Invitrogen).Briefly, a fixed amount of LSD1 was incubated on ice for 15 minutes, in the absence and/or in the presence of at least eight 3-fold serial dilutions of the respective test compound (e.g., from 0 to 75 μM, depending on the inhibitor strength). Tranylcypromine (Biomol International) was used as a control for inhibition. Within the experiment, each concentration of inhibitor was tested in duplicate. After leaving the enzyme interacting with the inhibitor, KM of di-methylated H3-K4 peptide was added to each reaction and the experiment was left for 30 minutes at 37° C. in the dark. The enzymatic reactions were set up in a 50 mM sodium phosphate, pH 7.4 buffer. At the end of the incubation, Amplex Red reagent and horseradish peroxidase (HPR) solution were added to the reaction according to the recommendations provided by the supplier (Invitrogen), and left to incubate for 5 extra minutes at room temperature in the dark. A 1 μM H2O2 solution was used as a control of the kit efficiency. The conversion of the Amplex Red reagent to resorufin due to the presence of H2O2 in the assay, was monitored by fluorescence (excitation at 540 nm, emission at 590 nm) using a microplate reader (Infinite 200, Tecan). Arbitrary units were used to measure level of H2O2 produced in the absence and/or in the presence of inhibitor.The maximum demethylase activity of LSD1 was obtained in the absence of inhibitor and corrected for background fluorescence in the absence of LSD1. The IC50 value of each inhibitor was calculated with GraphPad Prism Software. DAAO Enzymatic Assay The DAAO enzymatic activity assay was modified according to the report of Oguri et al (Oguri, S., Screening of d-amino acid oxidase inhibitor by a new multi-assay method. Food chemistry 2007, 100 (2), 616). The DAAO activity was measured by using substrate D-alanine reaction produced hydrogen peroxide (H2O2) to further react with 3-(4-hydroxyphenyl) propionic acid (HPPA). The HPPA were oxidized by H2O2 and peroxidase to become the fluorogenic dimer which was measured to represent the activity of DAAO. The substrate of DAAO was prepared in 50 mM D-alanine (dissolved in 0.2 M Tris-HCl buffer, pH 8.3). A 100 ul of D-alanine solution was mixed with 4 ul (in 100%) dimethyl sulfoxide, DMSO) of different concentrations of drugs ranging from 31.36 nM, 94.08 nM, 0.28 uM, 0.85 uM, 2.54 uM, 7.62 uM, 22.86 uM, 68.59 uM, 0.21 mM, 0.62 mM, 1.85 mM, 5.56 mM, 16.67 mM, and 50.00 mM with a final DMSO concentrations of 0.167% in each reaction concentration. A 10 ul of D-alanine and drug mixture was incubated with 220 ul of Reaction Master Mix in black 96 well plate at 37° C. for 5 min. The Reaction Master Mix contained 110 ul of 5 U/mL porcine kidney DAAO (Sigma-Aldrich, USA) solution (dissolved with 0.2 M Tris-HCl buffer, pH 8.3), 1.1 mL of 15 U/mL peroxidase solution (dissolved with 0.2 M Tris-HCl buffer, pH 8.3), 1.1 mL of 20 mM HPPA solution (dissolved with 0.2 M Tris-HCl buffer, pH 8.3), and 2.2 ml of 2 M Tris-HCl buffer (pH 8.3) for 110 reaction assays.Fluorescence intensity (Fs) was measured at 405 nm by irradiation excitation at 320 nm. The higher is the DAAO enzymatic activity, the higher is the fluorescence intensity. The fluorometric inhibition indicator (Fi) was obtained from the following equation: Fi=(Fs−FDrug)/(FDMSO). Where the fluorescent drug blank (FDrug) were measured in the drug mixture solution (using 0.2 M Tris HCl buffer, pH 8.3, without D-alanine). A DMSO blank (FDMSO) was measured under a 100% DMSO solution. Although, in the assay for D-amino acid oxidase, FAD was generally included in the reaction mixture because this co-factor is easily dissociated from the holoenzyme, the present method was performed without FAD. The inhibitory effect of DAAO inhibitors was compared by using inhibitory concentrations which reduce 50% of DAAO activity (IC50). The IC50 value was calculated by GraphPad Prism, version 5 software (GraphPad Software, Inc., La Jolla, Calif.) (GraphPad Prism 5, GraphPad software Inc: California, USA) through nonlinear regression model. Determining Endocannabinoid Hydrolase Activity In the Experimental example, endocannabinoid hydrolases used were Fatty Acid Amide Hydrolase (FAAH) and N-acylethanolamide hydrolyzing acid amidase (NAAA), which were prepared by the method described in the document (PMCID: PMC3423427, PMC3723234, PMC2692831, PMC3382457). The preparation method was as followed: a plasmid (pCDNA3.1/NAAA or pCDNA3.1/FAAH) carrying a whole NAAA/FAAH gene was constructed, wherein the plasmid carried Cytomegalovirus (CMV) promoter and Neomycin selectable gene; the plasmid was transformed into HEK-293 cell via lipid medium, stable cell lines expressing NAAA/FAAH at a high level were obtained by G418 screening and Western-blot method. HEK-293 recombinant cells were cultured and collected, washed with PBS for 2-3 times, and ultrasonically treated in 20 mM Tris-HCl containing 0.32 M sucrose, then repeatedly frozen and thawed twice, and then centrifuged at 4° C., 800 g for 15 min. The supernatant (i.e., the desired protein) was collected, the protein concentration was determined by BCA method, and the protein was diluted to a concentration of 1 mg/mL, and sub-packaged and stored in a refrigerator at −80° C. for further use.In the Experimental example, PBS solution used was prepared as followed: 8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, and 0.24 g KH2PO4 were dissolved in 1 L ultrapure water, and the resultant solution was subjected to moist heat sterilization and stored at 4° C.30 μL (1 mg/mL) endocannabinoid hydrolase was added to a sample vial, and 2 μL DMSO (Blank control group) or a different concentration of a test compound (Compounds 1-46 prepared in the Examples according to the invention) was further added. The reaction was carried out at 37° C. for 10 min. 170 μL buffer (the buffer consisted of 50 mM disodium Hydrogen Phosphate, 0.1% Triton X-100, 3 mM DTT, 150 μL) containing enzymatic hydrolysis substrate (the substrate was a heptadecenoyl ethanolamine containing a double bond and 17 carbon atoms, abbreviated as 17:1 FAE) was further added, wherein the concentration of 17:1 FAE was 5 μM. The reaction was carried out at 37° C. for 30 min, and 200 μL methanol solution containing internal standard (the internal standard was margaric acid, at a concentration of 1 nmol) was then added to stop the reaction. LC-MS was used to determine the yield of the hydrolysate 17:1 FA (i.e., a heptadecenoic acid containing a double bond) of 17:1 FAE, then a graph was plotted with Graphpad Prism 5. Thereby, the IC50 of the test compound on endocannabinoid hydrolase was determined.By the method above, the inhibitory effects of the Compounds 1-46 prepared in the invention on NAAA and FAAH were determined. The results were shown in Table 1, wherein IC50 (NAAA) represents a concentration that inhibits NAAA activity to 50% of the activity prior to inhibition, IC50 (FAAH) represents a concentration that inhibits FAAH activity to 50% of the activity prior to inhibition, and >100 μM represents that IC50 of a compound on a corresponding enzyme is above 100 μM, indicating that the compound has no inhibitory effect on the enzyme. Inhibition Assay A stock solution was prepared using a human MAO-B enzyme (purchased from Aldrich) and a Amplex Red monoamine oxidase assay kit according to a preparation manual. The kit includes a 5x reaction buffer, an Amplex red reagent (1 mg), HRP, DMSO, H2O2, p-tyramine (substrate of MAO-A, B), benzylamine (substrate of MAO-B), clorgiline (inhibitor of MAO-A), and pargyline (inhibitor of MAO-B). Among these reagents in the kit, benzylamine was used as a substrate for MAO-B, and pargyline was used as an MAO-B inhibitor. A solution as overall substrates was prepared as follows. 200 ul of a solution of 1 mg of Amplex red sufficiently dissolved in 200 ul of DMSO, 100 ul of a mixed solution of HRP and 1 ml of a 1x buffer, 200 ul of a solution of benzylamine dissolved in 1.2 ml of dH2O were added to 9.5 ml of a 1x buffer to reach a total volume of 10 mL, which is sufficient for 100 wells. 0.5 ul of a mixture of MAO-B inhibitor pargyline and 1 ml of dH2O was put into each well. First, the activity of MAO-B was determined using 10 uM of the synthesized compound. 96 wells were injected with positive and negative types, and the wile type. The positive type included only substrate and hydrogen peroxide, and the negative type included only substrate. For the wild type, corresponding wells were injected with the enzyme, substrate, and MAO-B inhibitor, but with no synthesized compound. Afterward, 2 ul of the synthesized compound (1 mM) was added into each well, and the human MAO-B enzyme was put only into the 1st row of wells. 0.5 ug of the human MAO-B was put into each well along with 100 ul of a 1x buffer. The human MAO-B enzyme was put into the 2nd row of the wells along with 0.5 ul of a pargyline, the MAO-B inhibitor. To reduce an experimental error for accuracy, the test was repeated three times for each compound. After 30 minutes, 100 ul of the substrate solution was added into each well in a darkroom. The test was performed in the darkroom due to light sensitivity of the Amplex reagent. Finally, a total volume of the reaction solution per well reached 200 ul. After about 2 to 3 hours, chromophoric degrees of the samples were measured. A variation in data values for the 1st and 2nd rows of the wells indicates the pure reaction activity of the MAO-B enzyme with the substrate. Using the samples with the synthesized compound the remaining activity of MAO-B after inhibited by the synthesized compound may be determined. This is because the activities of the other enzymes excluding the MAO-B enzyme may be excluded through this method. Compounds with high inhibitory activity at a concentration of 10 uM were screened from among the synthesized compounds at a compound concentration of 10 uM. Afterward, concentration-dependent IC50 values of these compounds may be obtained through an activity assay at different concentrations of 0.001 uM, 0.01 uM, 0.1 uM, 1 uM, and 10 uM. Fluorescence-based ELISA assay A variety of tissue culture cell lines, expressing endogenous levels of O-GlcNAcase, can be utilized; examples include rat PC-12, and human U-87, or SK-N-SH cells. In this assay, rat PC-12 cells were plated in 96-well plates with approximately 10,000 cells/well. Compounds to be tested were dissolved in DMSO, either 2 or 10 mM stock solution, and then diluted with DMSO and water in a two-step process using a Tecan workstation. Cells were treated with diluted compounds for 24 h (5.4 μL into 200 μL 1 well volume) to reach a final concentration of inhibitor desired to measure a compound concentration dependent response, typically, ten 3 fold dilution steps, starting at 10 μM were used to determine a concentration response curve. To prepare a cell lysate, the media from compound treated cells was removed, the cells were washed once with phosphate buffered saline (PBS) and then lysed for 5 minutes at room temperature in 50 μL of Phosphosafe reagent (Novagen Inc, Madison, Wis.) with protease inhibitors and PMSF. The cell lysate was collected and transferred to a new plate, which was then either coated to assay plates directly or frozen −80° C. until used in the ELISA procedure. If desired, the total protein concentration of samples was determined using 20 μL of the sample using the BCA method. The ELISA portion of the assay was performed in a black Maxisorp 96-well plate that was coated overnight at 4° C. with 100 μL/well of the cell lysate (1:10 dilution of the lysate with PBS containing protease inhibitors, phosphatase inhibitors, and PMSF). The following day the wells were washed 3 times with 300 μL/well of Wash buffer (Tris-buffered saline with 0.1% Tween 20). The wells were blocked with 100 μL/well Blocking buffer (Tris buffered saline w/0.05% Tween 20 and 2.5% Bovine serum albumin) Each well was then washed two times with 300 μL/well of wash buffer. The anti O-GlcNAc antibody RL-2 (Abcam, Cambridge, Mass.), diluted 1:1000 in blocking buffer, was added at 100 μL/well. The plate was sealed and incubated at 37° C. for 2 h with gentle shaking. The wells were then washed 3-times with 300 μL/well wash buffer. To detect the amount of RL-2 bound horse-radish peroxidase (HRP) conjugated goat anti-mouse secondary antibody (diluted 1:3000 in blocking buffer) was added at 100 μL/well. The plate was incubated for 60 min at 37° C. with gentle shaking. Each well was then washed 3-times with 300 μL/well wash buffer. The detection reagent was added, 100 μL/well of Amplex Ultra RED reagent (prepared by adding 30 μL of 10 mM Amplex Ultra Red stock solution to 10 mL PBS with 18 μL 3% hydrogen peroxide, H2O2). The detection reaction was incubated for 15 minutes at room temperature and then read with excitation at 530 nm and emission at 590 nm. The amount of O-GlcNAcylated protein, as detected by the ELISA assay, was plotted for each concentration of test compound using standard curve fitting algorithms for sigmoidal dose response curves. Inhibitory Action of PDHK1 Activity In Vitro In the case of human PDHK1 (hPDHK1, NCBI Reference Database Accession number NM_002610.3), a 1.3 kbp fragment encoding this protein was isolated from human liver cDNA by polymerase chain reaction (PCR). Modified hPDHK1 cDNA wherein FLAG-Tag sequence was added to the N terminus was prepared by PCR and ligated to the NdeI/EcoRI site of pET-17b vector (Merck MGaA, model number 69663-3). The recombinant construct was transformed into Escherichia coli DH5a (TOYOBO, model number DNA-903). The recombinant clones were identified, and plasmid DNA was isolated and subjected to the DNA sequence analysis. One clone which had the expected nucleic acid sequence was selected for expression work.For expression of hPDHK1 activity, Escherichia coli strain BL21(DE3) cells (Merck KGaA, model number 69450-4) were transformed with the pET17b vector containing modified hPDHK1 cDNA. The Escherichia coli were grown to an optical density 0.6 (600 nmol/L) at 30° C. Protein expression was induced by the addition of 500 μmol/L isopropyl-β-thiogalactopyranoside. The Escherichia coli were cultured at 20° C. for 17-18 hr and harvested by centrifugation.The harvested Escherichia coli was resuspended in a suspension buffer (20 mmol/L N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid-sodium hydroxide (HEPES-NaOH), 500 mmol/L sodium chloride, 1% ethylene glycol, and 0.1% polyoxyethylene-polyoxypropylene block copolymer (Pluronic F-68), complete, EDTA-free (pH 8.0)) and disrupted by a microfluidizer M-110H (MIZUHO INDUSTRIAL CO., LTD.) or ultrasonication. The precipitate was removed by centrifugation and the supernatant was added to DDDDK-tagged Protein PURIFICATION GEL (MBL, model number 3329). DDDDK-tagged Protein PURIFICATION GEL was washed with a washing buffer (20 mmol/L HEPES-NaOH, 500 mmol/L sodium chloride, 1% ethylene glycol, 0.1% pluronic F-68 (pH 8.0)) and the bound protein was eluted with elution buffer 1 (20 mmol/L HEPES-NaOH, 100 μg/mL peptide (amino acid sequence DYKDDDDK) (SEQ ID NO: 1), 500 mmol/L sodium chloride, 1% ethylene glycol, 0.1% pluronic F-68 (pH 8.0)).The eluted fractions containing FLAG-Tagged protein were pooled, concentrated by an ultrafiltration method, added to a gel filtration column (HiLoad 26/60 Superdex 200 (GE Healthcare, model number 17-1070-01)), and eluted with elution buffer 2 (20 mmol/L HEPES-NaOH, 150 mmol/L sodium chloride, 0.5 mmol/L ethylenediaminetetraacetic acid (EDTA), 1% ethylene glycol, 0.1% pluronic F-68 (pH 8.0)). The eluted fractions were pooled and preserved at −80° C.0.025 U/mL PDH (porcine heart PDH complex, Sigma P7032) and 0.5 μg/mL hPDHK1 were mixed in an assay buffer (50 mmol/L 3-morpholinopropanesulfonic acid (pH 7.0), 20 mmol/L dipotassium hydrogen phosphate, 60 mmol/L potassium chloride, 2 mmol/L magnesium chloride, 0.4 mmol/L EDTA, 0.2% poloxamer, 2 mmol/L dithiothreitol), and the mixture was incubated at 4° C. overnight to obtain a PDH/hPDHK1 complex solution. In the assay buffer, 0.025 U/mL PDH was mixed and incubated at 4° C. overnight to prepare a PDH solution.The test compounds were diluted with dimethyl sulfoxide (DMSO). To measure an inhibitory action of the test compound on the PDHK activity in the PDH/hPDHK1 complex solution, PDH/hPDHK1 complex solution (20 μL), test compound (1.5 μL) and 0.353 μmol/L ATP (diluted with assay buffer) (8.5 μL) were added to a 384 well microplate (Greiner Bio-One 781801) and PDHK reaction was performed at room temperature for 45 min (test compound well). DMSO (1.5 μL) was added to control wells instead of test compound. In addition, DMSO (1.5 μL) was added to blank wells instead of the test compound, and PDH solution was added instead of the PDH/hPDHK1 complex solution. Inhibitory Action of PDHK2 Activity In Vitro In the case of human PDHK2 (hPDHK2, NCBI Reference Database Accession number NM_002611.4), modified hPDHK2 cDNA wherein FLAG-Tag sequence was added to the N terminus of hPDHK2 cDNA clone (pReceiver-M01/PDK2-GeneCopoeia) as the base was prepared by PCR and ligated to the NdeI/EcoRI site of pET-17b vector. The recombinant construct was transformed into Escherichia coli DH5a. The recombinant clones were identified, and plasmid DNA was isolated and subjected to the DNA sequence analysis. One clone which had the expected nucleic acid sequence was selected for expression work.For expression of hPDHK2 activity, Escherichia coli strain BL21(DE3) cells were transformed with the pET17b vector containing modified hPDHK2 cDNA. The Escherichia coli were grown to an optical density 0.6 (600 nmol/L) at 30° C. Protein expression was induced by the addition of 500 μmol/L isopropyl-β-thiogalactopyranoside. The Escherichia coli were cultured at 20° C. for 17-18 hr and harvested by centrifugation. The harvested Escherichia coli was resuspended in a suspension buffer (20 mmol/L HEPES-NaOH, 500 mmol/L sodium chloride, 1% ethylene glycol, 0.1% pluronic F-68 (pH 8.0), cOmplete, EDTA-free (pH 8.0)), and disrupted by a microfluidizer. The precipitate was removed by centrifugation and the supernatant was added to DDDDK-tagged Protein PURIFICATION GEL. DDDDK-tagged Protein PURIFICATION GEL was washed with a washing buffer (20 mmol/L HEPES-NaOH, 500 mmol/L sodium chloride, 1% ethylene glycol, 0.1% pluronic F-68 (pH 8.0)) and the bound protein was eluted with elution buffer 1 (20 mmol/L HEPES-NaOH, 100 μg/mL peptide (amino acid sequence DYKDDDDK) (SEQ ID NO: 1), 500 mmol/L sodium chloride, 1% ethylene glycol, 0.1% pluronic F-68 (pH 8.0)). The eluted fractions containing FLAG-Tagged protein were pooled, concentrated by an ultrafiltration method, added to a gel filtration column (HiLoad 26/60 Superdex 200), and eluted with elution buffer 2 (20 mmol/L HEPES-NaOH, 150 mmol/L sodium chloride, 0.5 mmol/L ethylenediaminetetraacetic acid (EDTA), 1% ethylene glycol, 0.1% pluronic F-68 (pH 8.0)). The eluted fractions were pooled and preserved at −80° C.0.025 U/mL PDH and 0.5 μg/mL hPDHK2 were mixed in an assay buffer (50 mmol/L 3-morpholinopropanesulfonic acid (pH 7.0), 20 mmol/L dipotassium hydrogen phosphate, 60 mmol/L potassium chloride, 2 mmol/L magnesium chloride, 0.4 mmol/L EDTA, 0.2% poloxamer, 2 mmol/L dithiothreitol), and the mixture was incubated at 4° C. overnight to obtain a PDH/hPDHK2 complex solution. In the assay buffer, 0.025 U/mL PDH was mixed and incubated at 4° C. overnight to prepare a PDH solution.The test compounds were diluted with DMSO. To measure an inhibitory action of the test compound on the PDHK activity in the PDH/hPDHK2 complex solution, PDH/hPDHK2 complex solution (20 μL), test compound (1.5 μL) and 1.06 μmol/L ATP (diluted with assay buffer) (8.5 μL) were added to a 384 well microplate and PDHK reaction was performed at room temperature for 45 min (test compound well). DMSO (1.5 μL) was added to control wells instead of test compound. In addition, DMSO (1.5 μL) was added to blank wells instead of the test compound, and PDH solution was added instead of the PDH/hPDHK2 complex solution. To measure an inhibitory action of the test compound on the PDHK activity inherent in the PDH solution, a test compound was added and the PDH solution instead of the PDH/hPDHK2 complex solution was added to a blank+test compound well.Then, 10 μL of substrates (5 mmol/L sodium pyruvate, 5 mmol/L Coenzyme A, 12 mmol/L NAD, 5 mmol/L thiamine pyrophosphate, diluted with assay buffer) were added. The mixture was incubated at room temperature for 90 min, and the residual PDH activity was measured.The absorbance of each well at 340 nm was measured using a microplate reader to detect NADH produced by the PDH reaction.
 BDBM50086646 hydrogen (2-naphthyloxy)methylphosphonate
BDBM50086646 hydrogen (2-naphthyloxy)methylphosphonate BDBM50086647 hydrogen (1-phenanthryloxy)methylphosphonate
BDBM50086647 hydrogen (1-phenanthryloxy)methylphosphonate BDBM50086649 hydrogen (1-naphthyloxy)methylphosphonate
BDBM50086649 hydrogen (1-naphthyloxy)methylphosphonate cyclohexanaminium hydrogen (isobutylamino)carbonylphosphonate BDBM50146806
cyclohexanaminium hydrogen (isobutylamino)carbonylphosphonate BDBM50146806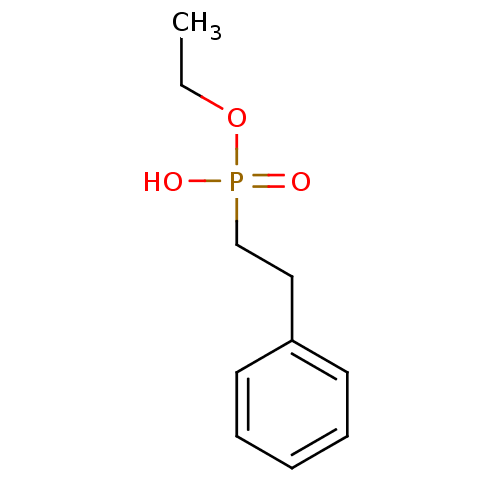 BDBM50195742 ethyl hydrogen 2-phenylethylphosphonate CHEMBL227517
BDBM50195742 ethyl hydrogen 2-phenylethylphosphonate CHEMBL227517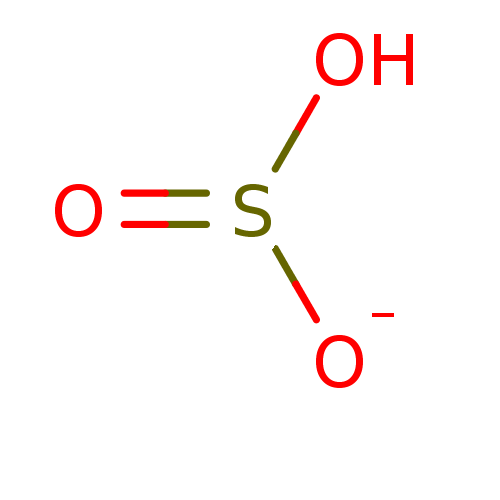 CHEMBL1689285 HSO3- bisulfite hydrogen sulfite BDBM26991
CHEMBL1689285 HSO3- bisulfite hydrogen sulfite BDBM26991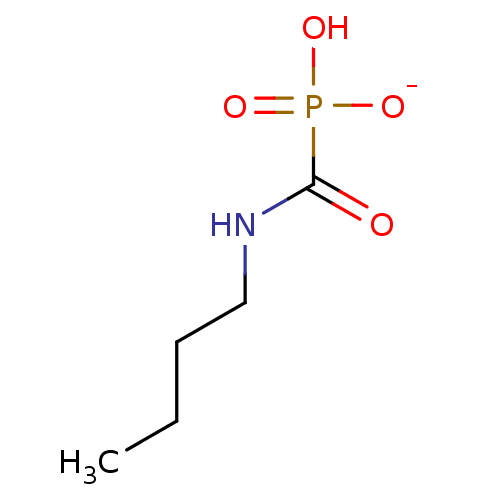 CHEMBL97389 sodium hydrogen (butylamino)carbonylphosphonate BDBM50146814
CHEMBL97389 sodium hydrogen (butylamino)carbonylphosphonate BDBM50146814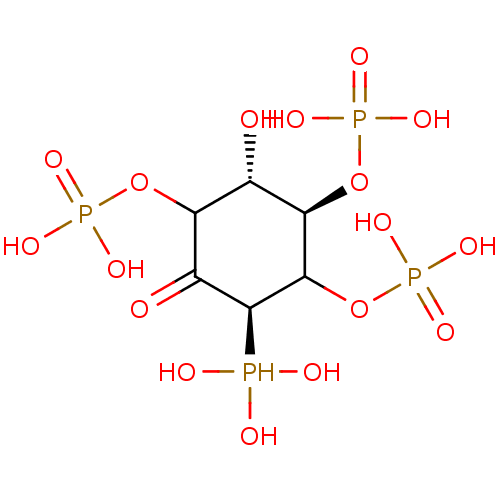 CHEMBL611046 BDBM50304359 (2R,3S,5S,6S)-6-(hydrogen phosphonato)-3,5-dihydroxycyclohexane-1,2,4-triyl tris(hydrogen phosphate)
CHEMBL611046 BDBM50304359 (2R,3S,5S,6S)-6-(hydrogen phosphonato)-3,5-dihydroxycyclohexane-1,2,4-triyl tris(hydrogen phosphate) BDBM50146812 cyclohexanaminium hydrogen [(3-methylbutyl)amino]carbonylphosphonate
BDBM50146812 cyclohexanaminium hydrogen [(3-methylbutyl)amino]carbonylphosphonate [NNNH] triazoic acid N3H CHEMBL186537 hydrogen azide BDBM50153977 hydrido-1kappaH-trinitrogen(2N--N)hydrogen trinitride(1-) hydrazoic acid
[NNNH] triazoic acid N3H CHEMBL186537 hydrogen azide BDBM50153977 hydrido-1kappaH-trinitrogen(2N--N)hydrogen trinitride(1-) hydrazoic acid BDBM50079022 HP2O7(3-) hydrogen diphosphate diphosphate diphosphate(3-)
BDBM50079022 HP2O7(3-) hydrogen diphosphate diphosphate diphosphate(3-) BDBM50198028 hydrogen 1-hydroxy-3-(hydroxyamino)-3-oxopropylphosphonate
BDBM50198028 hydrogen 1-hydroxy-3-(hydroxyamino)-3-oxopropylphosphonate CHEBI:28006 BDBM50487243 O,O-DIETHYL S-HYDROGEN PHOSPHOROTHIOATE
CHEBI:28006 BDBM50487243 O,O-DIETHYL S-HYDROGEN PHOSPHOROTHIOATE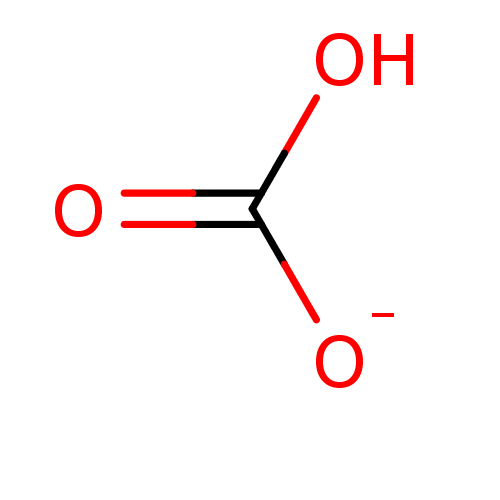 CHEMBL363707 SODIUM BICARBONATE bicarbonate ion hydrogen carbonate BDBM26986 HCO3-
CHEMBL363707 SODIUM BICARBONATE bicarbonate ion hydrogen carbonate BDBM26986 HCO3-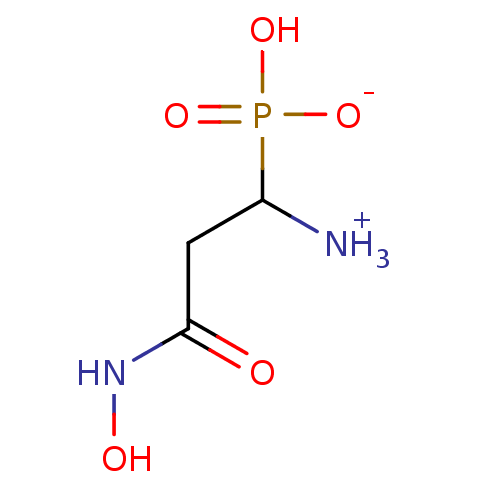 CHEMBL390655 BDBM50198029 hydrogen [1-azaniumyl-2-(hydroxycarbamoyl)ethyl]phosphonate
CHEMBL390655 BDBM50198029 hydrogen [1-azaniumyl-2-(hydroxycarbamoyl)ethyl]phosphonate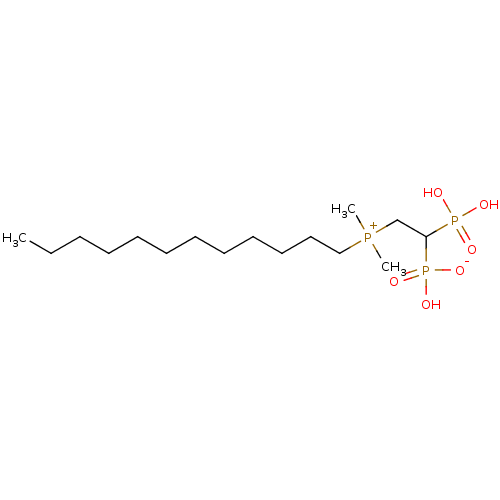 bisphosphonate, 9 hydrogen [2-(dodecyldimethylphosphanylium)-1-phosphonoethyl]phosphonate BDBM25256
bisphosphonate, 9 hydrogen [2-(dodecyldimethylphosphanylium)-1-phosphonoethyl]phosphonate BDBM25256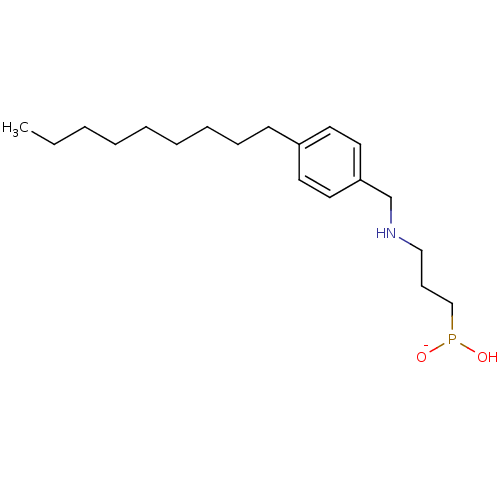 hydrogen (3-{[(4-nonylphenyl)methyl]amino}propyl)phosphonite BDBM50148402
hydrogen (3-{[(4-nonylphenyl)methyl]amino}propyl)phosphonite BDBM50148402 hydrogen 3-(dimethylammonio)-1-hydroxy-1-(hydroxyphosphinato)propylphosphonate BDBM50150877
hydrogen 3-(dimethylammonio)-1-hydroxy-1-(hydroxyphosphinato)propylphosphonate BDBM50150877 CHEMBL1096400 diammonium (2R,3S,5R)-2-[(hydrogen phosphonatooxy)methyl]-5-[6-(methylamino)-9H-purin-9-yl]oxolan-3-yl hydrogen phosphate BDBM50318029
CHEMBL1096400 diammonium (2R,3S,5R)-2-[(hydrogen phosphonatooxy)methyl]-5-[6-(methylamino)-9H-purin-9-yl]oxolan-3-yl hydrogen phosphate BDBM50318029 BDBM25309 hydrogen [1-phosphono-2-(trimethylphosphanylium)ethyl]phosphonate bisphosphonate, 58
BDBM25309 hydrogen [1-phosphono-2-(trimethylphosphanylium)ethyl]phosphonate bisphosphonate, 58 CHEMBL325304 Heptyl-phosphonic acid monoethyl ester BDBM50148588 ethyl hydrogen heptylphosphonate
CHEMBL325304 Heptyl-phosphonic acid monoethyl ester BDBM50148588 ethyl hydrogen heptylphosphonate Nonyl-phosphonic acid monoethyl ester CHEMBL119480 BDBM50148595 ethyl hydrogen nonylphosphonate
Nonyl-phosphonic acid monoethyl ester CHEMBL119480 BDBM50148595 ethyl hydrogen nonylphosphonate disodium hydrogen 3-carboxy-5-[(hydroxyphosphinato)methyl]benzylphosphonate BDBM50283584 CHEMBL88887
disodium hydrogen 3-carboxy-5-[(hydroxyphosphinato)methyl]benzylphosphonate BDBM50283584 CHEMBL88887 ethyl hydrogen butylphosphonate Butyl-phosphonic acid monoethyl ester CHEMBL118238 BDBM50148589
ethyl hydrogen butylphosphonate Butyl-phosphonic acid monoethyl ester CHEMBL118238 BDBM50148589 CHEMBL1094760 BDBM50318028 diammonium (2R,3S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-2-[(hydrogen phosphonatooxy)methyl]oxolan-3-yl hydrogen phosphate
CHEMBL1094760 BDBM50318028 diammonium (2R,3S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-2-[(hydrogen phosphonatooxy)methyl]oxolan-3-yl hydrogen phosphate 2-(hydroxy(3-hydroxypropyl)amino)-2-oxoethyl hydrogen phosphate BDBM50330436 CHEMBL1276292
2-(hydroxy(3-hydroxypropyl)amino)-2-oxoethyl hydrogen phosphate BDBM50330436 CHEMBL1276292 BDBM50147627 Bicarbonate Bicarbonate Ion CHEBI:28976 Carbonic Acid Anion Hydrogen Carbonate
BDBM50147627 Bicarbonate Bicarbonate Ion CHEBI:28976 Carbonic Acid Anion Hydrogen Carbonate BDBM50165348 hydrogen 1-hydroxy-2-isoquinolinium-2-yl-1-phosphonoethylphosphonate CHEMBL425896
BDBM50165348 hydrogen 1-hydroxy-2-isoquinolinium-2-yl-1-phosphonoethylphosphonate CHEMBL425896 bisphosphonate, 22 hydrogen {2-[dimethyl(octyl)phosphanylium]-1-phosphonoethyl}phosphonate BDBM25268
bisphosphonate, 22 hydrogen {2-[dimethyl(octyl)phosphanylium]-1-phosphonoethyl}phosphonate BDBM25268 hydrogen(sulfide)(-1) sulfanide BDBM26990 HS anion HS(-1) hydrosulfide CHEMBL1644699
hydrogen(sulfide)(-1) sulfanide BDBM26990 HS anion HS(-1) hydrosulfide CHEMBL1644699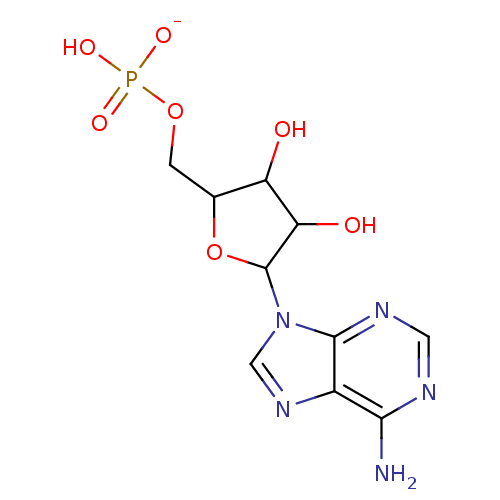 [5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl hydrogen phosphate;triethylazanium SMR000718807 [5-(6-aminopurin-9-yl)-3,4-dihydroxy-2-oxolanyl]methyl hydrogen phosphate;triethylammonium (5-adenin-9-yl-3,4-dihydroxy-tetrahydrofuran-2-yl)methyl hydrogen phosphate;triethylammonium BDBM61258 cid_16396156 MLS001306424 [5-(6-aminopurin-9-yl)-3,4-bis(oxidanyl)oxolan-2-yl]methyl hydrogen phosphate;triethylazanium
[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl hydrogen phosphate;triethylazanium SMR000718807 [5-(6-aminopurin-9-yl)-3,4-dihydroxy-2-oxolanyl]methyl hydrogen phosphate;triethylammonium (5-adenin-9-yl-3,4-dihydroxy-tetrahydrofuran-2-yl)methyl hydrogen phosphate;triethylammonium BDBM61258 cid_16396156 MLS001306424 [5-(6-aminopurin-9-yl)-3,4-bis(oxidanyl)oxolan-2-yl]methyl hydrogen phosphate;triethylazanium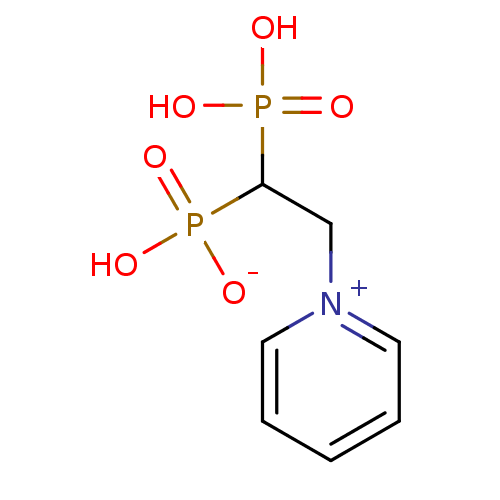 1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 60 BDBM25311
1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 60 BDBM25311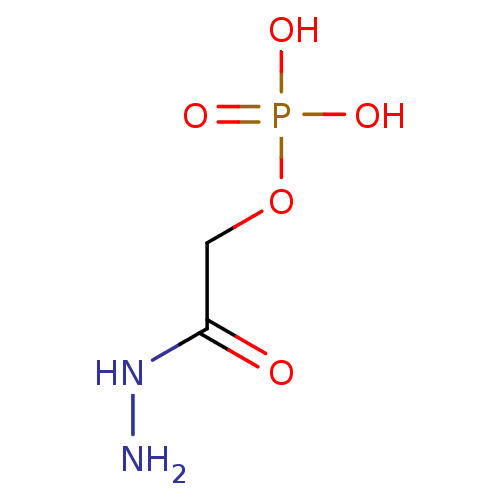 2-hydrazinyl-2-oxoethyl hydrogen phosphate BDBM50167772 Phosphoric acid monohydrazinocarbonylmethyl ester CHEMBL195520
2-hydrazinyl-2-oxoethyl hydrogen phosphate BDBM50167772 Phosphoric acid monohydrazinocarbonylmethyl ester CHEMBL195520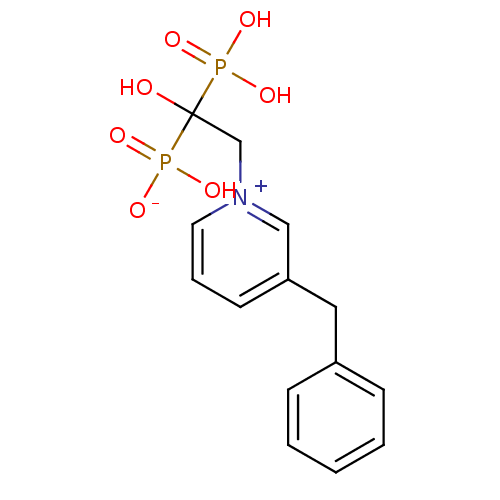 BDBM50165339 hydrogen 2-(3-benzylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL363434
BDBM50165339 hydrogen 2-(3-benzylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL363434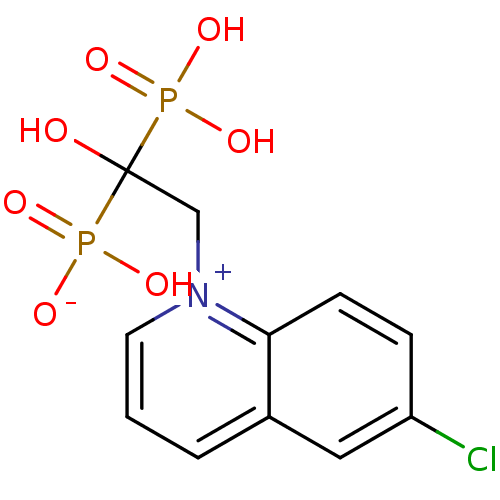 BDBM50165341 hydrogen 2-(6-chloroquinolinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL193131
BDBM50165341 hydrogen 2-(6-chloroquinolinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL193131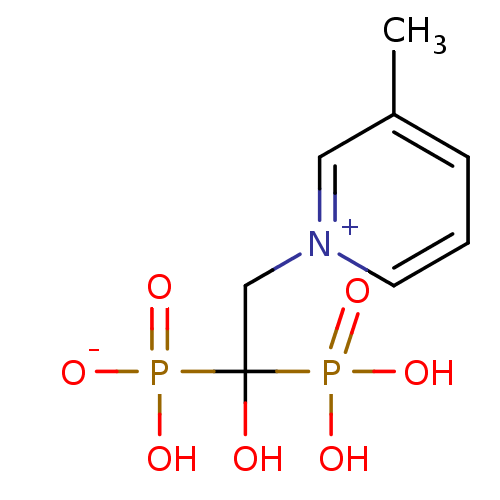 BDBM50165342 CHEMBL193356 hydrogen 1-hydroxy-2-(3-methylpyridinium-1-yl)-1-phosphonoethylphosphonate
BDBM50165342 CHEMBL193356 hydrogen 1-hydroxy-2-(3-methylpyridinium-1-yl)-1-phosphonoethylphosphonate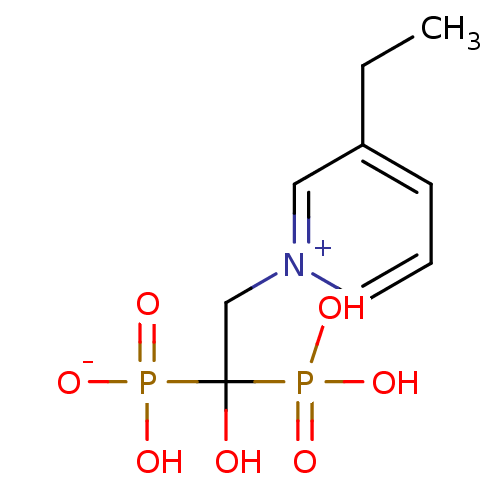 BDBM50165352 CHEMBL192043 hydrogen 2-(3-ethylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate
BDBM50165352 CHEMBL192043 hydrogen 2-(3-ethylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate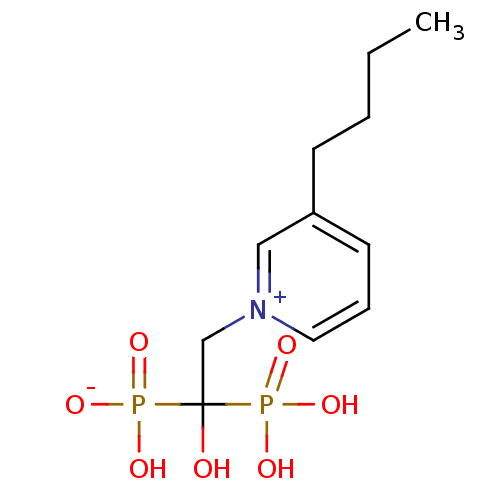 BDBM50165353 hydrogen 2-(3-butylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL373332
BDBM50165353 hydrogen 2-(3-butylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL373332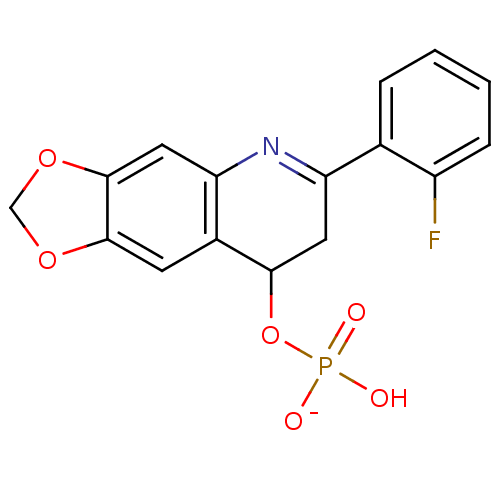 BDBM50312665 Sodium 2-(2-Fluorophenyl)-6,7-methylenedioxyquinolin-4-yl hydrogen phosphate CHEMBL1088572
BDBM50312665 Sodium 2-(2-Fluorophenyl)-6,7-methylenedioxyquinolin-4-yl hydrogen phosphate CHEMBL1088572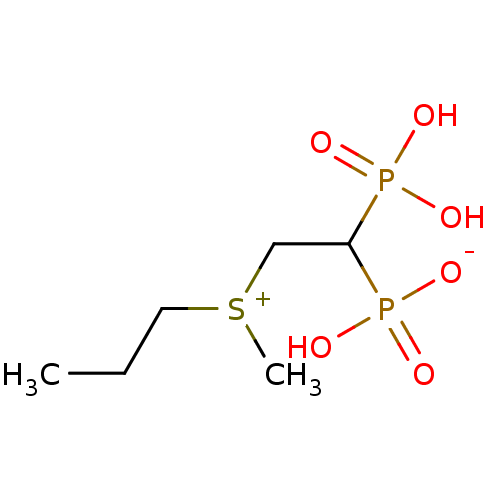 CHEMBL235059 bisphosphonate, 50 BDBM25303 hydrogen {2-[methyl(propyl)sulfanylium]-1-phosphonoethyl}phosphonate
CHEMBL235059 bisphosphonate, 50 BDBM25303 hydrogen {2-[methyl(propyl)sulfanylium]-1-phosphonoethyl}phosphonate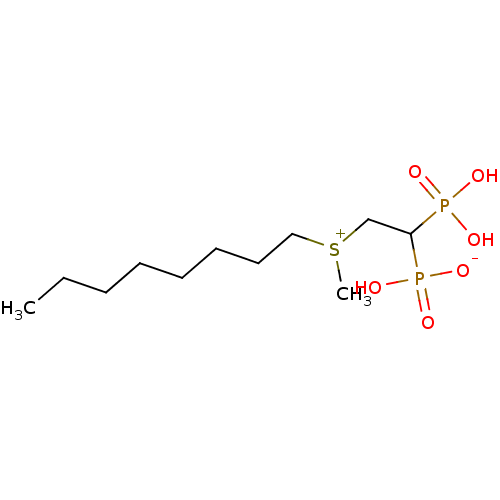 CHEMBL235690 hydrogen {2-[methyl(octyl)sulfanylium]-1-phosphonoethyl}phosphonate BDBM25287 bisphosphonate, 37
CHEMBL235690 hydrogen {2-[methyl(octyl)sulfanylium]-1-phosphonoethyl}phosphonate BDBM25287 bisphosphonate, 37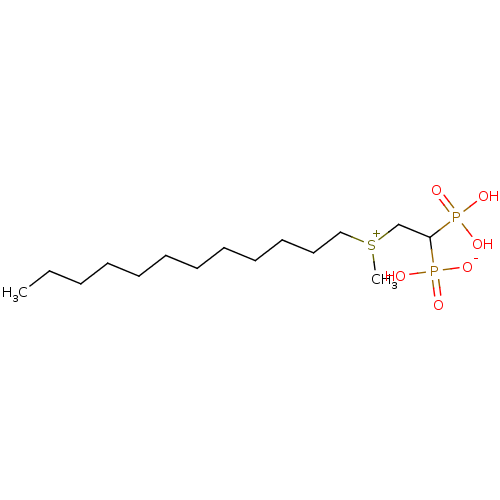 CHEMBL238046 BDBM25272 hydrogen {2-[dodecyl(methyl)sulfanylium]-1-phosphonoethyl}phosphonate bisphosphonate, 25
CHEMBL238046 BDBM25272 hydrogen {2-[dodecyl(methyl)sulfanylium]-1-phosphonoethyl}phosphonate bisphosphonate, 25 bisphosphonate, 43 BDBM25294 CHEMBL392884 hydrogen {2-[methyl(pentyl)sulfanylium]-1-phosphonoethyl}phosphonate
bisphosphonate, 43 BDBM25294 CHEMBL392884 hydrogen {2-[methyl(pentyl)sulfanylium]-1-phosphonoethyl}phosphonate bisphosphonate, 52 hydrogen {2-[(10-carboxydecyl)(methyl)sulfanylium]-1-phosphonoethyl}phosphonate BDBM25305
bisphosphonate, 52 hydrogen {2-[(10-carboxydecyl)(methyl)sulfanylium]-1-phosphonoethyl}phosphonate BDBM25305 hydrogen 1-hydroxy-2-(3-methoxypyridinium-1-yl)-1-phosphonoethylphosphonate BDBM50165349 CHEMBL363145
hydrogen 1-hydroxy-2-(3-methoxypyridinium-1-yl)-1-phosphonoethylphosphonate BDBM50165349 CHEMBL363145 hydrogen 2-(1,2-dihydropyridin-3-yl)-1-hydroxy-1-(hydroxyphosphinato)ethylphosphonate BDBM50150875
hydrogen 2-(1,2-dihydropyridin-3-yl)-1-hydroxy-1-(hydroxyphosphinato)ethylphosphonate BDBM50150875 hydrogen 2-(4-benzylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate BDBM50165338 CHEMBL190258
hydrogen 2-(4-benzylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate BDBM50165338 CHEMBL190258 hydrogen [2-(dimethylsulfanylium)-1-hydroxy-1-phosphonoethyl]phosphonate bisphosphonate, 49 CHEMBL277580 BDBM25302
hydrogen [2-(dimethylsulfanylium)-1-hydroxy-1-phosphonoethyl]phosphonate bisphosphonate, 49 CHEMBL277580 BDBM25302 hydrogen {2-[methyl(tetradecyl)sulfanylium]-1-phosphonoethyl}phosphonate CHEMBL237808 bisphosphonate, 28 BDBM25275
hydrogen {2-[methyl(tetradecyl)sulfanylium]-1-phosphonoethyl}phosphonate CHEMBL237808 bisphosphonate, 28 BDBM25275 (R)-Fluoro-phenyl-methanephosphonic acid anion BDBM50129059 hydrogen [(R)-fluoro(phenyl)methyl]phosphonate
(R)-Fluoro-phenyl-methanephosphonic acid anion BDBM50129059 hydrogen [(R)-fluoro(phenyl)methyl]phosphonate BDBM235706 trans-(1R(S),6R(S))-6-Hydroxycyclohex-3-enyl hydrogen sulfate (4)
BDBM235706 trans-(1R(S),6R(S))-6-Hydroxycyclohex-3-enyl hydrogen sulfate (4) BDBM25296 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-phenylpyridin-1-ium bisphosphonate, 45
BDBM25296 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-phenylpyridin-1-ium bisphosphonate, 45 BDBM25301 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-methylpyridin-1-ium bisphosphonate, 48
BDBM25301 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-methylpyridin-1-ium bisphosphonate, 48 Blausaeure formonitrile CHEMBL183419 hydrogen cyanide hydridonitridocarbonhydrogen(nitridocarbonate)methanenitrile Cyanwasserstoff BDBM50152968 [CHN] hydrocyanic acid
Blausaeure formonitrile CHEMBL183419 hydrogen cyanide hydridonitridocarbonhydrogen(nitridocarbonate)methanenitrile Cyanwasserstoff BDBM50152968 [CHN] hydrocyanic acid CHEMBL513014 ethyl hydrogen 4-(2-amino-5-(thiophen-2-yl)phenylcarbamoyl)benzylphosphonate BDBM50278306
CHEMBL513014 ethyl hydrogen 4-(2-amino-5-(thiophen-2-yl)phenylcarbamoyl)benzylphosphonate BDBM50278306 CHEMBL513255 ethyl hydrogen 4-(2-amino-5-(thiophen-2-yl)phenylcarbamoyl)phenylphosphonate BDBM50278250
CHEMBL513255 ethyl hydrogen 4-(2-amino-5-(thiophen-2-yl)phenylcarbamoyl)phenylphosphonate BDBM50278250 bisphosphonate, 20 BDBM25264 1-(2-hydrogen phosphonato-2-phosphonoethyl)-4-octylpyridin-1-ium
bisphosphonate, 20 BDBM25264 1-(2-hydrogen phosphonato-2-phosphonoethyl)-4-octylpyridin-1-ium (benzyloxy)(2-formylphenoxy)phosphinic acid PASBN BDBM14688 benzyl 2-formylphenyl hydrogen phosphate Fragment 18
(benzyloxy)(2-formylphenoxy)phosphinic acid PASBN BDBM14688 benzyl 2-formylphenyl hydrogen phosphate Fragment 18 3-(dodecyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25286 bisphosphonate, 36
3-(dodecyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25286 bisphosphonate, 36 3-bromo-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25307 bisphosphonate, 56
3-bromo-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25307 bisphosphonate, 56 BDBM235708 trans-(1R(S),8R(S),Z)-8-Hydroxycyclooct-4-enyl hydrogen sulfate (6)
BDBM235708 trans-(1R(S),8R(S),Z)-8-Hydroxycyclooct-4-enyl hydrogen sulfate (6) BDBM25261 3-decyl-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 17
BDBM25261 3-decyl-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 17 BDBM25273 3-(decylamino)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 26
BDBM25273 3-(decylamino)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 26 BDBM50287679 CHEMBL61555 Sodium; benzyl 3,4,5-trihydroxy-6-methyltetrahydro-2H-thiopyran-2-yl hydrogen phosphate
BDBM50287679 CHEMBL61555 Sodium; benzyl 3,4,5-trihydroxy-6-methyltetrahydro-2H-thiopyran-2-yl hydrogen phosphate BDBM50304360 BENZENE-1,2,3,4-TETRAYL TETRAKIS[DIHYDROGEN (PHOSPHATE)] benzene-1,2,3,4-tetrayl tetrakis(hydrogen phosphate) CHEMBL595349
BDBM50304360 BENZENE-1,2,3,4-TETRAYL TETRAKIS[DIHYDROGEN (PHOSPHATE)] benzene-1,2,3,4-tetrayl tetrakis(hydrogen phosphate) CHEMBL595349 CHEMBL119521 Hexyl-phosphonic acid monoethyl ester N-HEXYLPHOSPHONATE ETHYL ESTER BDBM50148593 ethyl hydrogen hexylphosphonate
CHEMBL119521 Hexyl-phosphonic acid monoethyl ester N-HEXYLPHOSPHONATE ETHYL ESTER BDBM50148593 ethyl hydrogen hexylphosphonate CHEMBL192938 BDBM50165350 hydrogen 1-hydroxy-2-[3-(3-methylbenzyl)pyridinium-1-yl]-1-phosphonoethylphosphonate
CHEMBL192938 BDBM50165350 hydrogen 1-hydroxy-2-[3-(3-methylbenzyl)pyridinium-1-yl]-1-phosphonoethylphosphonate CHEMBL193722 BPH-461 hydrogen 2-(3-fluoropyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate BDBM50165340
CHEMBL193722 BPH-461 hydrogen 2-(3-fluoropyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate BDBM50165340 bisphosphonate, 42 BDBM25293 3-hexyl-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium
bisphosphonate, 42 BDBM25293 3-hexyl-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)pyridin-1-ium CHEMBL191634 BDBM25310 bisphosphonate, 59
1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)pyridin-1-ium CHEMBL191634 BDBM25310 bisphosphonate, 59 3-(decyloxy)-5-(2-hydrogen phosphonato-2-phosphonoethyl)-1-methylpyridin-1-ium BDBM25258 bisphosphonate, 14
3-(decyloxy)-5-(2-hydrogen phosphonato-2-phosphonoethyl)-1-methylpyridin-1-ium BDBM25258 bisphosphonate, 14 3-(heptyloxy)-5-(2-hydrogen phosphonato-2-phosphonoethyl)-1-methylpyridin-1-ium bisphosphonate, 30 BDBM25278
3-(heptyloxy)-5-(2-hydrogen phosphonato-2-phosphonoethyl)-1-methylpyridin-1-ium bisphosphonate, 30 BDBM25278 BDBM12580 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-iodopyridin-1-ium Bisphosphonate 5
BDBM12580 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-iodopyridin-1-ium Bisphosphonate 5 BDBM25274 bisphosphonate, 27 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-octylpyridin-1-ium
BDBM25274 bisphosphonate, 27 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-octylpyridin-1-ium BDBM25283 3-(3-butoxyphenyl)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 35
BDBM25283 3-(3-butoxyphenyl)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 35 PAS219 (cyclohexylmethoxy)(2-formylphenoxy)phosphinic acid BDBM14685 CHEMBL24811 Fragment 15 cyclohexylmethyl 2-formylphenyl hydrogen phosphate
PAS219 (cyclohexylmethoxy)(2-formylphenoxy)phosphinic acid BDBM14685 CHEMBL24811 Fragment 15 cyclohexylmethyl 2-formylphenyl hydrogen phosphate sodium hydrogen 1-hydroxy-2-[3-(4-oxidophenyl)pyridinium-1-yl]-1-phosphonoethylphosphonate CHEMBL193619 BDBM50165351
sodium hydrogen 1-hydroxy-2-[3-(4-oxidophenyl)pyridinium-1-yl]-1-phosphonoethylphosphonate CHEMBL193619 BDBM50165351 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-phenylpyridin-1-ium BDBM25299 bisphosphonate, 46 CHEMBL192342
1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-phenylpyridin-1-ium BDBM25299 bisphosphonate, 46 CHEMBL192342 BDBM25259 Bisphpshonate-715 3-(decyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 15
BDBM25259 Bisphpshonate-715 3-(decyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 15 BDBM25262 bisphosphonate, 18 3-bromo-5-(decyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium
BDBM25262 bisphosphonate, 18 3-bromo-5-(decyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25271 3-[(3,7-dimethyloctyl)oxy]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 24
BDBM25271 3-[(3,7-dimethyloctyl)oxy]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 24 BDBM25282 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(octane-1-sulfonamido)pyridin-1-ium bisphosphonate, 34
BDBM25282 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(octane-1-sulfonamido)pyridin-1-ium bisphosphonate, 34 CHEMBL195976 Phosphoric acid mono-(N-hydroxycarbamimidoylmethyl) ester BDBM50167775 (Z)-2-amino-2-(hydroxyimino)ethyl hydrogen phosphate
CHEMBL195976 Phosphoric acid mono-(N-hydroxycarbamimidoylmethyl) ester BDBM50167775 (Z)-2-amino-2-(hydroxyimino)ethyl hydrogen phosphate (2R,3S,5S)-2-[(hydrogen phosphonatooxy)methyl]-5-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium BDBM50197422 CHEMBL247901
(2R,3S,5S)-2-[(hydrogen phosphonatooxy)methyl]-5-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium BDBM50197422 CHEMBL247901 (2S,3R,5R)-2-[(hydrogen phosphonatooxy)methyl]-5-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium CHEMBL247698 BDBM50197421
(2S,3R,5R)-2-[(hydrogen phosphonatooxy)methyl]-5-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium CHEMBL247698 BDBM50197421 (2S,3R,5R)-5-[(hydrogen phosphonatooxy)methyl]-2-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium CHEMBL393055 BDBM50197420
(2S,3R,5R)-5-[(hydrogen phosphonatooxy)methyl]-2-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium CHEMBL393055 BDBM50197420 3-(decyloxy)-5-(3,5-difluorophenyl)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25267 bisphosphonate, 13
3-(decyloxy)-5-(3,5-difluorophenyl)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25267 bisphosphonate, 13 3-(hydroxyamino)-3-oxopropylphosphonic acid BDBM50167776 hydrogen 3-(hydroxyamino)-3-oxopropylphosphonate (2-Hydroxycarbamoyl-ethyl)-phosphonic acid CHEMBL196442
3-(hydroxyamino)-3-oxopropylphosphonic acid BDBM50167776 hydrogen 3-(hydroxyamino)-3-oxopropylphosphonate (2-Hydroxycarbamoyl-ethyl)-phosphonic acid CHEMBL196442 CHEMBL247699 (2R,3S,5S)-5-[(hydrogen phosphonatooxy)methyl]-2-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium BDBM50197424
CHEMBL247699 (2R,3S,5S)-5-[(hydrogen phosphonatooxy)methyl]-2-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium BDBM50197424 bisphosphonate, 54 BDBM25306 3-[3-(2-ethoxyethoxy)propyl]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium
bisphosphonate, 54 BDBM25306 3-[3-(2-ethoxyethoxy)propyl]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 63 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(1H-imidazol-5-yl)pyridin-1-ium BDBM25312
bisphosphonate, 63 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(1H-imidazol-5-yl)pyridin-1-ium BDBM25312 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-(3-phenylphenyl)pyridin-1-ium CHEMBL192017 bisphosphonate, 7 BDBM25297
1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-(3-phenylphenyl)pyridin-1-ium CHEMBL192017 bisphosphonate, 7 BDBM25297 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(oct-1-yn-1-yl)pyridin-1-ium bisphosphonate, 29 BDBM25276
1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(oct-1-yn-1-yl)pyridin-1-ium bisphosphonate, 29 BDBM25276 BDBM50273310 CHEMBL508090 6-(2-Furyl)-2,4dioxo-1,3-dipropyl-2,3,4,5-tetrahydro-1H-pyrrolo[3,2-d]pyrimidin-7-yl hydrogen sulfate
BDBM50273310 CHEMBL508090 6-(2-Furyl)-2,4dioxo-1,3-dipropyl-2,3,4,5-tetrahydro-1H-pyrrolo[3,2-d]pyrimidin-7-yl hydrogen sulfate disodium monohydrogen phosphate Na2HPO4 disodium hydrogen phosphate Disodium phosphate disodium acid orthophosphate BDBM50080995 disodium hydrogenphosphate Dibasic sodium phosphate CHEMBL1060
disodium monohydrogen phosphate Na2HPO4 disodium hydrogen phosphate Disodium phosphate disodium acid orthophosphate BDBM50080995 disodium hydrogenphosphate Dibasic sodium phosphate CHEMBL1060 (2s)-2-ethoxy-n-hydroxy-3-[4-(pyridin-2-ylmethoxy)phenyl]propanamide hydrogen chloride (compound no. 1) US9562012, 5 BDBM228135
(2s)-2-ethoxy-n-hydroxy-3-[4-(pyridin-2-ylmethoxy)phenyl]propanamide hydrogen chloride (compound no. 1) US9562012, 5 BDBM228135 BDBM25295 bisphosphonate, 44 3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium
BDBM25295 bisphosphonate, 44 3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium {[(2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-methyloxolan-3-yl phosphonato]oxy}(hydrogen phosphonatooxy)phosphinate BDBM50262191
{[(2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-methyloxolan-3-yl phosphonato]oxy}(hydrogen phosphonatooxy)phosphinate BDBM50262191 N-[2-(2-Aminoethoxy)benzene-1-sulfonyl]-6-(dimethylamino)-1-benzofuran-2-carboxamide hydrogen chloride (1/1) BDBM757529 US12357603, Example 167
N-[2-(2-Aminoethoxy)benzene-1-sulfonyl]-6-(dimethylamino)-1-benzofuran-2-carboxamide hydrogen chloride (1/1) BDBM757529 US12357603, Example 167 (4aS,4bR,10bS,12aS)-12a-methyl-2-oxo-1,2,3,4,4a,4b,5,6,10b,11,12,12a-dodecahydronaphtho[2,1-f]quinolin-8-yl hydrogen sulfate CHEMBL1642920 BDBM50334791
(4aS,4bR,10bS,12aS)-12a-methyl-2-oxo-1,2,3,4,4a,4b,5,6,10b,11,12,12a-dodecahydronaphtho[2,1-f]quinolin-8-yl hydrogen sulfate CHEMBL1642920 BDBM50334791 BDBM429929 US10538497, Example 118 phenyl hydrogen [3-({4-[N-(3- bromo-4-fluorophenyl)-N'- hydroxycarbamimidoyl]-1,2,5- oxadiazol-3- yl}sulfanyl)propyl]phosphonate
BDBM429929 US10538497, Example 118 phenyl hydrogen [3-({4-[N-(3- bromo-4-fluorophenyl)-N'- hydroxycarbamimidoyl]-1,2,5- oxadiazol-3- yl}sulfanyl)propyl]phosphonate BDBM50308328 CHEMBL598208 (+/-)-Phenyl hydrogen{[6-(1H-indazol-3-ylmethyl)-5,7-dioxo-4-phenyl-4,5,6,7-tetrahydro-1H-1,4-diazepin-1-yl]methyl}phosphonate
BDBM50308328 CHEMBL598208 (+/-)-Phenyl hydrogen{[6-(1H-indazol-3-ylmethyl)-5,7-dioxo-4-phenyl-4,5,6,7-tetrahydro-1H-1,4-diazepin-1-yl]methyl}phosphonate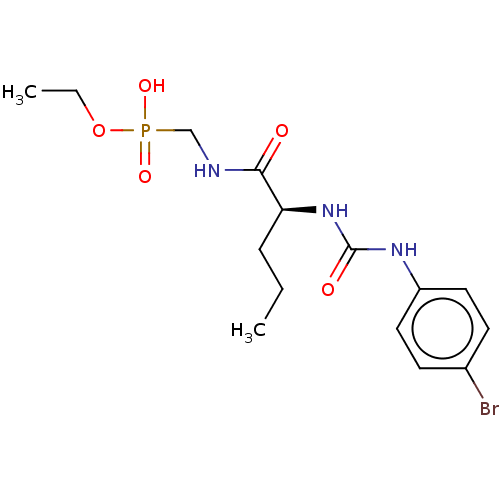 US10799518, Compound 123 US10434112, Compound 123 BDBM350661 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino} pentanoyl]amino}methyl) phosphonate US10208071, Compound 123
US10799518, Compound 123 US10434112, Compound 123 BDBM350661 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino} pentanoyl]amino}methyl) phosphonate US10208071, Compound 123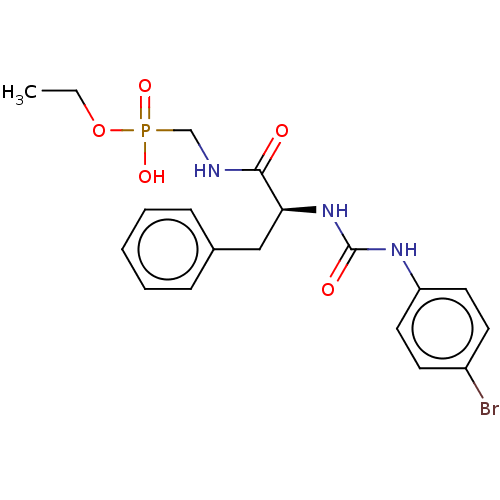 BDBM350664 US10208071, Compound 126 US10434112, Compound 126 US10799518, Compound 126 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino}- 3-phenylpropanoyl]amino} methyl)phosphonate
BDBM350664 US10208071, Compound 126 US10434112, Compound 126 US10799518, Compound 126 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino}- 3-phenylpropanoyl]amino} methyl)phosphonate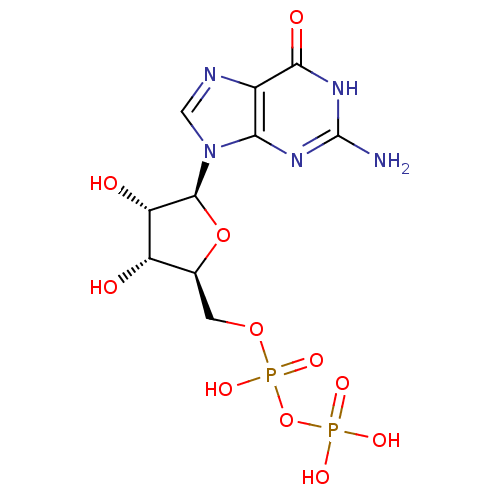 BDBM50300554 ((2S,3R,4S,5S)-5-(2-amino-6-oxo-1H-purin-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen diphosphate CHEMBL574200
BDBM50300554 ((2S,3R,4S,5S)-5-(2-amino-6-oxo-1H-purin-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen diphosphate CHEMBL574200 NaHCO3 sodium hydrogen carbonate bicarbonate of soda SODIUM BICARBONATE BDBM50155533 carbonic acid monosodium salt sodium hydrogencarbonate CHEMBL1353 sodium acid carbonate baking soda Natriumhydrogenkarbonat
NaHCO3 sodium hydrogen carbonate bicarbonate of soda SODIUM BICARBONATE BDBM50155533 carbonic acid monosodium salt sodium hydrogencarbonate CHEMBL1353 sodium acid carbonate baking soda Natriumhydrogenkarbonat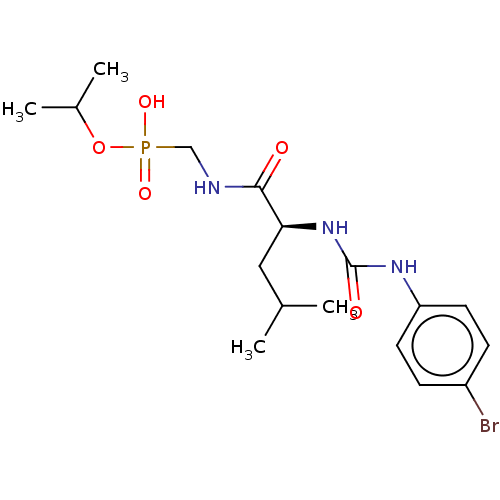 US10208071, Compound 129 BDBM350667 US10799518, Compound 129 US10434112, Compound 129 propan-2-yl hydrogen {[(2- {[(4- bromophenyl)carbamoyl]amino} pentanoyl)amino]methyl} phosphonate
US10208071, Compound 129 BDBM350667 US10799518, Compound 129 US10434112, Compound 129 propan-2-yl hydrogen {[(2- {[(4- bromophenyl)carbamoyl]amino} pentanoyl)amino]methyl} phosphonate US10799518, Compound 105 US10434112, Compound 105 BDBM350643 US10208071, Compound 105 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino}- 4-methylpentanoyl]amino} methyl)phosphonate
US10799518, Compound 105 US10434112, Compound 105 BDBM350643 US10208071, Compound 105 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino}- 4-methylpentanoyl]amino} methyl)phosphonate hydrogen ({[5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphonato}oxy)phosphonate BDBM50096295 Uridine dinucleoside 5'-polyphosphate analogue Uridine
hydrogen ({[5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphonato}oxy)phosphonate BDBM50096295 Uridine dinucleoside 5'-polyphosphate analogue Uridine 4-(2-Amino-propyl)-2,5-dimethoxy-benzoic acid propyl ester 4-(2-Amino-propyl)-2,5-dimethoxy-benzoic acid propyl ester : (Hydrogen oxalate) BDBM50005263 CHEMBL273251
4-(2-Amino-propyl)-2,5-dimethoxy-benzoic acid propyl ester 4-(2-Amino-propyl)-2,5-dimethoxy-benzoic acid propyl ester : (Hydrogen oxalate) BDBM50005263 CHEMBL273251 BDBM350653 US10208071, Compound 115 US10434112, Compound 115 US10799518, Compound 115 ethyl hydrogen ({[(2S,3S)-2- {[(4- bromophenyl)carbamoyl]amino}- 3-methylpentanoyl]amino} methyl)phosphonate
BDBM350653 US10208071, Compound 115 US10434112, Compound 115 US10799518, Compound 115 ethyl hydrogen ({[(2S,3S)-2- {[(4- bromophenyl)carbamoyl]amino}- 3-methylpentanoyl]amino} methyl)phosphonate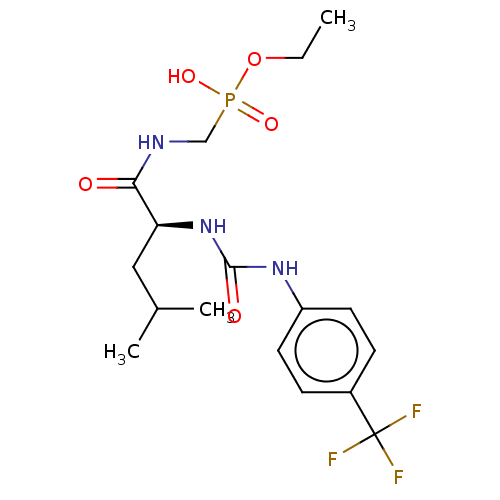 BDBM350673 US10434112, Compound 135 US10799518, Compound 135 US10208071, Compound 135 ethyl hydrogen ({[(2S)-4- methyl-2-({[4- (trifluoromethyl)phenyl]carbamoyl} amino)pentanoyl]amino} methyl)phosphonate
BDBM350673 US10434112, Compound 135 US10799518, Compound 135 US10208071, Compound 135 ethyl hydrogen ({[(2S)-4- methyl-2-({[4- (trifluoromethyl)phenyl]carbamoyl} amino)pentanoyl]amino} methyl)phosphonate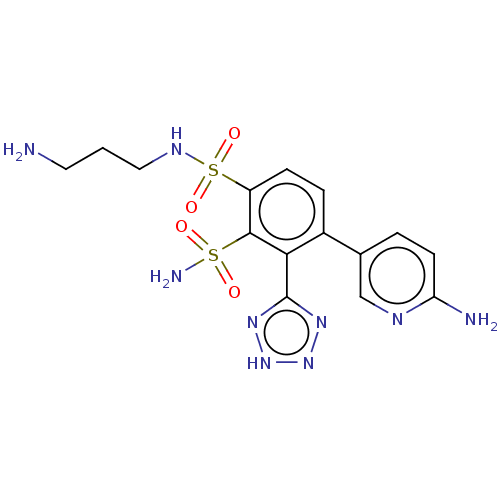 US10544130, Example 496 US10221163, Example 496 BDBM361339 N1-(3-aminopropyl)-4-(6- aminopyridin-3-yl)-3-(2H- tetrazol-5-yl)benzene-1,2- disulfonamide hydrogen chloride
US10544130, Example 496 US10221163, Example 496 BDBM361339 N1-(3-aminopropyl)-4-(6- aminopyridin-3-yl)-3-(2H- tetrazol-5-yl)benzene-1,2- disulfonamide hydrogen chloride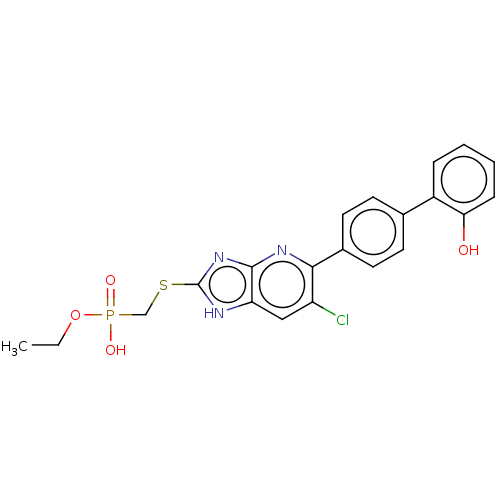 ethyl hydrogen (((6-chloro-5-(2'-hydroxy- [1,1'-biphenyl]-4-yl)-1H-imidazo[4,5- b]pyridin-2-yl)thio)methyl)phosphonate BDBM543472 US11279702, Compound 3
ethyl hydrogen (((6-chloro-5-(2'-hydroxy- [1,1'-biphenyl]-4-yl)-1H-imidazo[4,5- b]pyridin-2-yl)thio)methyl)phosphonate BDBM543472 US11279702, Compound 3 [5-[5-fluoranyl-2,4-bis(oxidanylidene)pyrimidin-1-yl]-3-oxidanyl-oxolan-2-yl]methyl [5-[4-(octadecylamino)-2-oxidanylidene-pyrimidin-1-yl]-3,4-bis(oxidanyl)oxolan-2-yl]methyl hydrogen phosphate cid_392613 SMR001565974 [3,4-dihydroxy-5-[2-keto-4-(stearylamino)pyrimidin-1-yl]tetrahydrofuran-2-yl]methyl [5-(5-fluoro-2,4-diketo-pyrimidin-1-yl)-3-hydroxy-tetrahydrofuran-2-yl]methyl hydrogen phosphate MLS002702412 [3,4-dihydroxy-5-[4-(octadecylamino)-2-oxopyrimidin-1-yl]oxolan-2-yl]methyl [5-(5-fluoro-2,4-dioxopyrimidin-1-yl)-3-hydroxyoxolan-2-yl]methyl hydrogen phosphate [3,4-dihydroxy-5-[4-(octadecylamino)-2-oxo-1-pyrimidinyl]-2-oxolanyl]methyl [5-(5-fluoro-2,4-dioxo-1-pyrimidinyl)-3-hydroxy-2-oxolanyl]methyl hydrogen phosphate BDBM80994
[5-[5-fluoranyl-2,4-bis(oxidanylidene)pyrimidin-1-yl]-3-oxidanyl-oxolan-2-yl]methyl [5-[4-(octadecylamino)-2-oxidanylidene-pyrimidin-1-yl]-3,4-bis(oxidanyl)oxolan-2-yl]methyl hydrogen phosphate cid_392613 SMR001565974 [3,4-dihydroxy-5-[2-keto-4-(stearylamino)pyrimidin-1-yl]tetrahydrofuran-2-yl]methyl [5-(5-fluoro-2,4-diketo-pyrimidin-1-yl)-3-hydroxy-tetrahydrofuran-2-yl]methyl hydrogen phosphate MLS002702412 [3,4-dihydroxy-5-[4-(octadecylamino)-2-oxopyrimidin-1-yl]oxolan-2-yl]methyl [5-(5-fluoro-2,4-dioxopyrimidin-1-yl)-3-hydroxyoxolan-2-yl]methyl hydrogen phosphate [3,4-dihydroxy-5-[4-(octadecylamino)-2-oxo-1-pyrimidinyl]-2-oxolanyl]methyl [5-(5-fluoro-2,4-dioxo-1-pyrimidinyl)-3-hydroxy-2-oxolanyl]methyl hydrogen phosphate BDBM80994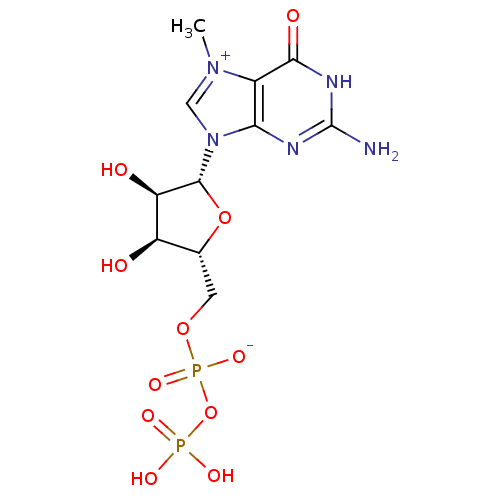 ((2R,3S,4R,5R)-5-(2-amino-7-methyl-6-oxo-1H-purin-1-ium-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen diphosphate CHEMBL1094974 BDBM50316303
((2R,3S,4R,5R)-5-(2-amino-7-methyl-6-oxo-1H-purin-1-ium-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen diphosphate CHEMBL1094974 BDBM50316303 BDBM268149 US9549932, 181 US10772893, Example 181 US11529356, Example 181 Methyl hydrogen {4-[2-(morpholin-4-yl)-8-(1H-pyrazol-5-yl)-1,7-naphthyridin-4-yl]phenyl}phosphonate
BDBM268149 US9549932, 181 US10772893, Example 181 US11529356, Example 181 Methyl hydrogen {4-[2-(morpholin-4-yl)-8-(1H-pyrazol-5-yl)-1,7-naphthyridin-4-yl]phenyl}phosphonate CHEMBL1094973 ((2R,3S,4R,5R)-5-(2-amino-7-methyl-6-oxo-1H-purin-1-ium-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen triphosphate BDBM50316302
CHEMBL1094973 ((2R,3S,4R,5R)-5-(2-amino-7-methyl-6-oxo-1H-purin-1-ium-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen triphosphate BDBM50316302 2-Amino-4-[1-({[3-(4-amino-butylamino)-propoxy]-hydroxy-phosphorylmethyl}-carbamoyl)-ethylcarbamoyl]-butyric acid BDBM50060962 Hydrogen O-[3-[N-(4-Aminobutyl)amino]propyl][[(glutamylalanyl) amino]methyl]phosphonate
2-Amino-4-[1-({[3-(4-amino-butylamino)-propoxy]-hydroxy-phosphorylmethyl}-carbamoyl)-ethylcarbamoyl]-butyric acid BDBM50060962 Hydrogen O-[3-[N-(4-Aminobutyl)amino]propyl][[(glutamylalanyl) amino]methyl]phosphonate CHEMBL1256967 BDBM50327179 (S)-3-(2-(2-aminothiazol-4-yl)-2-((1,5-dihydroxy-4-oxo-1,4-dihydropyridin-2-yl)methoxyimino)acetamido)-2,2-dimethyl-4-oxoazetidin-1-yl hydrogen sulfate
CHEMBL1256967 BDBM50327179 (S)-3-(2-(2-aminothiazol-4-yl)-2-((1,5-dihydroxy-4-oxo-1,4-dihydropyridin-2-yl)methoxyimino)acetamido)-2,2-dimethyl-4-oxoazetidin-1-yl hydrogen sulfate CHEMBL19259 CHEMBL1788135 BDBM50004337 (4-Fluoro-phenyl)-[1-(4-phenyl-butyl)-piperidin-4-yl]-methanone (4-Fluoro-phenyl)-[1-(4-phenyl-butyl)-piperidin-4-yl]-methanone; hydrogen oxalate salt
CHEMBL19259 CHEMBL1788135 BDBM50004337 (4-Fluoro-phenyl)-[1-(4-phenyl-butyl)-piperidin-4-yl]-methanone (4-Fluoro-phenyl)-[1-(4-phenyl-butyl)-piperidin-4-yl]-methanone; hydrogen oxalate salt US10544130, Example 497 US10221163, Example 497 4-(6-aminopyridin-3-yl)- 1-N-(3-methylazetidin-3- yl)-3-(2H-1,2,3,4-tetrazol- 5-yl)benzene-1,2- disulfonamide hydrogen BDBM361340
US10544130, Example 497 US10221163, Example 497 4-(6-aminopyridin-3-yl)- 1-N-(3-methylazetidin-3- yl)-3-(2H-1,2,3,4-tetrazol- 5-yl)benzene-1,2- disulfonamide hydrogen BDBM361340 ethyl hydrogen 1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)- 4-fluoropyrrolidin-1- yl)-2-oxoethyl)-1H- pyrazolo[3,4-e]pyridin- 3-ylphosphonate BDBM340257 US9758537, Compound 60
ethyl hydrogen 1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)- 4-fluoropyrrolidin-1- yl)-2-oxoethyl)-1H- pyrazolo[3,4-e]pyridin- 3-ylphosphonate BDBM340257 US9758537, Compound 60 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (2-(octyloxy)ethyl) hydrogen phosphate (26) WO2022081973, Example 26 BDBM534222
((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (2-(octyloxy)ethyl) hydrogen phosphate (26) WO2022081973, Example 26 BDBM534222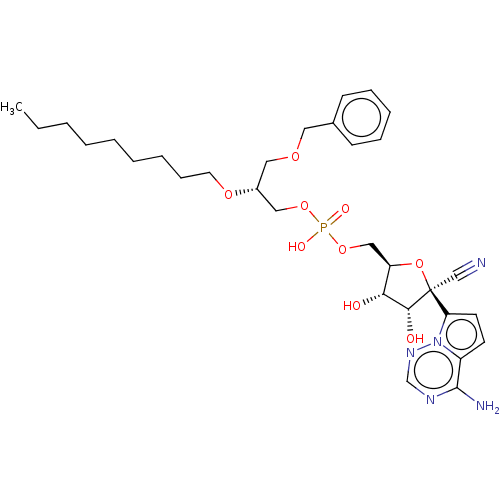 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (3-(nonyloxy)propyl) hydrogen phosphate (25) BDBM534221 WO2022081973, Example 25
((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (3-(nonyloxy)propyl) hydrogen phosphate (25) BDBM534221 WO2022081973, Example 25 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl (2-(octadecyloxy)ethyl) hydrogen phosphate (21) BDBM534217 WO2022081973, Example 21
((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl (2-(octadecyloxy)ethyl) hydrogen phosphate (21) BDBM534217 WO2022081973, Example 21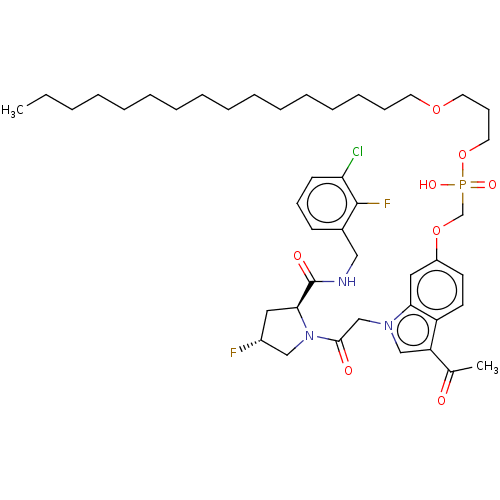 BDBM335530 US9732104, Compound 20 3-(hexadecyloxy)propyl hydrogen (3-acetyl-1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)- 4-fluoropyrrolidin-1-yl)- 2-oxoethyl)-1H-indol-6- yloxy)methylphosphonate
BDBM335530 US9732104, Compound 20 3-(hexadecyloxy)propyl hydrogen (3-acetyl-1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)- 4-fluoropyrrolidin-1-yl)- 2-oxoethyl)-1H-indol-6- yloxy)methylphosphonate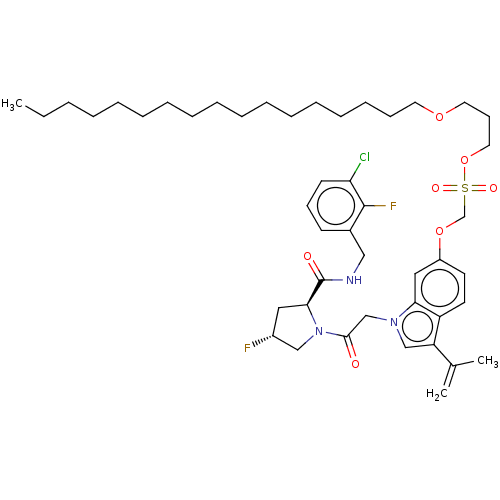 US10550140, Compound 20 BDBM431954 3- (hexadecyloxy)propyl hydrogen (3-acetyl-1- (2-((2S,4R)-2-(3- chloro-2- fluorobenzylcarbamoyl)- 4- fluoropyrrolidin-1- yl)-2-oxoethyl)-1H- indol-6- yloxy) methylphosphonate
US10550140, Compound 20 BDBM431954 3- (hexadecyloxy)propyl hydrogen (3-acetyl-1- (2-((2S,4R)-2-(3- chloro-2- fluorobenzylcarbamoyl)- 4- fluoropyrrolidin-1- yl)-2-oxoethyl)-1H- indol-6- yloxy) methylphosphonate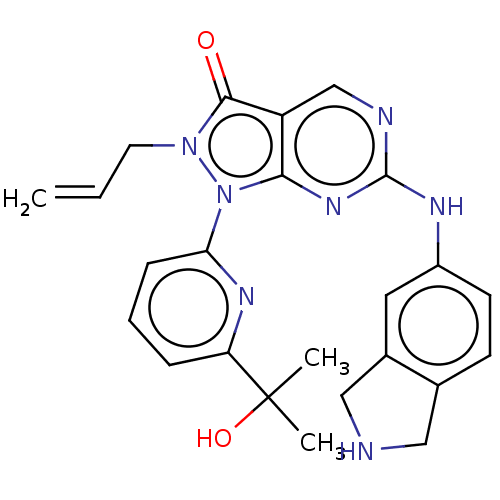 US11124518, Example 21 2-Allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-(isoindolin-5-ylamino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518547
US11124518, Example 21 2-Allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-(isoindolin-5-ylamino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518547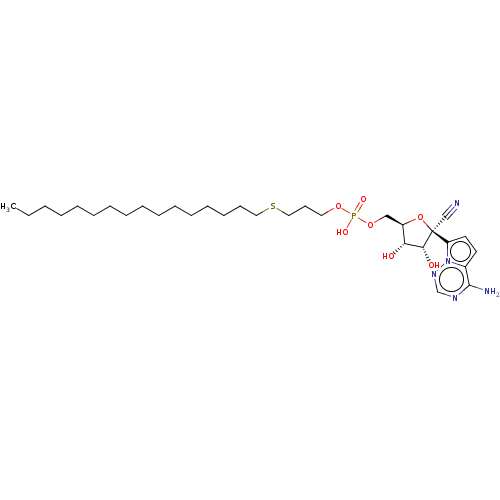 WO2022081973, Example 19 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (3-(hexadecylthio)propyl) hydrogen phosphate (19) BDBM534215
WO2022081973, Example 19 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (3-(hexadecylthio)propyl) hydrogen phosphate (19) BDBM534215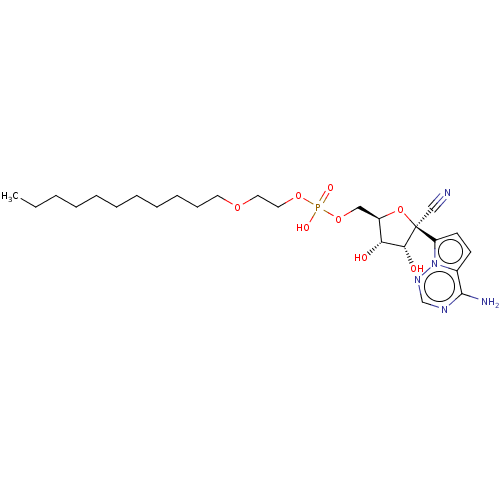 WO2022081973, Example 27 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (2-(undecyloxy)ethyl) hydrogen phosphate (27) BDBM534223
WO2022081973, Example 27 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (2-(undecyloxy)ethyl) hydrogen phosphate (27) BDBM534223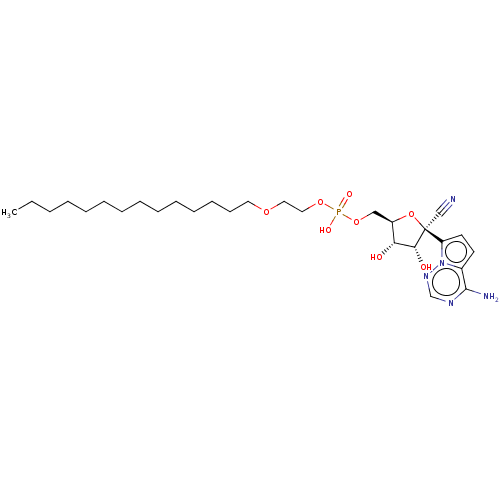 WO2022081973, Example 59 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-tetradecoxyethyl hydrogen phosphate (59) BDBM534255
WO2022081973, Example 59 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-tetradecoxyethyl hydrogen phosphate (59) BDBM534255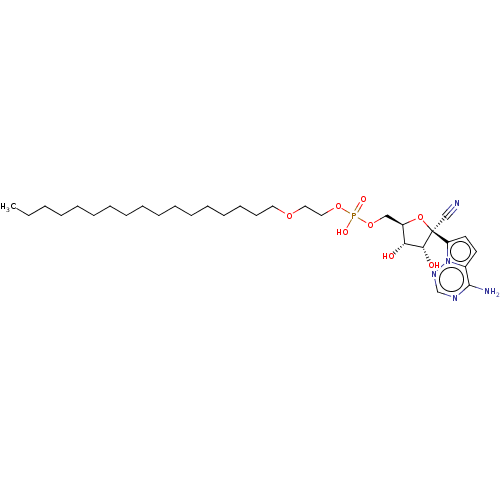 WO2022081973, Example 60 BDBM534256 [(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-heptadecoxyethyl hydrogen phosphate (60)
WO2022081973, Example 60 BDBM534256 [(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-heptadecoxyethyl hydrogen phosphate (60)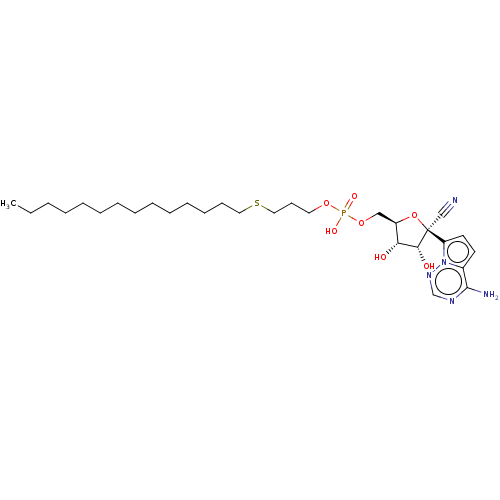 WO2022081973, Example 61 [(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-tetradecylsulfanylpropyl hydrogen phosphate (61) BDBM534257
WO2022081973, Example 61 [(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-tetradecylsulfanylpropyl hydrogen phosphate (61) BDBM534257 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-hexadecoxyethyl hydrogen phosphate (66) BDBM534262 WO2022081973, Example 66
[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-hexadecoxyethyl hydrogen phosphate (66) BDBM534262 WO2022081973, Example 66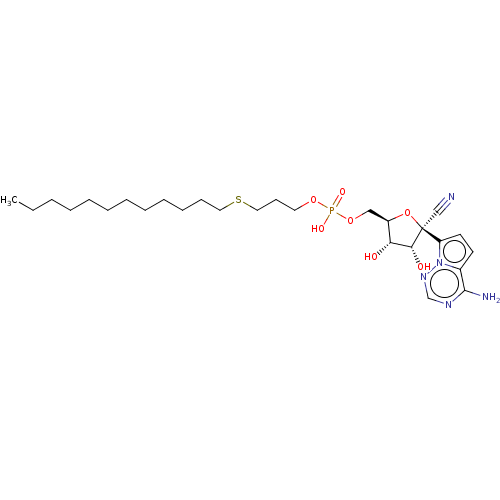 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-dodecylsulfanylpropyl hydrogen phosphate (63) BDBM534259 WO2022081973, Example 63
[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-dodecylsulfanylpropyl hydrogen phosphate (63) BDBM534259 WO2022081973, Example 63 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-hexadecylsulfanylpropyl hydrogen phosphate (65) BDBM534261 WO2022081973, Example 65
[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-hexadecylsulfanylpropyl hydrogen phosphate (65) BDBM534261 WO2022081973, Example 65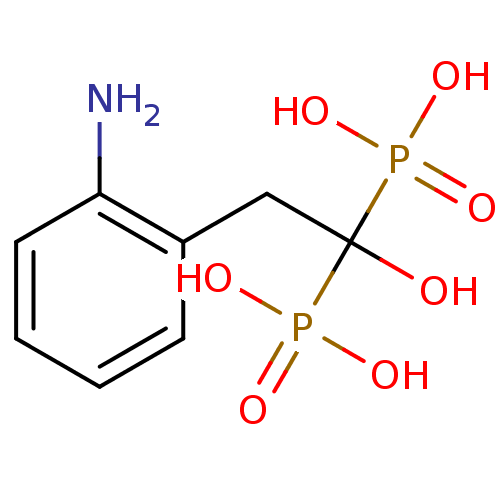 [2-(2-amino-phenyl)-1-hydroxy-1-phosphono-ethyl]-phosphonic acid CHEMBL414849 2-(2-aminophenyl)-1-hydroxyethane-1,1-diyldiphosphonic acid BDBM50173792 hydrogen [2-(2-azaniumylphenyl)-1-hydroxy-1-phosphonatoethyl]phosphonate
[2-(2-amino-phenyl)-1-hydroxy-1-phosphono-ethyl]-phosphonic acid CHEMBL414849 2-(2-aminophenyl)-1-hydroxyethane-1,1-diyldiphosphonic acid BDBM50173792 hydrogen [2-(2-azaniumylphenyl)-1-hydroxy-1-phosphonatoethyl]phosphonate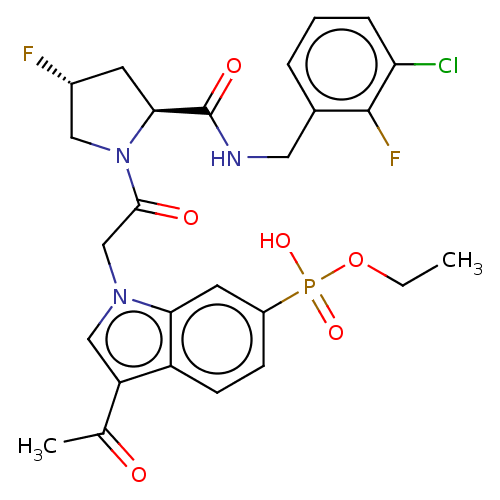 BDBM330226 ethyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate US9663543, Compound 2 US10301336, Comp No. 2
BDBM330226 ethyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate US9663543, Compound 2 US10301336, Comp No. 2 BDBM517113 2-((4-(2-fluoro-4-(2-(4-fluorophenyl)-3-oxo-2,3-dihydropyridazine-4-carboxamido)phenoxy)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)-2-methylpropyl hydrogen sulfate US11104676, Example 183
BDBM517113 2-((4-(2-fluoro-4-(2-(4-fluorophenyl)-3-oxo-2,3-dihydropyridazine-4-carboxamido)phenoxy)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)-2-methylpropyl hydrogen sulfate US11104676, Example 183 WO2022081973, Example 72 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyloxy)henicosyl) hydrogen phosphate (72) BDBM534269
WO2022081973, Example 72 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyloxy)henicosyl) hydrogen phosphate (72) BDBM534269 butyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate BDBM330245 US10301336, Comp No. 21 US9663543, Compound 21
butyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate BDBM330245 US10301336, Comp No. 21 US9663543, Compound 21 ethyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-5- ylphosphonate US10301336, Comp No. 9 BDBM330233 US9663543, Compound 9
ethyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-5- ylphosphonate US10301336, Comp No. 9 BDBM330233 US9663543, Compound 9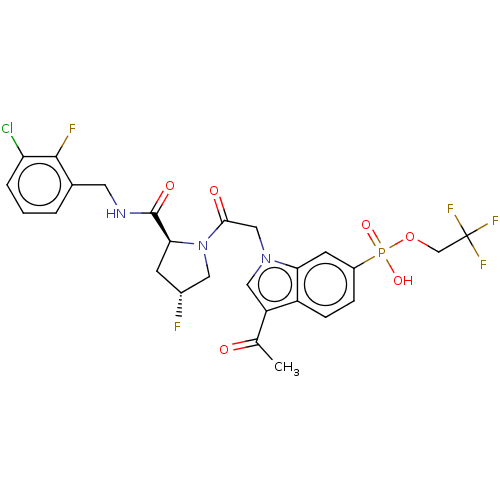 2,2,2-trifluoroethyl hydrogen 3-acetyl-1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate BDBM330253 US10301336, Comp No. 29 US9663543, Compound 29
2,2,2-trifluoroethyl hydrogen 3-acetyl-1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate BDBM330253 US10301336, Comp No. 29 US9663543, Compound 29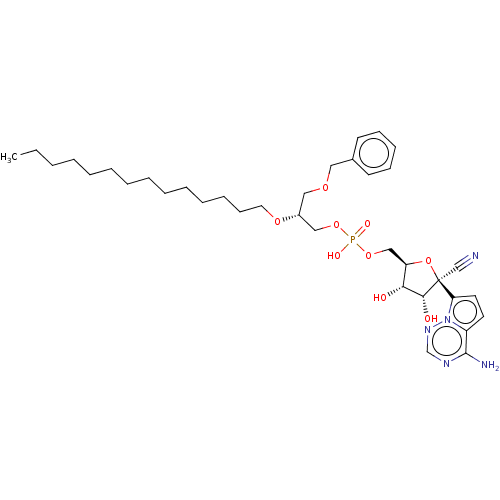 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(tetradecyloxy)propyl) hydrogen phosphate (24) BDBM534220 WO2022081973, Example 24
((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(tetradecyloxy)propyl) hydrogen phosphate (24) BDBM534220 WO2022081973, Example 24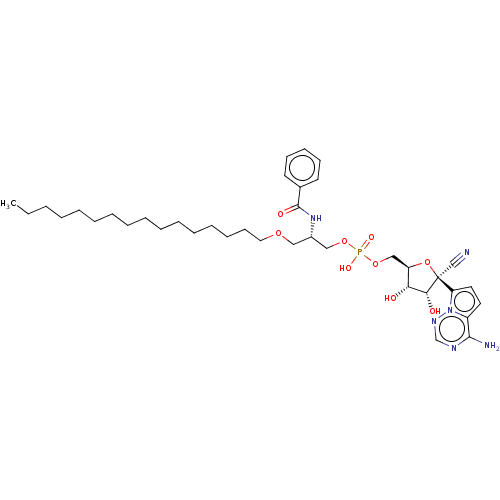 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-benzamido-3-(octadecyloxy)propyl) hydrogen phosphate (30) BDBM534226 WO2022081973, Example 30
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-benzamido-3-(octadecyloxy)propyl) hydrogen phosphate (30) BDBM534226 WO2022081973, Example 30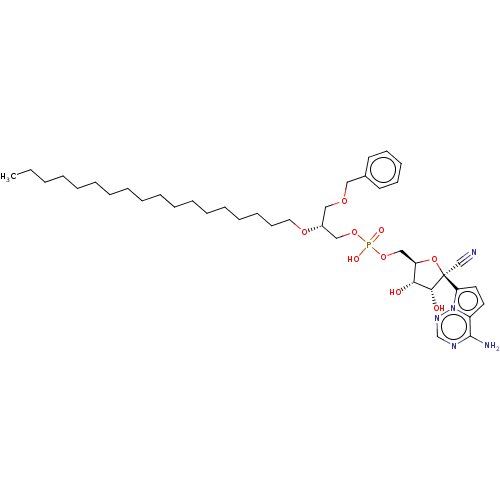 BDBM534218 WO2022081973, Example 22 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(octadecyloxy)propyl) hydrogen phosphate (22)
BDBM534218 WO2022081973, Example 22 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(octadecyloxy)propyl) hydrogen phosphate (22)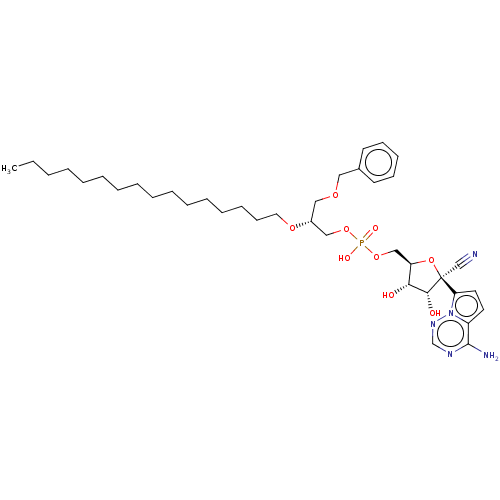 BDBM534219 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(hexadecyloxy)propyl) hydrogen phosphate (23) WO2022081973, Example 23
BDBM534219 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(hexadecyloxy)propyl) hydrogen phosphate (23) WO2022081973, Example 23 US20230357239, Example 26 1-{4-[2-methyl-4-({(1R)-1-[2-methyl-3-(trifluoromethyl)phenyl]ethyl}amino)pyrido[2,3-d]pyrimidin-6-yl]-3,6-dihydropyridin-1(2H)-yl}ethan-1-one hydrogen BDBM634058
US20230357239, Example 26 1-{4-[2-methyl-4-({(1R)-1-[2-methyl-3-(trifluoromethyl)phenyl]ethyl}amino)pyrido[2,3-d]pyrimidin-6-yl]-3,6-dihydropyridin-1(2H)-yl}ethan-1-one hydrogen BDBM634058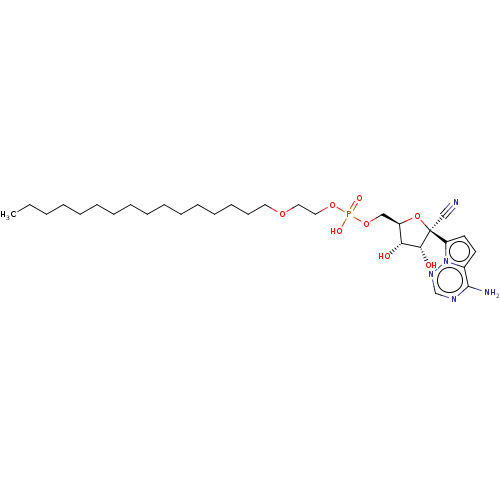 WO2022081973, Example 66 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl (2-(hexadecyloxy)ethyl) hydrogen phosphate (20) BDBM534216 WO2022081973, Example 20
WO2022081973, Example 66 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl (2-(hexadecyloxy)ethyl) hydrogen phosphate (20) BDBM534216 WO2022081973, Example 20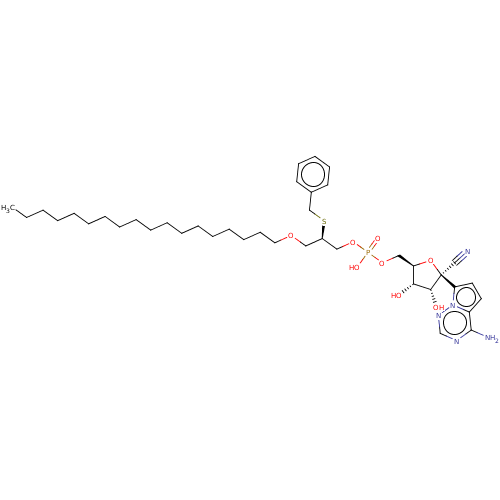 WO2022081973, Example 7 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzylthio)-3-(octadecyloxy)propyl) hydrogen phosphate: (7) BDBM534203
WO2022081973, Example 7 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzylthio)-3-(octadecyloxy)propyl) hydrogen phosphate: (7) BDBM534203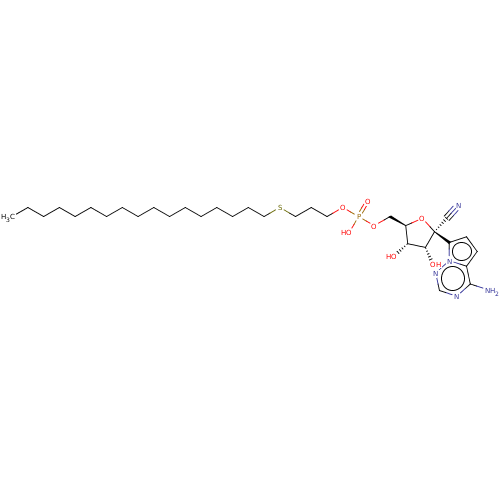 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-heptadecylsulfanylpropyl hydrogen phosphate (62) BDBM534258 WO2022081973, Example 62 WO2022081973, Example 65
[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-heptadecylsulfanylpropyl hydrogen phosphate (62) BDBM534258 WO2022081973, Example 62 WO2022081973, Example 65 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((2S,3R)-2-(benzyloxy)-3-(octadecyloxy)butyl) hydrogen phosphate (8) BDBM534204 WO2022081973, Example 8
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((2S,3R)-2-(benzyloxy)-3-(octadecyloxy)butyl) hydrogen phosphate (8) BDBM534204 WO2022081973, Example 8 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-(octadecyloxy)-3-phenoxypropan-2-yl) hydrogen phosphate (18) WO2022081973, Example 18 BDBM534214
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-(octadecyloxy)-3-phenoxypropan-2-yl) hydrogen phosphate (18) WO2022081973, Example 18 BDBM534214 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(3-cyanobenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (57) BDBM534253 WO2022081973, Example 57
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(3-cyanobenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (57) BDBM534253 WO2022081973, Example 57 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-1-(octadecyloxy)-3-phenylpropan-2-yl) hydrogen phosphate (40) BDBM534236 WO2022081973, Example 40
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-1-(octadecyloxy)-3-phenylpropan-2-yl) hydrogen phosphate (40) BDBM534236 WO2022081973, Example 40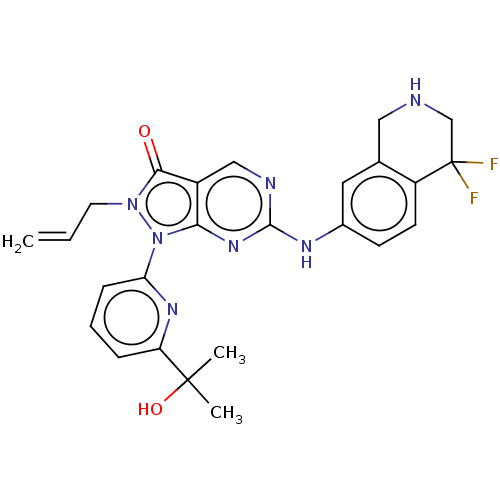 2-Allyl-6-((4,4-difluoro-1,2,3,4-tetrahydroisoquinolin-7-yl)amino)-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518554 US11124518, Example 28
2-Allyl-6-((4,4-difluoro-1,2,3,4-tetrahydroisoquinolin-7-yl)amino)-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518554 US11124518, Example 28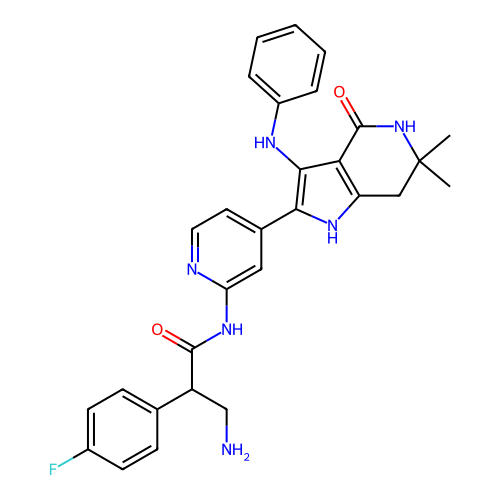 3-amino-N-[4-(3-anilino-6,6-dimethyl-4-oxo-4,5,6,7-tetrahydro-1H-pyrrolo[3,2-c]pyridin-2-yl)pyridin-2-yl]-2-(4-fluorophenyl)propanamide-hydrogen chloride salt (Racemate) BDBM721181 US12227501, Example 139
3-amino-N-[4-(3-anilino-6,6-dimethyl-4-oxo-4,5,6,7-tetrahydro-1H-pyrrolo[3,2-c]pyridin-2-yl)pyridin-2-yl]-2-(4-fluorophenyl)propanamide-hydrogen chloride salt (Racemate) BDBM721181 US12227501, Example 139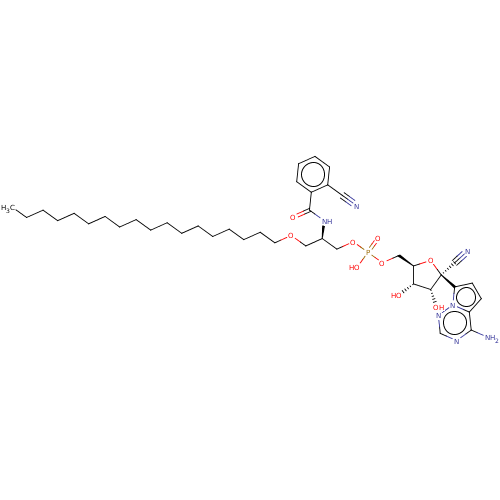 BDBM534254 WO2022081973, Example 58 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(2-cyanobenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (58)
BDBM534254 WO2022081973, Example 58 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(2-cyanobenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (58)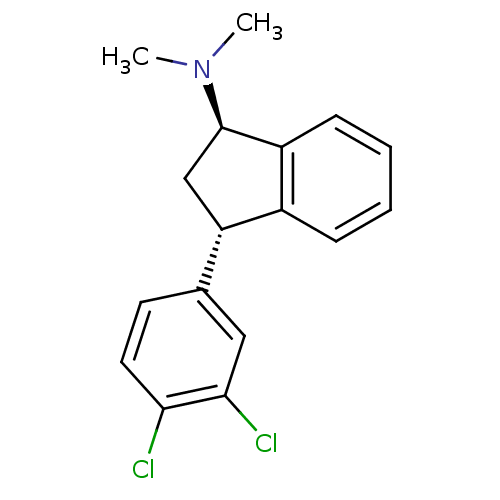 CHEMBL356750 [3-(3,4-Dichloro-phenyl)-indan-1-yl]-ethyl-amine;chloride [3-(3,4-Dichloro-phenyl)-indan-1-yl]-dimethyl-amine;hydrogen maleate tert-Butyl-[3-(3,4-dichloro-phenyl)-indan-1-yl]-amine;chloride BDBM50095611
CHEMBL356750 [3-(3,4-Dichloro-phenyl)-indan-1-yl]-ethyl-amine;chloride [3-(3,4-Dichloro-phenyl)-indan-1-yl]-dimethyl-amine;hydrogen maleate tert-Butyl-[3-(3,4-dichloro-phenyl)-indan-1-yl]-amine;chloride BDBM50095611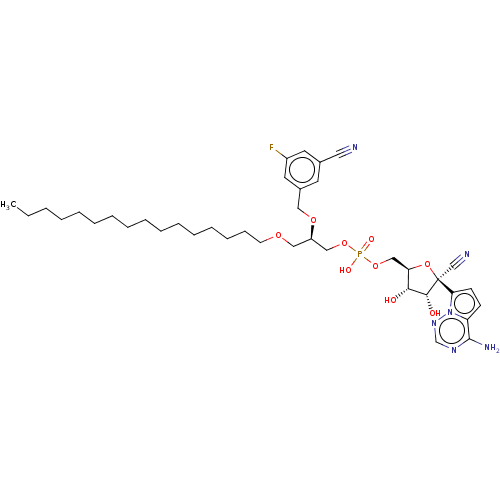 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)henicosyl) hydrogen phosphate (35) BDBM534231 WO2022081973, Example 35
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)henicosyl) hydrogen phosphate (35) BDBM534231 WO2022081973, Example 35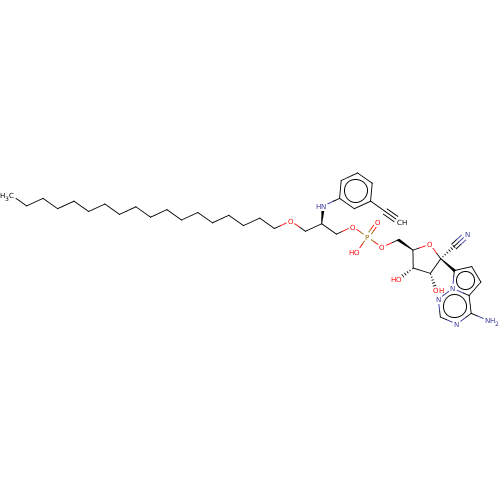 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyanophenyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (16) WO2022081973, Example 16 BDBM534212
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyanophenyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (16) WO2022081973, Example 16 BDBM534212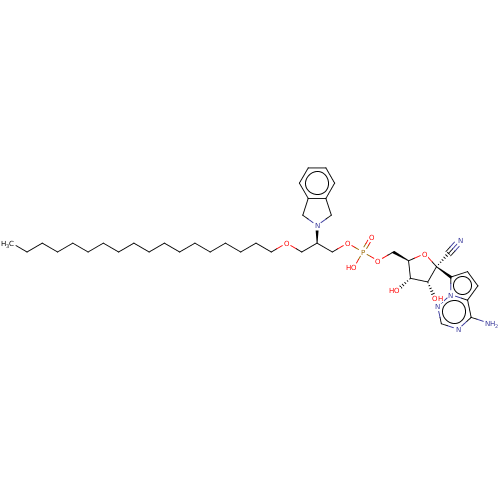 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(isoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (1) WO2022081973, Example 1 BDBM534197
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(isoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (1) WO2022081973, Example 1 BDBM534197 (methylidyneammoniumyl)oxidanide H-C#NO formonitrile oxideformonitrile-N-oxidehydrido(nitrosyl-kappaN)carbonhydrido(oxidonitrato-N)carbon [C(H)NO] hydrogen cyanide N-oxide fulminic acid methylidyne(oxo)-lambda(5)-azane BDBM50152965 [CH(NO)] CHEMBL185198 HCNO Knallsaeure
(methylidyneammoniumyl)oxidanide H-C#NO formonitrile oxideformonitrile-N-oxidehydrido(nitrosyl-kappaN)carbonhydrido(oxidonitrato-N)carbon [C(H)NO] hydrogen cyanide N-oxide fulminic acid methylidyne(oxo)-lambda(5)-azane BDBM50152965 [CH(NO)] CHEMBL185198 HCNO Knallsaeure BDBM534198 WO2022081973, Example 2 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyl(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (2)
BDBM534198 WO2022081973, Example 2 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyl(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (2) BDBM534208 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((3R)-3-(benzyloxy)-4-(octadecyloxy)butan-2-yl) hydrogen phosphate (12) WO2022081973, Example 12
BDBM534208 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((3R)-3-(benzyloxy)-4-(octadecyloxy)butan-2-yl) hydrogen phosphate (12) WO2022081973, Example 12 BDBM534209 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((3R)-3-(benzyloxy)-4-(octadecyloxy)butan-2-yl) hydrogen phosphate (13) WO2022081973, Example 13
BDBM534209 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((3R)-3-(benzyloxy)-4-(octadecyloxy)butan-2-yl) hydrogen phosphate (13) WO2022081973, Example 13 BDBM534270 WO2022081973, Example 73 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyloxy)nonadec-4-yn-1-yl) hydrogen phosphate (73)
BDBM534270 WO2022081973, Example 73 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyloxy)nonadec-4-yn-1-yl) hydrogen phosphate (73)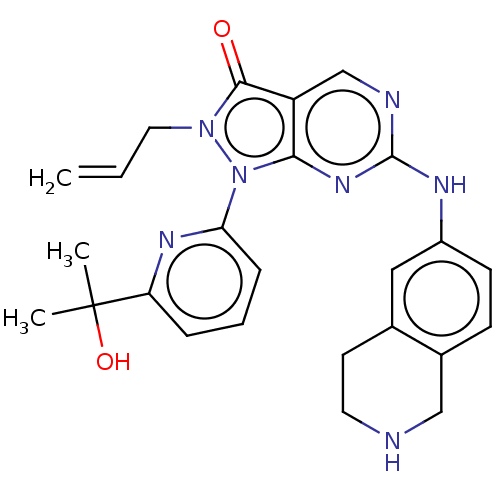 US11124518, Example 15 2-Allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-((1,2,3,4-tetrahydroisoquinolin-6-yl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518539 US11299493, Compound 1.8
US11124518, Example 15 2-Allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-((1,2,3,4-tetrahydroisoquinolin-6-yl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518539 US11299493, Compound 1.8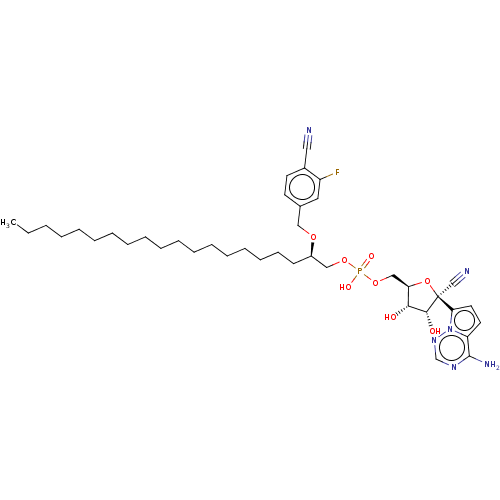 WO2022081973, Example 71 BDBM534268 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3-fluorobenzyl)oxy)henicosyl) hydrogen phosphate (71)
WO2022081973, Example 71 BDBM534268 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3-fluorobenzyl)oxy)henicosyl) hydrogen phosphate (71)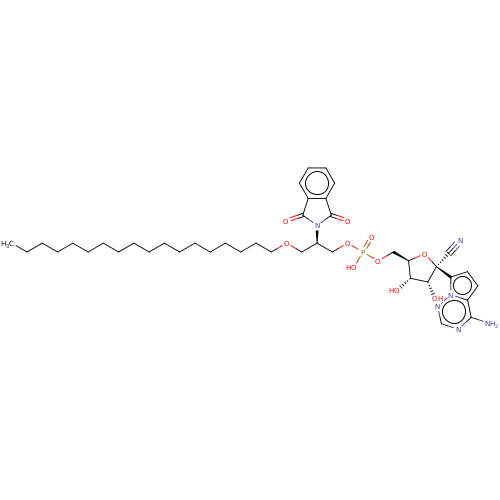 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(1,3-dioxoisoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (15) WO2022081973, Example 15 BDBM534211
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(1,3-dioxoisoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (15) WO2022081973, Example 15 BDBM534211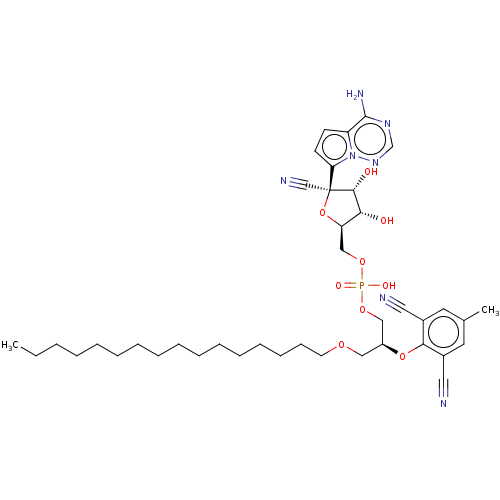 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(2,6-dicyano-4-methylphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (29) WO2022081973, Example 29 BDBM534225
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(2,6-dicyano-4-methylphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (29) WO2022081973, Example 29 BDBM534225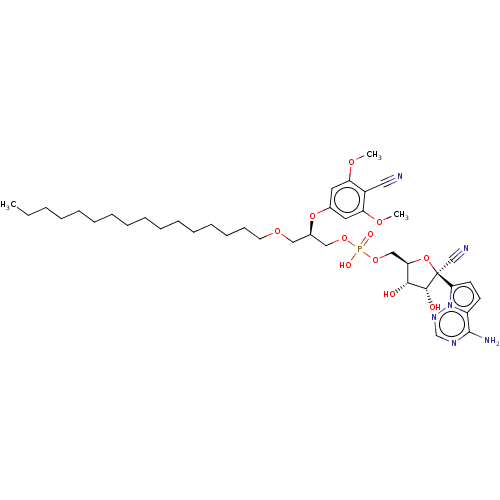 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-3,5-dimethoxyphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (34) BDBM534230 WO2022081973, Example 34
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-3,5-dimethoxyphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (34) BDBM534230 WO2022081973, Example 34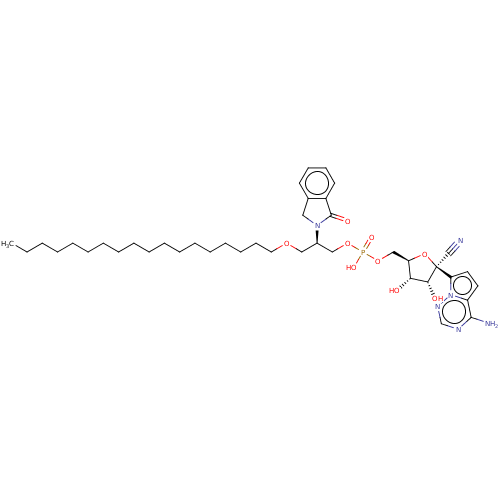 BDBM534207 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(octadecyloxy)-2-(1-oxoisoindolin-2-yl)propyl) hydrogen phosphate (11) WO2022081973, Example 11
BDBM534207 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(octadecyloxy)-2-(1-oxoisoindolin-2-yl)propyl) hydrogen phosphate (11) WO2022081973, Example 11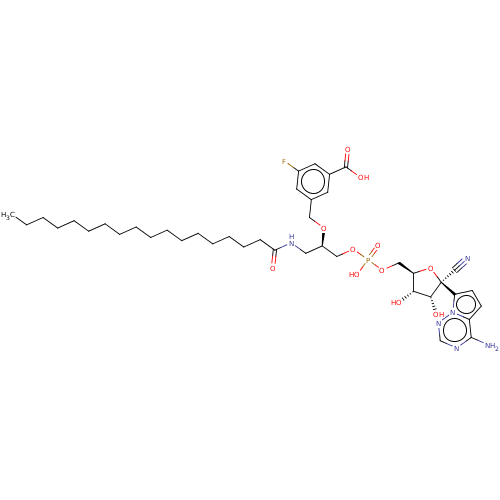 BDBM534241 WO2022081973, Example 45 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)-3-stearamidopropyl) hydrogen phosphate (45)
BDBM534241 WO2022081973, Example 45 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)-3-stearamidopropyl) hydrogen phosphate (45)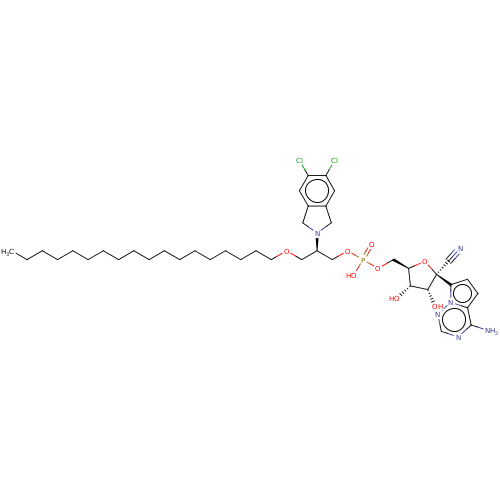 WO2022081973, Example 9 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(5,6-dichloroisoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (9) BDBM534205
WO2022081973, Example 9 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(5,6-dichloroisoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (9) BDBM534205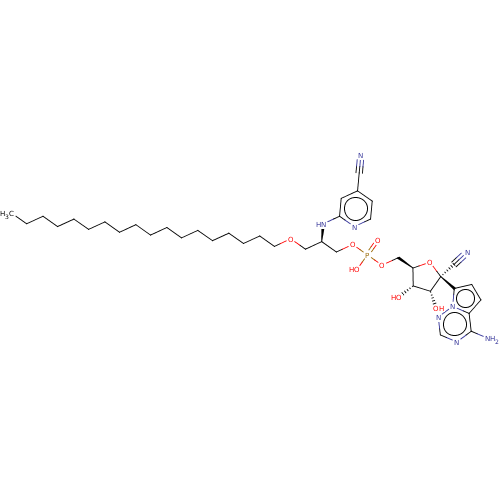 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((5-cyanopyridin-2-yl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (42) BDBM534238 WO2022081973, Example 42
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((5-cyanopyridin-2-yl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (42) BDBM534238 WO2022081973, Example 42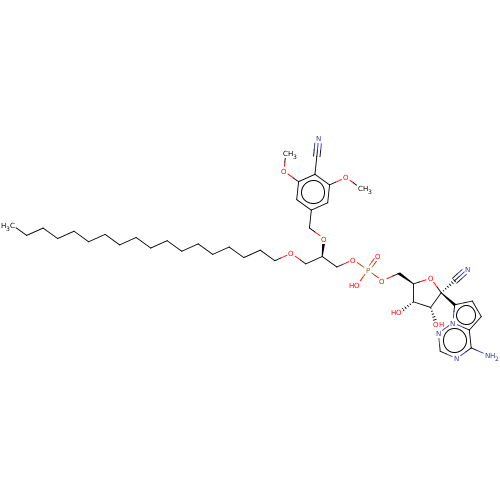 BDBM534251 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3,5-dimethoxybenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (55) WO2022081973, Example 55
BDBM534251 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3,5-dimethoxybenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (55) WO2022081973, Example 55 BDBM534263 WO2022081973, Example 67 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) O-hydrogen (R)-phosphorothioate (67)
BDBM534263 WO2022081973, Example 67 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) O-hydrogen (R)-phosphorothioate (67)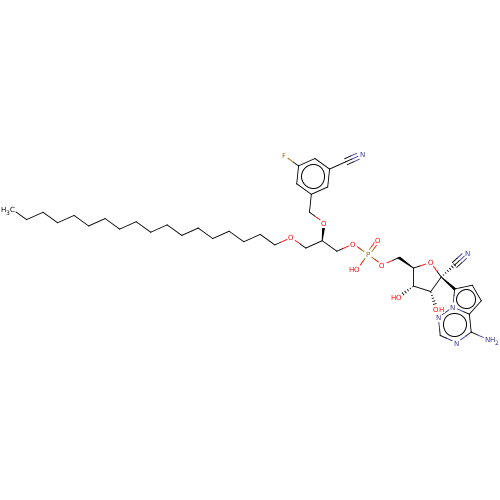 BDBM534265 WO2022081973, Example 68 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) O-hydrogen (S)-phosphorothioate (68)
BDBM534265 WO2022081973, Example 68 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) O-hydrogen (S)-phosphorothioate (68)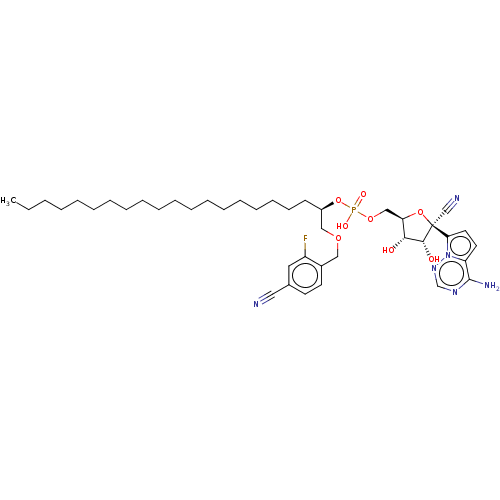 BDBM534266 WO2022081973, Example 69 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((4-cyano-2-fluorobenzyl)oxy)henicosan-2-yl) hydrogen phosphate (69)
BDBM534266 WO2022081973, Example 69 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((4-cyano-2-fluorobenzyl)oxy)henicosan-2-yl) hydrogen phosphate (69) BDBM634045 1-{6-[2-methyl-4-({(1R)-1-[2-methyl-3-(trifluoromethyl)phenyl]ethyl}amino)pyrido[2,3-d]pyrimidin-6-yl]-2,6-diazaspiro[3.3]heptan-2-yl}ethan-1-one-hydrogen chloride (1/1) US20230357239, Example 13
BDBM634045 1-{6-[2-methyl-4-({(1R)-1-[2-methyl-3-(trifluoromethyl)phenyl]ethyl}amino)pyrido[2,3-d]pyrimidin-6-yl]-2,6-diazaspiro[3.3]heptan-2-yl}ethan-1-one-hydrogen chloride (1/1) US20230357239, Example 13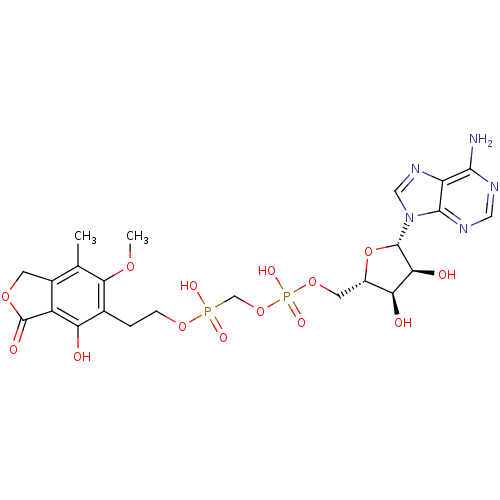 CHEMBL1683742 ((2S,3R,4S,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl(hydroxy(2-(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)ethoxy)phosphoryl)methyl hydrogen phosphate BDBM50338540
CHEMBL1683742 ((2S,3R,4S,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl(hydroxy(2-(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)ethoxy)phosphoryl)methyl hydrogen phosphate BDBM50338540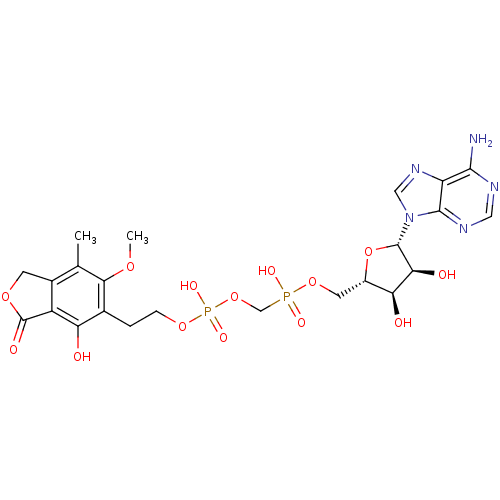 CHEMBL1683743 ((((2S,3R,4S,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(hydroxy)phosphoryl)methyl 2-(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)ethyl hydrogen phosphate BDBM50338541
CHEMBL1683743 ((((2S,3R,4S,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(hydroxy)phosphoryl)methyl 2-(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)ethyl hydrogen phosphate BDBM50338541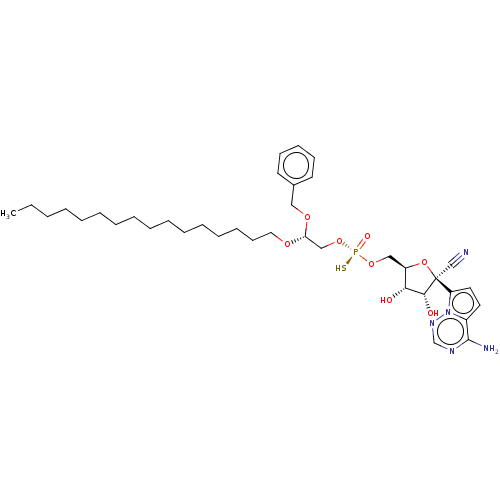 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) S- hydrogen (R)-phosphorothioate (28) WO2022081973, Example 28 BDBM534224
O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) S- hydrogen (R)-phosphorothioate (28) WO2022081973, Example 28 BDBM534224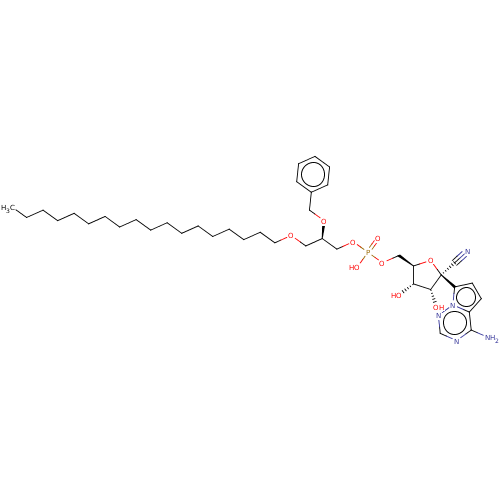 WO2022081973, Example 67 WO2022081973, Example 5 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((2S,3S)-2-(benzyloxy)-3-(octadecyloxy)butyl) hydrogen phosphate (5) BDBM534201
WO2022081973, Example 67 WO2022081973, Example 5 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((2S,3S)-2-(benzyloxy)-3-(octadecyloxy)butyl) hydrogen phosphate (5) BDBM534201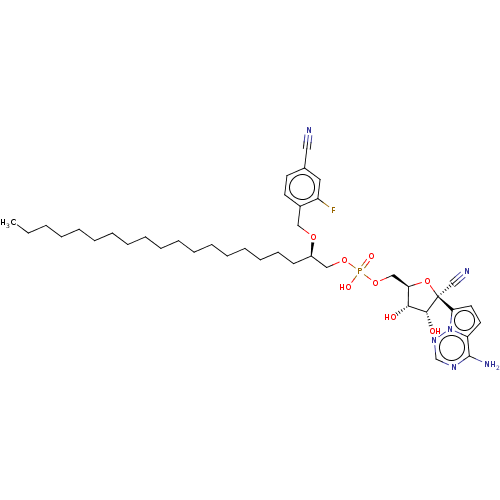 WO2022081973, Example 70 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((4-cyano-2-fluorobenzyl)oxy)henicosan-2-yl) hydrogen phosphate (70) BDBM534267
WO2022081973, Example 70 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((4-cyano-2-fluorobenzyl)oxy)henicosan-2-yl) hydrogen phosphate (70) BDBM534267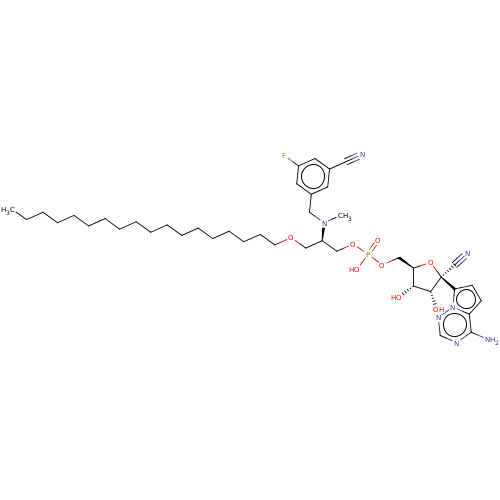 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (6) BDBM534202 WO2022081973, Example 6
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (6) BDBM534202 WO2022081973, Example 6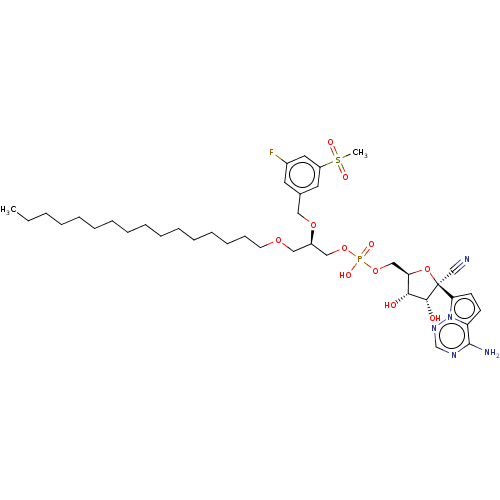 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-fluoro-5-(methylsulfonyl)benzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (32) BDBM534228 WO2022081973, Example 32
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-fluoro-5-(methylsulfonyl)benzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (32) BDBM534228 WO2022081973, Example 32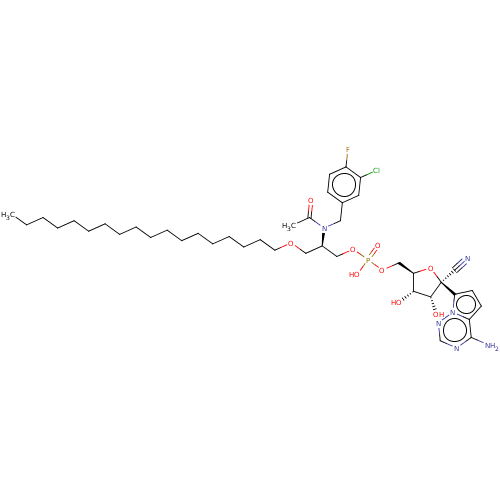 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(N-(3-chloro-4-fluorobenzyl)acetamido)-3-(octadecyloxy)propyl) hydrogen phosphate (10) BDBM534206 WO2022081973, Example 10
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(N-(3-chloro-4-fluorobenzyl)acetamido)-3-(octadecyloxy)propyl) hydrogen phosphate (10) BDBM534206 WO2022081973, Example 10 BDBM534200 WO2022081973, Example 4 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(3-chloro-4-fluoro-N-methylbenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (4)
BDBM534200 WO2022081973, Example 4 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(3-chloro-4-fluoro-N-methylbenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (4) BDBM534239 WO2022081973, Example 43 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((5-(methylsulfonyl)pyridin-3-yl)methoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (43)
BDBM534239 WO2022081973, Example 43 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((5-(methylsulfonyl)pyridin-3-yl)methoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (43) BDBM534243 WO2022081973, Example 47 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-2-((3-cyano-5-fluorobenzyl)oxy)-3-(octadecylamino)-3-oxopropyl) hydrogen phosphate (47)
BDBM534243 WO2022081973, Example 47 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-2-((3-cyano-5-fluorobenzyl)oxy)-3-(octadecylamino)-3-oxopropyl) hydrogen phosphate (47) WO2022081973, Example 3 BDBM534199 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-chloro-4-fluorobenzyl)(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (3)
WO2022081973, Example 3 BDBM534199 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-chloro-4-fluorobenzyl)(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (3)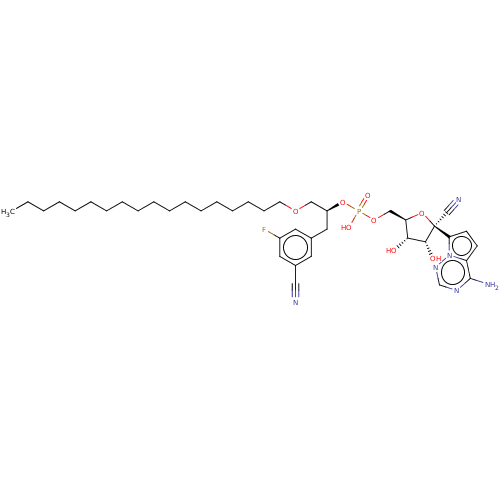 WO2022081973, Example 39 BDBM534235 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-(3-cyano-5-fluorophenoxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (39)
WO2022081973, Example 39 BDBM534235 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-(3-cyano-5-fluorophenoxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (39) WO2022081973, Example 48 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-2-fluoro-3-methoxyphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (48) BDBM534244
WO2022081973, Example 48 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-2-fluoro-3-methoxyphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (48) BDBM534244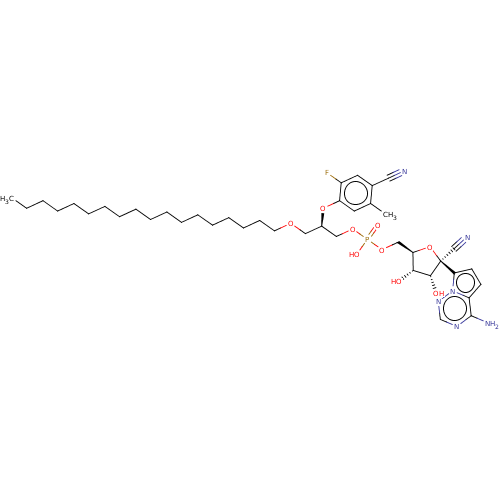 WO2022081973, Example 49 BDBM534245 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-2-fluoro-5-methylphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (49)
WO2022081973, Example 49 BDBM534245 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-2-fluoro-5-methylphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (49)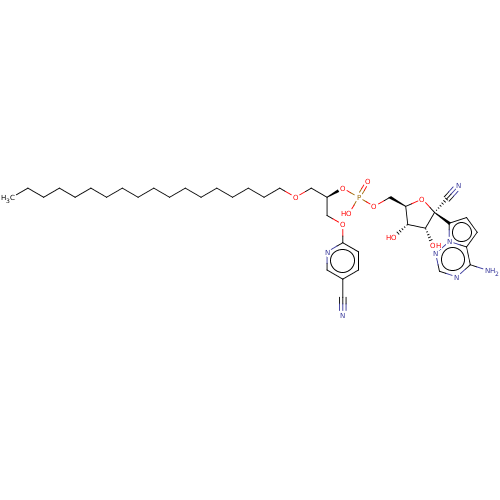 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((5-cyanopyridin-2-yl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (41) BDBM534237 WO2022081973, Example 41
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((5-cyanopyridin-2-yl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (41) BDBM534237 WO2022081973, Example 41 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluoro-4-methylbenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (53) BDBM534249 WO2022081973, Example 53
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluoro-4-methylbenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (53) BDBM534249 WO2022081973, Example 53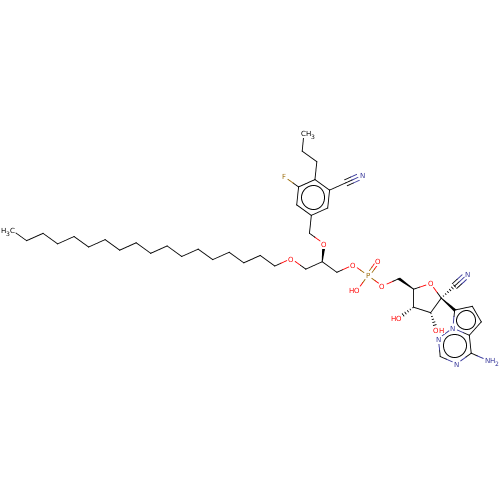 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluoro-4-propylbenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (51) BDBM534247 WO2022081973, Example 51
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluoro-4-propylbenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (51) BDBM534247 WO2022081973, Example 51 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3-fluorobenzyl)oxy)nonadec-4-yn-1-yl) hydrogen phosphate (75) BDBM534272 WO2022081973, Example 75
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3-fluorobenzyl)oxy)nonadec-4-yn-1-yl) hydrogen phosphate (75) BDBM534272 WO2022081973, Example 75 (3R,6R)-6-((3S,5R,7R,8R,9S,10S,13R,14S,17R)-3-(3-(4-(3-aminopropylamino)butylamino)propylamino)-7-hydroxy-10,13-dimethyl-hexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-methylheptan-3-yl hydrogen sulfate CHEMBL508583 BDBM50333649 trodusquemine
(3R,6R)-6-((3S,5R,7R,8R,9S,10S,13R,14S,17R)-3-(3-(4-(3-aminopropylamino)butylamino)propylamino)-7-hydroxy-10,13-dimethyl-hexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-methylheptan-3-yl hydrogen sulfate CHEMBL508583 BDBM50333649 trodusquemine BDBM534232 WO2022081973, Example 36 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((3-cyano-5-fluorobenzyl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (36)
BDBM534232 WO2022081973, Example 36 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((3-cyano-5-fluorobenzyl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (36) BDBM534242 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((6-cyanopyridazin-3-yl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (46) WO2022081973, Example 46
BDBM534242 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((6-cyanopyridazin-3-yl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (46) WO2022081973, Example 46 BDBM534250 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-chloro-3-cyano-5-methoxybenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (54) WO2022081973, Example 54
BDBM534250 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-chloro-3-cyano-5-methoxybenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (54) WO2022081973, Example 54 BDBM534260 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl [(2R)-2-[(3-chloro-2,4-difluoro-phenyl)methoxy]-3-octadecoxy-propyl] hydrogen phosphate (64) WO2022081973, Example 64
BDBM534260 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl [(2R)-2-[(3-chloro-2,4-difluoro-phenyl)methoxy]-3-octadecoxy-propyl] hydrogen phosphate (64) WO2022081973, Example 64 BDBM534271 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)nonadec-4-yn-1-yl) hydrogen phosphate (74) WO2022081973, Example 74
BDBM534271 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)nonadec-4-yn-1-yl) hydrogen phosphate (74) WO2022081973, Example 74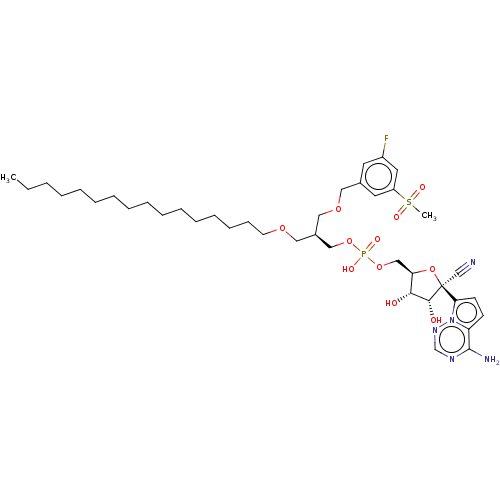 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((3-fluoro-5-(methylsulfonyl)benzyl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (33) BDBM534229 WO2022081973, Example 33
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((3-fluoro-5-(methylsulfonyl)benzyl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (33) BDBM534229 WO2022081973, Example 33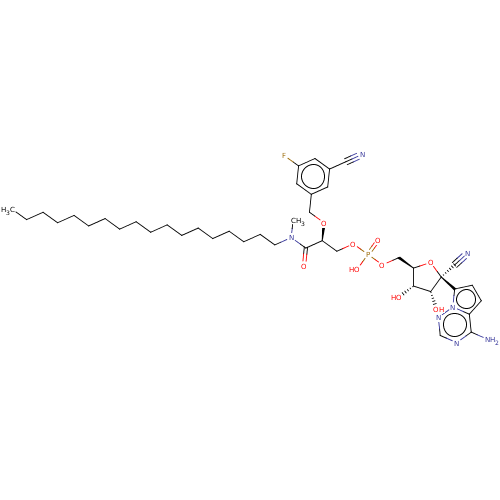 BDBM534252 WO2022081973, Example 56 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-2-((3-cyano-5-fluorobenzyl)oxy)-3-(methyl(octadecyl)amino)-3-oxopropyl) hydrogen phosphate (56)
BDBM534252 WO2022081973, Example 56 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-2-((3-cyano-5-fluorobenzyl)oxy)-3-(methyl(octadecyl)amino)-3-oxopropyl) hydrogen phosphate (56) erythro-Phenyl-2-piperidyl-carbinol,(-) trans-N-Methylphenylcyclopropylamine 3,4-DNH Benzine Cc-34,(+/-) benzene Benzol phenyl hydride CHEMBL277500 Pyrobenzol 5-Hydroxy Tryptamine Mineral naphtha Coal naphtha Bicarburet of hydrogen cyclohexatriene Benzen benzole [6]annulene trans-N, N-Dimethylphenylcyclopropylamine Phene Pyrobenzole BDBM50167939
erythro-Phenyl-2-piperidyl-carbinol,(-) trans-N-Methylphenylcyclopropylamine 3,4-DNH Benzine Cc-34,(+/-) benzene Benzol phenyl hydride CHEMBL277500 Pyrobenzol 5-Hydroxy Tryptamine Mineral naphtha Coal naphtha Bicarburet of hydrogen cyclohexatriene Benzen benzole [6]annulene trans-N, N-Dimethylphenylcyclopropylamine Phene Pyrobenzole BDBM50167939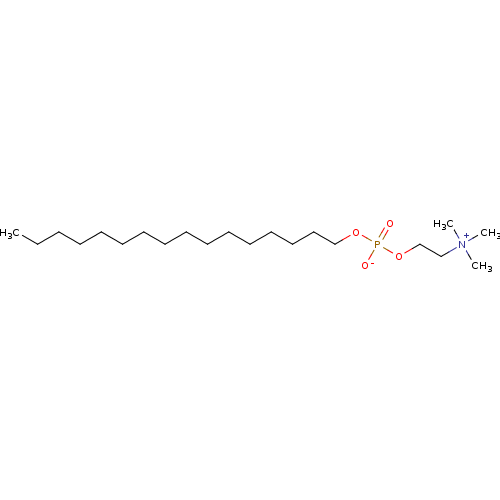 hexadecyl 2-(trimethylammonio)ethyl phosphate [2-(Hexadecyloxy-hydroxy-phosphoryloxy)-ethyl]-trimethyl-ammonium hexadecylphosphocholine hexadecyl 2-(trimethyl-lambda~5~-azanyl)ethyl hydrogen phosphate hexadecylphosphocholine, miltefosine MILTEFOSINE CHEMBL125 hexadecyloxy-2-trimethylammonioethylphosphorate 2-(((Hexadecyloxy)hydroxyphosphinyl)oxy)-N,N,N-trimethylethanaminium hydroxide BDBM50034220 n-hexadecylphosphocholine
hexadecyl 2-(trimethylammonio)ethyl phosphate [2-(Hexadecyloxy-hydroxy-phosphoryloxy)-ethyl]-trimethyl-ammonium hexadecylphosphocholine hexadecyl 2-(trimethyl-lambda~5~-azanyl)ethyl hydrogen phosphate hexadecylphosphocholine, miltefosine MILTEFOSINE CHEMBL125 hexadecyloxy-2-trimethylammonioethylphosphorate 2-(((Hexadecyloxy)hydroxyphosphinyl)oxy)-N,N,N-trimethylethanaminium hydroxide BDBM50034220 n-hexadecylphosphocholine 2-[(Bis-phosphono-methyl)-amino]-pyridinium CHEMBL291736 2-{[hydrogen phosphonato(phosphonato)methyl]amino}pyridin-1-ium [Phosphono-(pyridin-2-ylamino)-methyl]-phosphonic acid BDBM50115104 (pyridin-2-ylamino)methylenediphosphonic acid [Phosphono-(pyridin-2-ylamino)-methyl]-phosphonic acid(NE11807) [(2-pyridinylamino)methylene]-1,1-bisphosphonate
2-[(Bis-phosphono-methyl)-amino]-pyridinium CHEMBL291736 2-{[hydrogen phosphonato(phosphonato)methyl]amino}pyridin-1-ium [Phosphono-(pyridin-2-ylamino)-methyl]-phosphonic acid BDBM50115104 (pyridin-2-ylamino)methylenediphosphonic acid [Phosphono-(pyridin-2-ylamino)-methyl]-phosphonic acid(NE11807) [(2-pyridinylamino)methylene]-1,1-bisphosphonate WO2022081973, Example 50 BDBM534246 WO2022081973, Example 54 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-chloro-3-cyano-5-fluorobenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (50)
WO2022081973, Example 50 BDBM534246 WO2022081973, Example 54 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-chloro-3-cyano-5-fluorobenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (50) BDBM50098390 2-[(Bis-phosphono-methyl)-amino]-3-methyl-pyridinium [(3-Methyl-pyridin-2-ylamino)-phosphono-methyl]-phosphonic acid(NE97200) (3-methylpyridin-2-ylamino)methylenediphosphonic acid CHEMBL55140 [(3-methyl-pyridin-2-ylamino)-phosphono-methyl]-phosphonic acid 2-{[hydrogen phosphonato(phosphonato)methyl]amino}-3-methylpyridin-1-ium
BDBM50098390 2-[(Bis-phosphono-methyl)-amino]-3-methyl-pyridinium [(3-Methyl-pyridin-2-ylamino)-phosphono-methyl]-phosphonic acid(NE97200) (3-methylpyridin-2-ylamino)methylenediphosphonic acid CHEMBL55140 [(3-methyl-pyridin-2-ylamino)-phosphono-methyl]-phosphonic acid 2-{[hydrogen phosphonato(phosphonato)methyl]amino}-3-methylpyridin-1-ium {4-[5-bromo-3-(sulfooxy)-1H-indol-2-yl]-13-chloro-3-thiatricyclo[7.4.0.0^{2,6}]trideca-1(9),2(6),4,7,10,12-hexaen-5-yl}oxidanesulfonic acid 5-bromo-2-[9-chloro-3-(sulfooxy)naphtho[1,2-b]thiophen-2-yl]-1H-indol-3-yl hydrogen sulfate SALOR2 BDBM22584
{4-[5-bromo-3-(sulfooxy)-1H-indol-2-yl]-13-chloro-3-thiatricyclo[7.4.0.0^{2,6}]trideca-1(9),2(6),4,7,10,12-hexaen-5-yl}oxidanesulfonic acid 5-bromo-2-[9-chloro-3-(sulfooxy)naphtho[1,2-b]thiophen-2-yl]-1H-indol-3-yl hydrogen sulfate SALOR2 BDBM22584 Zaditor 4-(1-Methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one(Ketotifen hydrogen malate) 4-(1-methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one CHEMBL534 BDBM50002087 KETOTIFEN 4-(1-Methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one(Ketotifen)
Zaditor 4-(1-Methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one(Ketotifen hydrogen malate) 4-(1-methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one CHEMBL534 BDBM50002087 KETOTIFEN 4-(1-Methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one(Ketotifen) {1-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperidin-4-yl}-phosphonic acid monoethyl ester CHEMBL245315 ethyl hydrogen 1-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperidin-4-ylphosphonate BDBM50241302
{1-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperidin-4-yl}-phosphonic acid monoethyl ester CHEMBL245315 ethyl hydrogen 1-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperidin-4-ylphosphonate BDBM50241302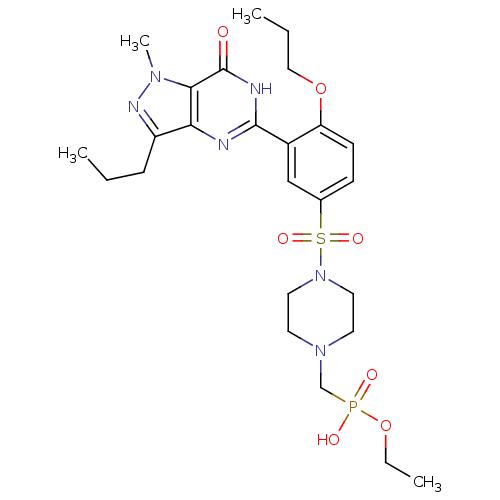 ethyl hydrogen (4-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperazin-1-yl)methylphosphonate {4-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperazin-1-ylmethyl}-phosphonic acid monoethyl ester BDBM50241300 CHEMBL245517
ethyl hydrogen (4-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperazin-1-yl)methylphosphonate {4-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperazin-1-ylmethyl}-phosphonic acid monoethyl ester BDBM50241300 CHEMBL245517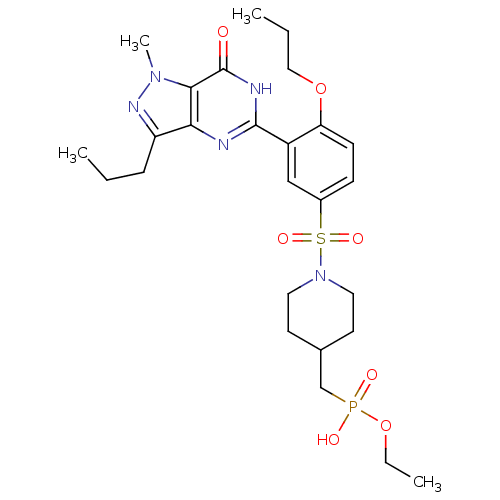 {1-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperidin-4-ylmethyl}-phosphonic acid monoethyl ester ethyl hydrogen (1-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperidin-4-yl)methylphosphonate CHEMBL245317 BDBM50241303
{1-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperidin-4-ylmethyl}-phosphonic acid monoethyl ester ethyl hydrogen (1-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperidin-4-yl)methylphosphonate CHEMBL245317 BDBM50241303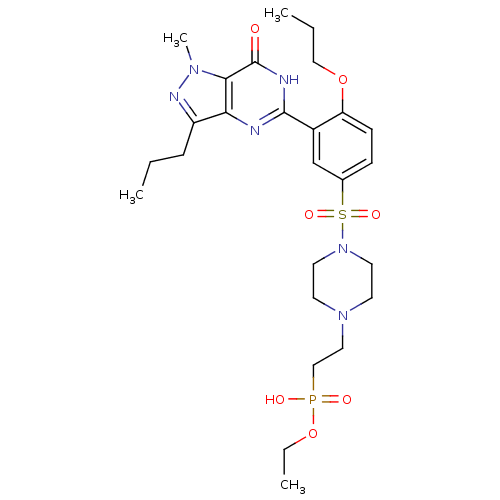 CHEMBL541468 ethyl hydrogen 2-(4-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperazin-1-yl)ethylphosphonate (2-{4-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperazin-1-yl}-ethyl)-phosphonic acid monoethyl ester BDBM50241301
CHEMBL541468 ethyl hydrogen 2-(4-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperazin-1-yl)ethylphosphonate (2-{4-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperazin-1-yl}-ethyl)-phosphonic acid monoethyl ester BDBM50241301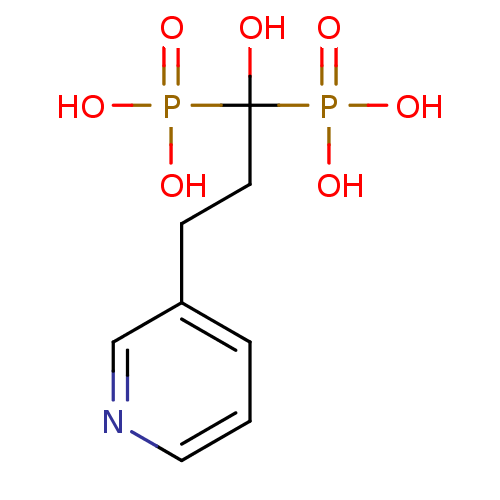 (1-Hydroxy-1-phosphono-3-pyridin-3-yl-propyl)-phosphonic acid(NE58051,homorisedronate) 3-(3-Hydroxy-3,3-bis-phosphono-propyl)-pyridinium (1-Hydroxy-1-phosphono-3-pyridin-3-yl-propyl)-phosphonic acid BDBM50098382 CHEMBL293522 1-hydroxy-3-(pyridin-3-yl)propane-1,1-diyldiphosphonic acid 3-(3-pyridyl)-1-hydroxy-propane-1,1-bisphosphonate 3-(3-hydrogen phosphonato-3-hydroxy-3-phosphonatopropyl)pyridin-1-ium
(1-Hydroxy-1-phosphono-3-pyridin-3-yl-propyl)-phosphonic acid(NE58051,homorisedronate) 3-(3-Hydroxy-3,3-bis-phosphono-propyl)-pyridinium (1-Hydroxy-1-phosphono-3-pyridin-3-yl-propyl)-phosphonic acid BDBM50098382 CHEMBL293522 1-hydroxy-3-(pyridin-3-yl)propane-1,1-diyldiphosphonic acid 3-(3-pyridyl)-1-hydroxy-propane-1,1-bisphosphonate 3-(3-hydrogen phosphonato-3-hydroxy-3-phosphonatopropyl)pyridin-1-ium BDBM50057660 CHEMBL1116 Keoxifene Hydrochloride Evista RALOXIFENE HYDROCHLORIDE 1-(2-{4-[6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophene-3-carbonyl]-phenoxy}-ethyl)-piperidinium; chloride Raloxifene LY-156758 [6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone; Hydrogen chloride [6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone; hydrochloride
BDBM50057660 CHEMBL1116 Keoxifene Hydrochloride Evista RALOXIFENE HYDROCHLORIDE 1-(2-{4-[6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophene-3-carbonyl]-phenoxy}-ethyl)-piperidinium; chloride Raloxifene LY-156758 [6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone; Hydrogen chloride [6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone; hydrochloride [(2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-[({[(2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}phosphinato)oxy]-3-hydroxyoxolan-2-yl]methyl (2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-({[(2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-[(hydrogen phosphonatooxy)methyl]-4-hydroxyoxolan-3-yl phosphonato]oxy}methyl)-4-hydroxyoxolan-3-yl phosphate 2''-5--oligoadenylate derivative BDBM50152834
[(2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-[({[(2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}phosphinato)oxy]-3-hydroxyoxolan-2-yl]methyl (2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-({[(2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-[(hydrogen phosphonatooxy)methyl]-4-hydroxyoxolan-3-yl phosphonato]oxy}methyl)-4-hydroxyoxolan-3-yl phosphate 2''-5--oligoadenylate derivative BDBM50152834 CHEMBL394276 BDBM19253 NSC 358285 [(2R,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [[(2R,3S,4R,5R)-5-(4-carbamoyl-1,3-thiazol-2-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]hydrogen phosphate tiazofurin adenine dinucleotide Tiazofurin Adenine Dinucleotide (TAD) [({[(2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)oxy]({[(2R,3S,4R,5R)-5-(4-carbamoyl-1,3-thiazol-2-yl)-3,4-dihydroxyoxolan-2-yl]methoxy})phosphinic acid
CHEMBL394276 BDBM19253 NSC 358285 [(2R,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [[(2R,3S,4R,5R)-5-(4-carbamoyl-1,3-thiazol-2-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]hydrogen phosphate tiazofurin adenine dinucleotide Tiazofurin Adenine Dinucleotide (TAD) [({[(2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)oxy]({[(2R,3S,4R,5R)-5-(4-carbamoyl-1,3-thiazol-2-yl)-3,4-dihydroxyoxolan-2-yl]methoxy})phosphinic acid (3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyl-tetrahydro-2H-pyran-2-yl hydrogen (S)-1-((S)-1-(1H-indol-3-yl)-3-oxobutan-2-ylamino)-4-methyl-1-oxopentan-2-ylphosphoramidate BDBM50251742 (S)-3-(1-Hydroxy-1H-indol-3-yl)-2-{(S)-2-[hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-propionic acid (S)-2-{(S)-2-[Hydroxy-((2S,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid 2-{2-[Hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid Phosphoramidon Phosporamidon (S)-2-{(S)-2-[Hydroxy-((3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid phosphramidon N-alpha-L-rhamnopyranosyloxy(hydroxyphosphinyl)-L-Leucyl-L-Tryptophan CHEMBL479579 (phosphoramidon) 2-{2-[Hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid 2-{2-[Hydroxy-(3,4,6-trihydroxy-5-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid( Phosphoramidon) (S)-2-{(S)-2-[Hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid (S)-2-{(R)-2-[Hydroxy-((2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid (S)-2-{(S)-2-[Hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-1-oxo-pentylamino}-3-(1H-indol-3-yl)-propionic acid
(3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyl-tetrahydro-2H-pyran-2-yl hydrogen (S)-1-((S)-1-(1H-indol-3-yl)-3-oxobutan-2-ylamino)-4-methyl-1-oxopentan-2-ylphosphoramidate BDBM50251742 (S)-3-(1-Hydroxy-1H-indol-3-yl)-2-{(S)-2-[hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-propionic acid (S)-2-{(S)-2-[Hydroxy-((2S,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid 2-{2-[Hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid Phosphoramidon Phosporamidon (S)-2-{(S)-2-[Hydroxy-((3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid phosphramidon N-alpha-L-rhamnopyranosyloxy(hydroxyphosphinyl)-L-Leucyl-L-Tryptophan CHEMBL479579 (phosphoramidon) 2-{2-[Hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid 2-{2-[Hydroxy-(3,4,6-trihydroxy-5-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid( Phosphoramidon) (S)-2-{(S)-2-[Hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid (S)-2-{(R)-2-[Hydroxy-((2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-pentanoylamino}-3-(1H-indol-3-yl)-propionic acid (S)-2-{(S)-2-[Hydroxy-(3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-phosphorylamino]-4-methyl-1-oxo-pentylamino}-3-(1H-indol-3-yl)-propionic acid