Target (3)
Compound (239)
Article Title (233)
Article Author (58)
Assay (316)
Marutani, E; Sakaguchi, M; Chen, W; Sasakura, K; Liu, J; Xian, M; Hanaoka, K; Nagano, T; Ichinose, F Cytoprotective effects of hydrogen sulfide-releasing Medchemcomm 5: 1577 -1583 (2014) Silverman, RB; Xue, F Intramolecular hydrogen-bonded nitric oxide synthase inhibitors US Patent US8927730 (2015) Gooyit, M; Lee, M; Schroeder, VA; Ikejiri, M; Suckow, MA; Mobashery, S; Chang, M Selective water-soluble gelatinase inhibitor prodrugs. J Med Chem 54: 6676 -90 (2011) Singh, D; Silakari, O Sodium hydrogen exchanger inhibitory activity of benzotriazole derivatives. Eur J Med Chem 126: 183 -189 (2017) Lowe, JA; Drozda, SE; McLean, S; Bryce, DK; Crawford, RT; Zorn, S; Morrone, J; Appleton, TA; Lombardo, F A water soluble benzazepine cholecystokinin-B receptor antagonist Bioorg Med Chem Lett 5: 1933 -1936 (1995) Göring, S; Taymans, JM; Baekelandt, V; Schmidt, B Indolinone based LRRK2 kinase inhibitors with a key hydrogen bond. Bioorg Med Chem Lett 24: 4630 -7 (2014) Ross, PD; Rekharsky, MV Thermodynamics of hydrogen bond and hydrophobic interactions in cyclodextrin complexes. Biophys J 71: 2144 -54 (1996) Nakamura, M; Inoue, J Exploration of peptidyl hydrazones as water-soluble calpain inhibitors. Bioorg Med Chem Lett 12: 1603 -6 (2002) Kato, E; Yama, M; Nakagomi, R; Shibata, T; Hosokawa, K; Kawabata, J Substrate-like water soluble lipase inhibitors from Filipendula kamtschatica. Bioorg Med Chem Lett 22: 6410 -2 (2012) Park, H; McEachon, JD; Pollock, JA Synthesis and characterization of hydrogen peroxide activated estrogen receptor beta ligands. Bioorg Med Chem 27: 2075 -2082 (2019) Donkor, IO; Zheng, X; Han, J; Lacy, C; Miller, DD Significance of hydrogen bonding at the S(1)' subsite of calpain I. Bioorg Med Chem Lett 11: 1753 -5 (2001) Schumann, NC; Bruning, J; Marshall, AC; Abell, AD The role of N-terminal heterocycles in hydrogen bonding to α-chymotrypsin. Bioorg Med Chem Lett 29: 396 -399 (2019) Nakagawa-Goto, K; Chen, CX; Hamel, E; Wu, CC; Bastow, KF; Brossi, A; Lee, KH Antitumor agents. Part 236: Synthesis of water-soluble colchicine derivatives. Bioorg Med Chem Lett 15: 235 -8 (2004) Chenera, B; DesJarlais, RL; Finkelstein, JA; Eggleston, DS; Meek, TD; Tomaszek, TA; Dreyer, GB Nonpeptide HIV protease inhibitors designed to replace a bound water Bioorg Med Chem Lett 3: 2717 -2722 (1993) Vartak, AP; Gabriela Deaciuc, A; Dwoskin, LP; Crooks, PA Quinlobelane: a water-soluble lobelane analogue and inhibitor of VMAT2. Bioorg Med Chem Lett 20: 3584 -7 (2010) Sheppard, GS; Davidsen, SK; Carrera, GM; Pireh, D; Holms, JH; Heyman, HR; Steinman, DH; Curtin, ML; Conway, RG; Rhein, DA; Albert, DH; Tapang, P; Magoc, TJ; Summers, JB Synthesis and evaluation of water soluble indole pyrrolothiazole paf antagonists Bioorg Med Chem Lett 5: 2913 -2918 (1995) Govek, SP; Oshiro, G; Anzola, JV; Beauregard, C; Chen, J; Coyle, AR; Gamache, DA; Hellberg, MR; Hsien, JN; Lerch, JM; Liao, JC; Malecha, JW; Staszewski, LM; Thomas, DJ; Yanni, JM; Noble, SA; Shiau, AK Water-soluble PDE4 inhibitors for the treatment of dry eye. Bioorg Med Chem Lett 20: 2928 -32 (2010) Mazurov, AA; Kombo, DC; Akireddy, S; Murthy, S; Hauser, TA; Jordan, KG; Gatto, GJ; Yohannes, D Novel nicotinic acetylcholine receptor agonists containing carbonyl moiety as a hydrogen bond acceptor. Bioorg Med Chem Lett 23: 3927 -34 (2013) Liu, KK; Huang, X; Bagrodia, S; Chen, JH; Greasley, S; Cheng, H; Sun, S; Knighton, D; Rodgers, C; Rafidi, K; Zou, A; Xiao, J; Yan, S Quinazolines with intra-molecular hydrogen bonding scaffold (iMHBS) as PI3K/mTOR dual inhibitors. Bioorg Med Chem Lett 21: 1270 -4 (2011) Sciabola, S; Goetz, GH; Bai, G; Rogers, BN; Gray, DL; Duplantier, A; Fonseca, KR; Vanase-Frawley, MA; Kablaoui, NM Systematic N-methylation of oxytocin: Impact on pharmacology and intramolecular hydrogen bonding network. Bioorg Med Chem 24: 3513 -20 (2016) Meibom, D; Meyer, J; von Buehler, CJ; Collins, KD; Maassen, S; Gericke, KM; Hüser, J; Mittendorf, J; Ortega Hernandez, N; Schamberger, J; Stampfuss, J; Straub, A; Torge, A; Witowski, N; Wunder, F BAY-6096: A Potent, Selective, and Highly Water-Soluble Adrenergic α J Med Chem 66: 4659 -4670 (2023) Xu, L; Farthing, AK; Dropinski, JF; Meinke, PT; McCallum, C; Leavitt, PS; Hickey, EJ; Colwell, L; Barrett, J; Liu, K Nocathiacin analogs: Synthesis and antibacterial activity of novel water-soluble amides. Bioorg Med Chem Lett 19: 3531 -5 (2009) Ettmayer, P; Billich, A; Hecht, P; Rosenwirth, B; Gstach, H Paracyclophanes: a novel class of water-soluble inhibitors of HIV proteinase. J Med Chem 39: 3291 -9 (1996) Attolino, E; Calderone, V; Dragoni, E; Fragai, M; Richichi, B; Luchinat, C; Nativi, C Structure-based approach to nanomolar, water soluble matrix metalloproteinases inhibitors (MMPIs). Eur J Med Chem 45: 5919 -25 (2010) Leung, IK; Flashman, E; Yeoh, KK; Schofield, CJ; Claridge, TD Using NMR solvent water relaxation to investigate metalloenzyme-ligand binding interactions. J Med Chem 53: 867 -75 (2010) Lackey, K; Sternbach, DD; Croom, DK; Emerson, DL; Evans, MG; Leitner, PL; Luzzio, MJ; McIntyre, G; Vuong, A; Yates, J; Besterman, JM Water soluble inhibitors of topoisomerase I: quaternary salt derivatives of camptothecin. J Med Chem 39: 713 -9 (1996) Yang, CY; Phillips, JG; Stuckey, JA; Bai, L; Sun, H; Delproposto, J; Brown, WC; Chinnaswamy, K Buried Hydrogen Bond Interactions Contribute to the High Potency of Complement Factor D Inhibitors. ACS Med Chem Lett 7: 1092 -1096 (2016) Böhm, M; Klebe, G Development of new hydrogen-bond descriptors and their application to comparative molecular field analyses. J Med Chem 45: 1585 -97 (2002) Mao, F; Chen, J; Zhou, Q; Luo, Z; Huang, L; Li, X Novel tacrine-ebselen hybrids with improved cholinesterase inhibitory, hydrogen peroxide and peroxynitrite scavenging activity. Bioorg Med Chem Lett 23: 6737 -42 (2013) Géraldy, M; Morgen, M; Sehr, P; Steimbach, RR; Moi, D; Ridinger, J; Oehme, I; Witt, O; Malz, M; Nogueira, MS; Koch, O; Gunkel, N; Miller, AK Selective Inhibition of Histone Deacetylase 10: Hydrogen Bonding to the Gatekeeper Residue is Implicated. J Med Chem 62: 4426 -4443 (2019) Soares, J; Keppler, BR; Wang, X; Lee, KH; Jarstfer, MB ortho-Quinone tanshinones directly inhibit telomerase through an oxidative mechanism mediated by hydrogen peroxide. Bioorg Med Chem Lett 21: 7474 -8 (2011) Bhat, TN; Bentley, GA; Boulot, G; Greene, MI; Tello, D; Dall'Acqua, W; Souchon, H; Schwarz, FP; Mariuzza, RA; Poljak, RJ Bound water molecules and conformational stabilization help mediate an antigen-antibody association. Proc Natl Acad Sci U S A 91: 1089 -93 (1994) Bock, MG; DiPardo, RM; Rittle, KE; Evans, BE; Freidinger, RM; Veber, DF; Chang, RS; Chen, TB; Keegan, ME; Lotti, VJ Cholecystokinin antagonists. Synthesis of asperlicin analogues with improved potency and water solubility. J Med Chem 29: 1941 -5 (1986) Shadrick, WR; Slavish, PJ; Chai, SC; Waddell, B; Connelly, M; Low, JA; Tallant, C; Young, BM; Bharatham, N; Knapp, S; Boyd, VA; Morfouace, M; Roussel, MF; Chen, T; Lee, RE; Kiplin Guy, R; Shelat, AA; Potter, PM Exploiting a water network to achieve enthalpy-driven, bromodomain-selective BET inhibitors. Bioorg Med Chem 26: 25 -36 (2018) Badarau, E; Mongeot, A; Collighan, R; Rathbone, D; Griffin, M Imidazolium-based warheads strongly influence activity of water-soluble peptidic transglutaminase inhibitors. Eur J Med Chem 66: 526 -30 (2013) Lloyd, MG; Huckvale, R; Cheung, KJ; Rodrigues, MJ; Collie, GW; Pierrat, OA; Gatti Iou, M; Carter, M; Davis, OA; McAndrew, PC; Gunnell, E; Le Bihan, YV; Talbot, R; Henley, AT; Johnson, LD; Hayes, A; Bright, MD; Raynaud, FI; Meniconi, M; Burke, R; van Montfort, RLM; Rossanese, OW; Bellenie, BR; Hoelder, S Into Deep Water: Optimizing BCL6 Inhibitors by Growing into a Solvated Pocket. J Med Chem 64: 17079 -17097 (2021) Zafrir, E; Carmeli, S Micropeptins from an Israeli fishpond water bloom of the cyanobacterium Microcystis sp. J Nat Prod 73: 352 -8 (2010) Hale, JJ; Mills, SG; MacCoss, M; Dorn, CP; Finke, PE; Budhu, RJ; Reamer, RA; Huskey, SE; Luffer-Atlas, D; Dean, BJ; McGowan, EM; Feeney, WP; Chiu, SH; Cascieri, MA; Chicchi, GG; Kurtz, MM; Sadowski, S; Ber, E; Tattersall, FD; Rupniak, NM; Williams, AR; Rycroft, W; Hargreaves, R; Metzger, JM; MacIntyre, DE Phosphorylated morpholine acetal human neurokinin-1 receptor antagonists as water-soluble prodrugs. J Med Chem 43: 1234 -41 (2000) Carta, F; Temperini, C; Innocenti, A; Scozzafava, A; Kaila, K; Supuran, CT Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 53: 5511 -22 (2010) Patil, RV; Xu, S; van Hoek, AN; Rusinko, A; Feng, Z; May, J; Hellberg, M; Sharif, NA; Wax, MB; Irigoyen, M; Carr, G; Brittain, T; Brown, P; Colbert, D; Kumari, S; Varadaraj, K; Mitra, AK Rapid Identification of Novel Inhibitors of the Human Aquaporin-1 Water Channel. Chem Biol Drug Des 87: 794 -805 (2016) Wheelock, CE; Severson, TF; Hammock, BD Synthesis of new carboxylesterase inhibitors and evaluation of potency and water solubility. Chem Res Toxicol 14: 1563 -72 (2001) Sonawane, ND; Verkman, AS Thiazolidinone CFTR inhibitors with improved water solubility identified by structure-activity analysis. Bioorg Med Chem 16: 8187 -95 (2008) Ponticello, GS; Freedman, MB; Habecker, CN; Lyle, PA; Schwam, H; Varga, SL; Christy, ME; Randall, WC; Baldwin, JJ Thienothiopyran-2-sulfonamides: a novel class of water-soluble carbonic anhydrase inhibitors. J Med Chem 30: 591 -7 (1987) Antoni, F; Wifling, D; Bernhardt, G Water-soluble inhibitors of ABCG2 (BCRP) - A fragment-based and computational approach. Eur J Med Chem 210: (2021) Oguma, T; Uehara, S; Nakahara, K; Okuyama, Y; Fuchino, K; Suzuki, N; Kan, Y; Kanegawa, N; Ogata, Y; Kusakabe, KI A Quantum Mechanics-Based Method to Predict Intramolecular Hydrogen Bond Formation Reflecting P-glycoprotein Recognition. ACS Med Chem Lett 14: 223 -228 (2023) Ahmad, S; Doweyko, LM; Dugar, S; Grazier, N; Ngu, K; Wu, SC; Yost, KJ; Chen, BC; Gougoutas, JZ; DiMarco, JD; Lan, SJ; Gavin, BJ; Chen, AY; Dorso, CR; Serafino, R; Kirby, M; Atwal, KS Arylcyclopropanecarboxyl guanidines as novel, potent, and selective inhibitors of the sodium hydrogen exchanger isoform-1. J Med Chem 44: 3302 -10 (2001) Zheng, YG; Zhang, WQ; Meng, L; Wu, XQ; Zhang, L; An, L; Li, CL; Gao, CY; Xu, L; Liu, Y Design, synthesis and biological evaluation of 4-aniline quinazoline derivatives conjugated with hydrogen sulfide (H Eur J Med Chem 202: (2020) Katragadda, M; Magotti, P; Sfyroera, G; Lambris, JD Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J Med Chem 49: 4616 -22 (2006) Labby, KJ; Xue, F; Kraus, JM; Ji, H; Mataka, J; Li, H; Martásek, P; Roman, LJ; Poulos, TL; Silverman, RB Intramolecular hydrogen bonding: a potential strategy for more bioavailable inhibitors of neuronal nitric oxide synthase. Bioorg Med Chem 20: 2435 -43 (2012) Watterson, SH; Dhar, TG; Ballentine, SK; Shen, Z; Barrish, JC; Cheney, D; Fleener, CA; Rouleau, KA; Townsend, R; Hollenbaugh, DL; Iwanowicz, EJ Novel indole-based inhibitors of IMPDH: introduction of hydrogen bond acceptors at indole C-3. Bioorg Med Chem Lett 13: 1273 -6 (2003) Gonzalez, AZ; Li, Z; Beck, HP; Canon, J; Chen, A; Chow, D; Duquette, J; Eksterowicz, J; Fox, BM; Fu, J; Huang, X; Houze, J; Jin, L; Li, Y; Ling, Y; Lo, MC; Long, AM; McGee, LR; McIntosh, J; Oliner, JD; Osgood, T; Rew, Y; Saiki, AY; Shaffer, P; Wortman, S; Yakowec, P; Yan, X; Ye, Q; Yu, D; Zhao, X; Zhou, J; Olson, SH; Sun, D; Medina, JC Novel inhibitors of the MDM2-p53 interaction featuring hydrogen bond acceptors as carboxylic acid isosteres. J Med Chem 57: 2963 -88 (2014) Wiedeman, PE; Fesik, SW; Petros, AM; Nettesheim, DG; Mollison, KW; Lane, BC; Or, YS; Luly, JR Retention of immunosuppressant activity in an ascomycin analogue lacking a hydrogen-bonding interaction with FKBP12. J Med Chem 42: 4456 -61 (1999) Foloppe, N; Fisher, LM; Howes, R; Kierstan, P; Potter, A; Robertson, AG; Surgenor, AE Structure-based design of novel Chk1 inhibitors: insights into hydrogen bonding and protein-ligand affinity. J Med Chem 48: 4332 -45 (2005) Moss, RJ; Petrie, CR; Meyer, RB; Nord, LD; Willis, RC; Smith, RA; Larson, SB; Kini, GD; Robins, RK Synthesis, intramolecular hydrogen bonding, and biochemical studies of clitocine, a naturally occurring exocyclic amino nucleoside. J Med Chem 31: 786 -90 (1988) Tardia, P; Stefanachi, A; Niso, M; Stolfa, DA; Mangiatordi, GF; Alberga, D; Nicolotti, O; Lattanzi, G; Carotti, A; Leonetti, F; Perrone, R; Berardi, F; Azzariti, A; Colabufo, NA; Cellamare, S Trimethoxybenzanilide-based P-glycoprotein modulators: an interesting case of lipophilicity tuning by intramolecular hydrogen bonding. J Med Chem 57: 6403 -18 (2014) Ettorre, A; D'Andrea, P; Mauro, S; Porcelloni, M; Rossi, C; Altamura, M; Catalioto, RM; Giuliani, S; Maggi, CA; Fattori, D hNK2 receptor antagonists. The use of intramolecular hydrogen bonding to increase solubility and membrane permeability. Bioorg Med Chem Lett 21: 1807 -9 (2011) Liu, DG; Gao, Y; Voigt, JH; Lee, K; Nicklaus, MC; Wu, L; Zhang, ZY; Burke, TR Acylsulfonamide-containing PTP1B inhibitors designed to mimic an enzyme-bound water of hydration. Bioorg Med Chem Lett 13: 3005 -7 (2003) Jordan, DB; Basarab, GS Binding dynamics of two water molecules constrained within the scytalone dehydratase binding pocket. Bioorg Med Chem Lett 10: 23 -6 (2000) Randad, RS; Pan, W; Gulnik, SV; Burt, S; Erickson, JW De novo design of nonpeptidic HIV-1 protease inhibitors: Incorporation of structural water. Bioorg Med Chem Lett 4: 1247 -1252 (1994) Kemami Wangun, HV; Wood, A; Fiorilla, C; Reed, JK; McCarthy, PJ; Wright, AE Gymnochromes E and F, cytotoxic phenanthroperylenequinones from a deep-water crinoid, Holopus rangii. J Nat Prod 73: 712 -5 (2010) Carpenter, RD; Andrei, M; Lau, EY; Lightstone, FC; Liu, R; Lam, KS; Kurth, MJ Highly Potent, Water Soluble Benzimidazole Antagonist for Activated alpha(4)beta(1) Integrin. J Med Chem 50: 5863 -5867 (2007) Kim, IH; Heirtzler, FR; Morisseau, C; Nishi, K; Tsai, HJ; Hammock, BD Optimization of amide-based inhibitors of soluble epoxide hydrolase with improved water solubility. J Med Chem 48: 3621 -9 (2005) Bedeschi, A; Zarini, F; Cabri, W; Candiani, I; Penco, S; Capolongo, L; Ciomei, M; Farao, M; Grandi, M Synthesis and antitumor activity of a new class of water soluble camptothecin derivatives Bioorg Med Chem Lett 6: 671 -674 (1996) Boyd, HF; Hammond, B; Hickey, DM; Ife, RJ; Leach, CA; Lewis, VA; Macphee, CH; Milliner, KJ; Pinto, IL; Smith, SA; Stansfield, IG; Theobald, CJ; Whittaker, CM The identification of a potent, water soluble inhibitor of lipoprotein-associated phospholipase A2. Bioorg Med Chem Lett 11: 701 -4 (2001) Dawson, TK; Dziedzic, P; Robertson, MJ; Cisneros, JA; Krimmer, SG; Newton, AS; Tirado-Rives, J; Jorgensen, WL Adding a Hydrogen Bond May Not Help: Naphthyridinone vs Quinoline Inhibitors of Macrophage Migration Inhibitory Factor. ACS Med Chem Lett 8: 1287 -1291 (2017) Katz, BA; Elrod, K; Verner, E; Mackman, RL; Luong, C; Shrader, WD; Sendzik, M; Spencer, JR; Sprengeler, PA; Kolesnikov, A; Tai, VW; Hui, HC; Breitenbucher, JG; Allen, D; Janc, JW Elaborate manifold of short hydrogen bond arrays mediating binding of active site-directed serine protease inhibitors. J Mol Biol 329: 93 -120 (2003) Klumb, LA; Chu, V; Stayton, PS Energetic roles of hydrogen bonds at the ureido oxygen binding pocket in the streptavidin-biotin complex. Biochemistry 37: 7657 -63 (1998) Harada, T; Nakagawa, Y; Akamatsu, M; Miyagawa, H Evaluation of hydrogen bonds of ecdysteroids in the ligand-receptor interactions using a protein modeling system. Bioorg Med Chem 17: 5868 -73 (2009) Bao, Q; Zhang, L; Wang, N; Gabet, B; Yang, W; Gao, X; You, Q; Jiang, Z Hydrogen Peroxide Inducible JAK3 Covalent Inhibitor: Prodrug for the Treatment of RA with Enhanced Safety Profile. ACS Med Chem Lett 11: 2182 -2189 (2020) Wu, WL; Burnett, DA; Clader, J; Greenlee, WJ; Jiang, Q; Hyde, LA; Del Vecchio, RA; Cohen-Williams, ME; Song, L; Lee, J; Terracina, G; Zhang, Q; Nomeir, A; Parker, EM; Zhang, L Design and synthesis of water solubleß-aminosulfone analogues of SCH 900229 as¿-secretase inhibitors. Bioorg Med Chem Lett 26: 5836 -5841 (2016) He, H; Zatorska, D; Kim, J; Aguirre, J; Llauger, L; She, Y; Wu, N; Immormino, RM; Gewirth, DT; Chiosis, G Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J Med Chem 49: 381 -90 (2006) Heider, F; Pantsar, T; Kudolo, M; Ansideri, F; De Simone, A; Pruccoli, L; Schneider, T; Goettert, MI; Tarozzi, A; Andrisano, V; Laufer, SA; Koch, P Pyridinylimidazoles as GSK3β Inhibitors: The Impact of Tautomerism on Compound Activity via Water Networks. ACS Med Chem Lett 10: 1407 -1414 (2019) Chiari, A; Fancelli, D; Lovisolo, PP; Radice, A; Savoia, L; Severino, D; Ghiselli, G Synthesis and pharmacological profile of FCE 28654: A water-soluble and injectable ACAT inhibitor Bioorg Med Chem Lett 5: 1581 -1586 (1995) Paulsen, ES; Villadsen, J; Tenori, E; Liu, H; Bonde, DF; Lie, MA; Bublitz, M; Olesen, C; Autzen, HE; Dach, I; Sehgal, P; Nissen, P; Møller, JV; Schiøtt, B; Christensen, SB Water-mediated interactions influence the binding of thapsigargin to sarco/endoplasmic reticulum calcium adenosinetriphosphatase. J Med Chem 56: 3609 -19 (2013) Hodge, C; Pierce, J A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of scytalone dehydratase Bioorg Med Chem Lett 3: 1605 -1608 (1993) Vaz, RJ; Gao, Z; Pribish, J; Chen, X; Levell, J; Davis, L; Albert, E; Brollo, M; Ugolini, A; Cramer, DM; Cairns, J; Sides, K; Liu, F; Kwong, J; Kang, J; Rebello, S; Elliot, M; Lim, H; Chellaraj, V; Singleton, RW; Li, Y Design of bivalent ligands using hydrogen bond linkers: synthesis and evaluation of inhibitors for human beta-tryptase. Bioorg Med Chem Lett 14: 6053 -6 (2004) Kuo, PY; Shie, TL; Chen, YS; Lai, JT; Yang, DY Enzyme inhibition potency enhancement by active site metal chelating and hydrogen bonding induced conformation-restricted cyclopropanecarbonyl derivatives. Bioorg Med Chem Lett 16: 6024 -7 (2006) Rusere, LN; Lockbaum, GJ; Lee, SK; Henes, M; Kosovrasti, K; Spielvogel, E; Nalivaika, EA; Swanstrom, R; Yilmaz, NK; Schiffer, CA; Ali, A HIV-1 Protease Inhibitors Incorporating Stereochemically Defined P2' Ligands To Optimize Hydrogen Bonding in the Substrate Envelope. J Med Chem 62: 8062 -8079 (2019) Mahanti, M; Pal, KB; Kumar, R; Schulze, M; Leffler, H; Logan, DT; Nilsson, UJ Ligand Sulfur Oxidation State Progressively Alters Galectin-3-Ligand Complex Conformations To Induce Affinity-Influencing Hydrogen Bonds. J Med Chem 66: 14716 -14723 (2023) Rosenberg, SH; Dellaria, JF; Kempf, DJ; Hutchins, CW; Woods, KW; Maki, RG; de Lara, E; Spina, KP; Stein, HH; Cohen, J Potent, low molecular weight renin inhibitors containing a C-terminal heterocycle: hydrogen bonding at the active site. J Med Chem 33: 1582 -90 (1990) Zhang, W; Zhang, D; Stashko, MA; DeRyckere, D; Hunter, D; Kireev, D; Miley, MJ; Cummings, C; Lee, M; Norris-Drouin, J; Stewart, WM; Sather, S; Zhou, Y; Kirkpatrick, G; Machius, M; Janzen, WP; Earp, HS; Graham, DK; Frye, SV; Wang, X Pseudo-cyclization through intramolecular hydrogen bond enables discovery of pyridine substituted pyrimidines as new Mer kinase inhibitors. J Med Chem 56: 9683 -92 (2014) Cowan, JA; Ohyama, T; Wang, D; Natarajan, K Recognition of a cognate RNA aptamer by neomycin B: quantitative evaluation of hydrogen bonding and electrostatic interactions. Nucleic Acids Res 28: 2935 -42 (2000) Ashwood, VA; Field, MJ; Horwell, DC; Julien-Larose, C; Lewthwaite, RA; McCleary, S; Pritchard, MC; Raphy, J; Singh, L Utilization of an intramolecular hydrogen bond to increase the CNS penetration of an NK(1) receptor antagonist. J Med Chem 44: 2276 -85 (2001) Meurer, LC; Finke, PE; Owens, KA; Tsou, NN; Ball, RG; Mills, SG; Maccoss, M; Sadowski, S; Cascieri, MA; Tsao, KL; Chicchi, GG; Egger, LA; Luell, S; Metzger, JM; Macintyre, DE; Rupniak, NM; Williams, AR; Hargreaves, RJ Cyclopentane-based human NK1 antagonists. Part 2: development of potent, orally active, water-soluble derivatives. Bioorg Med Chem Lett 16: 4504 -11 (2006) Kuroda, S; Akahane, A; Itani, H; Nishimura, S; Durkin, K; Kinoshita, T; Tenda, Y; Sakane, K Discovery of FR166124, a novel water-soluble pyrazolo-[1,5-a]pyridine adenosine A1 receptor antagonist. Bioorg Med Chem Lett 9: 1979 -84 (1999) Crawford, TD; Tsui, V; Flynn, EM; Wang, S; Taylor, AM; Côté, A; Audia, JE; Beresini, MH; Burdick, DJ; Cummings, R; Dakin, LA; Duplessis, M; Good, AC; Hewitt, MC; Huang, HR; Jayaram, H; Kiefer, JR; Jiang, Y; Murray, J; Nasveschuk, CG; Pardo, E; Poy, F; Romero, FA; Tang, Y; Wang, J; Xu, Z; Zawadzke, LE; Zhu, X; Albrecht, BK; Magnuson, SR; Bellon, S; Cochran, AG Diving into the Water: Inducible Binding Conformations for BRD4, TAF1(2), BRD9, and CECR2 Bromodomains. J Med Chem 59: 5391 -402 (2016) Bock, MG; DiPardo, RM; Mellin, EC; Newton, RC; Veber, DF; Freedman, SB; Smith, AJ; Patel, S; Kemp, JA; Marshall, GR Second-generation benzodiazepine CCK-B antagonists. Development of subnanomolar analogs with selectivity and water solubility. J Med Chem 37: 722 -4 (1994) Sant' Anna, Gda S; Machado, P; Sauzem, PD; Rosa, FA; Rubin, MA; Ferreira, J; Bonacorso, HG; Zanatta, N; Martins, MA Ultrasound promoted synthesis of 2-imidazolines in water: a greener approach toward monoamine oxidase inhibitors. Bioorg Med Chem Lett 19: 546 -9 (2008) Rosenberg, SH; Woods, KW; Sham, HL; Kleinert, HD; Martin, DL; Stein, H; Cohen, J; Egan, DA; Bopp, B; Merits, I Water-soluble renin inhibitors: design of a subnanomolar inhibitor with a prolonged duration of action. J Med Chem 33: 1962 -9 (1990) Katz, BA; Elrod, K; Luong, C; Rice, MJ; Mackman, RL; Sprengeler, PA; Spencer, J; Hataye, J; Janc, J; Link, J; Litvak, J; Rai, R; Rice, K; Sideris, S; Verner, E; Young, W A novel serine protease inhibition motif involving a multi-centered short hydrogen bonding network at the active site. J Mol Biol 307: 1451 -86 (2001) Patterson, JR; Terrell, LR; Donatelli, CA; Holt, DA; Jolivette, LJ; Rivero, RA; Roethke, TJ; Shu, A; Stoy, P; Ye, G; Youngman, M; Lawhorn, BG Design and Optimization of an Acyclic Amine Series of TRPV4 Antagonists by Electronic Modulation of Hydrogen Bond Interactions. J Med Chem 63: 14867 -14884 (2020) Development of UTX-143, a selective sodium-hydrogen exchange subtype 5 inhibitor, using amiloride as a lead compound. Dichiara, M; Artacho-Cordón, A; Turnaturi, R; Santos-Caballero, M; González-Cano, R; Pasquinucci, L; Barbaraci, C; Rodríguez-Gómez, I; Gómez-Guzmán, M; Marrazzo, A; Cobos, EJ; Amata, E Dual Sigma-1 receptor antagonists and hydrogen sulfide-releasing compounds for pain treatment: Design, synthesis, and pharmacological evaluation. Eur J Med Chem 230: (2022) Napoletano, M; Norcini, G; Pellacini, F; Marchini, F; Morazzoni, G; Fattori, R; Ferlenga, P; Pradella, L Phthalazine PDE4 inhibitors. Part 3: the synthesis and in vitro evaluation of derivatives with a hydrogen bond acceptor. Bioorg Med Chem Lett 12: 5 -8 (2002) Zheng, B; D'Andrea, SV; Sun, LQ; Wang, AX; Chen, Y; Hrnciar, P; Friborg, J; Falk, P; Hernandez, D; Yu, F; Sheaffer, AK; Knipe, JO; Mosure, K; Rajamani, R; Good, AC; Kish, K; Tredup, J; Klei, HE; Paruchuri, M; Ng, A; Gao, Q; Rampulla, RA; Mathur, A; Meanwell, NA; McPhee, F; Scola, PM Potent Inhibitors of Hepatitis C Virus NS3 Protease: Employment of a Difluoromethyl Group as a Hydrogen-Bond Donor. ACS Med Chem Lett 9: 143 -148 (2018) Daly, JW; Padgett, W; Shamim, MT; Butts-Lamb, P; Waters, J 1,3-Dialkyl-8-(p-sulfophenyl)xanthines: potent water-soluble antagonists for A1- and A2-adenosine receptors. J Med Chem 28: 487 -92 (1985) Yan, F; Cao, XX; Jiang, HX; Zhao, XL; Wang, JY; Lin, YH; Liu, QL; Zhang, C; Jiang, B; Guo, F A novel water-soluble gossypol derivative increases chemotherapeutic sensitivity and promotes growth inhibition in colon cancer. J Med Chem 53: 5502 -10 (2010) Showell, GA; Bourrain, S; Fletcher, SR; Neduvelil, JG; Fletcher, AE; Freedman, SB; Patel, S; Smith, AJ; Marshall, GR; Graham, MI; Sohal, B; Matassa, VG C5-piperazinyl-1,4-benzodiazepines, water-soluble, orally bioa vailable CCKB /gastrin receptor antagonists Bioorg Med Chem Lett 5: 3023 -3026 (1995) Käck, H; Doyle, K; Hughes, SJ; Bodnarchuk, MS; Lönn, H; Van De Poël, A; Palmer, N DPP1 Inhibitors: Exploring the Role of Water in the S2 Pocket of DPP1 with Substituted Pyrrolidines. ACS Med Chem Lett 10: 1222 -1227 (2019) Gunasekera, SP; McCarthy, PJ; Longley, RE; Pomponi, SA; Wright, AE; Lobkovsky, E; Clardy, J Discorhabdin P, a new enzyme inhibitor from a deep-water Caribbean sponge of the genus Batzella. J Nat Prod 62: 173 -5 (1999) Dong, J; Huang, G; Cui, Q; Meng, Q; Li, S; Cui, J Discovery of heterocycle-containing α-naphthoflavone derivatives as water-soluble, highly potent and selective CYP1B1 inhibitors. Eur J Med Chem 209: (2021) Nishiwaki, K; Nakatani, S; Nakamura, S; Yoshioka, K; Nakagawa, E; Tsuyuguchi, M; Kinoshita, T; Nakanishi, I Enhanced inhibitory activity of compounds containing purine scaffolds compared to protein kinase CK2α considering crystalline water. RSC Med Chem 15: 1274 -1282 (2024) Larraufie, MH; Yang, WS; Jiang, E; Thomas, AG; Slusher, BS; Stockwell, BR Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorg Med Chem Lett 25: 4787 -92 (2015) Biela, A; Sielaff, F; Terwesten, F; Heine, A; Steinmetzer, T; Klebe, G Ligand binding stepwise disrupts water network in thrombin: enthalpic and entropic changes reveal classical hydrophobic effect. J Med Chem 55: 6094 -110 (2012) Lodin-Friedman, A; Carmeli, S Metabolites from Microcystis aeruginosa bloom material collected at a water reservoir near Kibbutz Hafetz Haim, Israel. J Nat Prod 76: 1196 -200 (2013) Palin, R; Clark, JK; Cowley, P; Muir, AW; Pow, E; Prosser, AB; Taylor, R; Zhang, MQ Novel piperidinium and pyridinium agents as water-soluble acetylcholinesterase inhibitors for the reversal of neuromuscular blockade. Bioorg Med Chem Lett 12: 2569 -72 (2002) Gunasekera, SP; Sennett, SH; Kelly-Borges, M; Bryant, RW Ophirapstanol trisulfate, a new biologically active steroid sulfate from the deep water marine sponge Topsentia ophiraphidites. J Nat Prod 57: 1751 -4 (1994) Persch, E; Basile, T; Bockelmann, S; Huss, M; Wieczorek, H; Carlomagno, T; Menche, D Synthesis and biological evaluation of a water-soluble derivative of the potent V-ATPase inhibitor archazolid. Bioorg Med Chem Lett 22: 7735 -8 (2012) DeNinno, MP; Masamune, H; Chenard, LK; DiRico, KJ; Eller, C; Etienne, JB; Tickner, JE; Kennedy, SP; Knight, DR; Kong, J; Oleynek, JJ; Tracey, WR; Hill, RJ The synthesis of highly potent, selective, and water-soluble agonists at the human adenosine A3 receptor. Bioorg Med Chem Lett 16: 2525 -7 (2006) Sauer, R; Maurinsh, J; Reith, U; Fülle, F; Klotz, KN; Müller, CE Water-soluble phosphate prodrugs of 1-propargyl-8-styrylxanthine derivatives, A(2A)-selective adenosine receptor antagonists. J Med Chem 43: 440 -8 (2000) Baraldi, PG; Saponaro, G; Romagnoli, R; Aghazadeh Tabrizi, M; Baraldi, S; Moorman, AR; Cosconati, S; Di Maro, S; Marinelli, L; Gessi, S; Merighi, S; Varani, K; Borea, PA; Preti, D Water-soluble pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidines as human A3 adenosine receptor antagonists. J Med Chem 55: 5380 -90 (2012) Ahmad, S; Ngu, K; Combs, DW; Wu, SC; Weinstein, DS; Liu, W; Chen, BC; Chandrasena, G; Dorso, CR; Kirby, M; Atwal, KS Aminoimidazoles as bioisosteres of acylguanidines: novel, potent, selective and orally bioavailable inhibitors of the sodium hydrogen exchanger isoform-1. Bioorg Med Chem Lett 14: 177 -80 (2003) El-Gamil, DS; ElHady, AK; Chen, PJ; Hwang, TL; Abadi, AH; Abdel-Halim, M; Engel, M Development of novel conformationally restricted selective Clk1/4 inhibitors through creating an intramolecular hydrogen bond involving an imide linker. Eur J Med Chem 238: (2022) Shore, DGM; Sweeney, ZK; Beresford, A; Chan, BK; Chen, H; Drummond, J; Gill, A; Kleinheinz, T; Liu, X; Medhurst, AD; McIver, EG; Moffat, JG; Zhu, H; Estrada, AA Discovery of potent azaindazole leucine-rich repeat kinase 2 (LRRK2) inhibitors possessing a key intramolecular hydrogen bond - Part 2. Bioorg Med Chem Lett 29: 674 -680 (2019) Guzman-Perez, A; Wester, RT; Allen, MC; Brown, JA; Buchholz, AR; Cook, ER; Day, WW; Hamanaka, ES; Kennedy, SP; Knight, DR; Kowalczyk, PJ; Marala, RB; Mularski, CJ; Novomisle, WA; Ruggeri, RB; Tracey, WR; Hill, RJ Discovery of zoniporide: a potent and selective sodium-hydrogen exchanger type 1 (NHE-1) inhibitor with high aqueous solubility. Bioorg Med Chem Lett 11: 803 -7 (2001) Mellon, C; Aspiotis, R; Black, CW; Bayly, CI; Grimm, EL; Giroux, A; Han, Y; Isabel, E; McKay, DJ; Nicholson, DW; Rasper, DM; Roy, S; Tam, J; Thornberry, NA; Vaillancourt, JP; Xanthoudakis, S; Zamboni, R Lipophilic versus hydrogen-bonding effect in P3 on potency and selectivity of valine aspartyl ketones as caspase 3 inhibitors. Bioorg Med Chem Lett 15: 3886 -90 (2005) Gemma, S; Camodeca, C; Brindisi, M; Brogi, S; Kukreja, G; Kunjir, S; Gabellieri, E; Lucantoni, L; Habluetzel, A; Taramelli, D; Basilico, N; Gualdani, R; Tadini-Buoninsegni, F; Bartolommei, G; Moncelli, MR; Martin, RE; Summers, RL; Lamponi, S; Savini, L; Fiorini, I; Valoti, M; Novellino, E; Campiani, G; Butini, S Mimicking the intramolecular hydrogen bond: synthesis, biological evaluation, and molecular modeling of benzoxazines and quinazolines as potential antimalarial agents. J Med Chem 55: 10387 -404 (2012) Lange, JH; van der Neut, MA; den Hartog, AP; Wals, HC; Hoogendoorn, J; van Stuivenberg, HH; van Vliet, BJ; Kruse, CG Synthesis, SAR and intramolecular hydrogen bonding pattern of 1,3,5-trisubstituted 4,5-dihydropyrazoles as potent cannabinoid CB(1) receptor antagonists. Bioorg Med Chem Lett 20: 1752 -7 (2010) Brunschweiger, A; Koch, P; Schlenk, M; Rafehi, M; Radjainia, H; Küppers, P; Hinz, S; Pineda, F; Wiese, M; Hockemeyer, J; Heer, J; Denonne, F; Müller, CE 8-Substituted 1,3-dimethyltetrahydropyrazino[2,1-f]purinediones: Water-soluble adenosine receptor antagonists and monoamine oxidase B inhibitors. Bioorg Med Chem 24: 5462 -5480 (2016) Harrison, T; Owens, AP; Williams, BJ; Swain, CJ; Williams, A; Carlson, EJ; Rycroft, W; Tattersall, FD; Cascieri, MA; Chicchi, GG; Sadowski, S; Rupniak, NM; Hargreaves, RJ An orally active, water-soluble neurokinin-1 receptor antagonist suitable for both intravenous and oral clinical administration. J Med Chem 44: 4296 -9 (2001) Watts, KR; Ratnam, J; Ang, KH; Tenney, K; Compton, JE; McKerrow, J; Crews, P Assessing the trypanocidal potential of natural and semi-synthetic diketopiperazines from two deep water marine-derived fungi. Bioorg Med Chem 18: 2566 -74 (2010) Lubisch, W; Beckenbach, E; Bopp, S; Hofmann, HP; Kartal, A; Kästel, C; Lindner, T; Metz-Garrecht, M; Reeb, J; Regner, F; Vierling, M; Möller, A Benzoylalanine-derived ketoamides carrying vinylbenzyl amino residues: discovery of potent water-soluble calpain inhibitors with oral bioavailability. J Med Chem 46: 2404 -12 (2003) Burmistrov, V; Morisseau, C; Karlov, D; Pitushkin, D; Vernigora, A; Rasskazova, E; Butov, GM; Hammock, BD Bioisosteric substitution of adamantane with bicyclic lipophilic groups improves water solubility of human soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett 30: (2020) Casini, A; Scozzafava, A; Mincione, F; Menabuoni, L; Ilies, MA; Supuran, CT Carbonic anhydrase inhibitors: water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J Med Chem 43: 4884 -92 (2000) Chambers, MS; Hobbs, SC; Graham, MI; Watt, AP; Fletcher, SR; Baker, R; Freedman, SB; Patel, S; Smith, AJ; Matassa, VG Potent, selective, water-soluble benzodiazepine-based CCKB receptor antagonists that contain lipophilic carboxylate surrogates Bioorg Med Chem Lett 5: 2303 -2308 (1995) Luzzio, MJ; Besterman, JM; Emerson, DL; Evans, MG; Lackey, K; Leitner, PL; McIntyre, G; Morton, B; Myers, PL; Peel, M Synthesis and antitumor activity of novel water soluble derivatives of camptothecin as specific inhibitors of topoisomerase I. J Med Chem 38: 395 -401 (1995) Yang, J; Li, LL; Li, JR; Yang, JX; Zhang, F; Chen, G; Yu, R; Ouyang, WJ; Wu, SW Synthesis and biological evaluation of water-soluble derivatives of chiral gossypol as HIV fusion inhibitors targeting gp41. Bioorg Med Chem Lett 28: 49 -52 (2018) Bavetsias, V; Skelton, LA; Yafai, F; Mitchell, F; Wilson, SC; Allan, B; Jackman, AL The design and synthesis of water-soluble analogues of CB30865, a quinazolin-4-one-based antitumor agent. J Med Chem 45: 3692 -702 (2002) Pierce, AC; ter Haar, E; Binch, HM; Kay, DP; Patel, SR; Li, P CH...O and CH...N hydrogen bonds in ligand design: a novel quinazolin-4-ylthiazol-2-ylamine protein kinase inhibitor. J Med Chem 48: 1278 -81 (2005) Liu, C; Li, H; Xu, F; Jiang, X; Ma, H; Seeram, NP Cannabidiol Protects Human Skin Keratinocytes from Hydrogen-Peroxide-Induced Oxidative Stress via Modulation of the Caspase-1-IL-1β Axis. J Nat Prod 84: 1563 -1572 (2021) Hong, L; Fast, W Inhibition of human dimethylarginine dimethylaminohydrolase-1 by S-nitroso-L-homocysteine and hydrogen peroxide. Analysis, quantification, and implications for hyperhomocysteinemia. J Biol Chem 282: 34684 -92 (2007) Venables, BL; Sin, N; Wang, AX; Sun, LQ; Tu, Y; Hernandez, D; Sheaffer, A; Lee, M; Dunaj, C; Zhai, G; Barry, D; Friborg, J; Yu, F; Knipe, J; Sandquist, J; Falk, P; Parker, D; Good, AC; Rajamani, R; McPhee, F; Meanwell, NA; Scola, PM P3-P4 ureas and reverse carbamates as potent HCV NS3 protease inhibitors: Effective transposition of the P4 hydrogen bond donor. Bioorg Med Chem Lett 28: 1853 -1859 (2018) Anderson, A; Belelli, D; Bennett, DJ; Buchanan, KI; Casula, A; Cooke, A; Feilden, H; Gemmell, DK; Hamilton, NM; Hutchinson, EJ; Lambert, JJ; Maidment, MS; McGuire, R; McPhail, P; Miller, S; Muntoni, A; Peters, JA; Sansbury, FH; Stevenson, D; Sundaram, H Alpha-amino acid phenolic ester derivatives: novel water-soluble general anesthetic agents which allosterically modulate GABA(A) receptors. J Med Chem 44: 3582 -91 (2001) Sun, H; Xi, M; Jin, Q; Zhu, Z; Zhang, Y; Jia, G; Zhu, G; Sun, M; Zhang, H; Ren, X; Zhang, Y; Xu, Z; Huang, H; Shen, J; Li, B; Ge, G; Chen, K; Zhu, W Chemo- and Site-Selective Lysine Modification of Peptides and Proteins under Native Conditions Using the Water-Soluble Zolinium. J Med Chem 65: 11840 -11853 (2022) Blanco, B; Sedes, A; Peón, A; Otero, JM; van Raaij, MJ; Thompson, P; Hawkins, AR; González-Bello, C Exploring the water-binding pocket of the type II dehydroquinase enzyme in the structure-based design of inhibitors. J Med Chem 57: 3494 -510 (2014) Burmistrov, V; Morisseau, C; D'yachenko, V; Karlov, D; Butov, GM; Hammock, BD Imidazolidine-2,4,5- and pirimidine-2,4,6-triones - New primary pharmacophore for soluble epoxide hydrolase inhibitors with enhanced water solubility. Bioorg Med Chem Lett 30: (2020) Vullo, D; Nishimori, I; Scozzafava, A; Köhler, S; Winum, JY; Supuran, CT Inhibition studies of a beta-carbonic anhydrase from Brucella suis with a series of water soluble glycosyl sulfanilamides. Bioorg Med Chem Lett 20: 2178 -82 (2010) Horbert, R; Pinchuk, B; Johannes, E; Schlosser, J; Schmidt, D; Cappel, D; Totzke, F; Schächtele, C; Peifer, C Optimization of potent DFG-in inhibitors of platelet derived growth factor receptorß (PDGF-Rß) guided by water thermodynamics. J Med Chem 58: 170 -82 (2015) Gunasekera, SP; McCarthy, PJ; Longley, RE; Pomponi, SA; Wright, AE Secobatzellines A and B, two new enzyme inhibitors from a deep-water Caribbean sponge of the genus Batzella. J Nat Prod 62: 1208 -11 (1999) Cozzini, P; Fornabaio, M; Marabotti, A; Abraham, DJ; Kellogg, GE; Mozzarelli, A Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 1. Models without explicit constrained water. J Med Chem 45: 2469 -83 (2002) Nakamura, T; Wada, H; Kurebayashi, H; McInally, T; Bonnert, R; Isobe, Y Synthesis and evaluation of 8-oxoadenine derivatives as potent Toll-like receptor 7 agonists with high water solubility. Bioorg Med Chem Lett 23: 669 -72 (2013) Smith, ND; Reger, TS; Payne, J; Zunic, J; Lorrain, D; Correa, L; Stock, N; Cramer, M; Chen, W; Yang, J; Prasit, P; Munoz, B Water soluble prodrug of a COX-2 selective inhibitor suitable for intravenous administration in models of cerebral ischemia. Bioorg Med Chem Lett 15: 3197 -200 (2005) Parrino, B; Carbone, A; Ciancimino, C; Spanò, V; Montalbano, A; Barraja, P; Cirrincione, G; Diana, P; Sissi, C; Palumbo, M; Pinato, O; Pennati, M; Beretta, G; Folini, M; Matyus, P; Balogh, B; Zaffaroni, N Water-soluble isoindolo[2,1-a]quinoxalin-6-imines: in vitro antiproliferative activity and molecular mechanism(s) of action. Eur J Med Chem 94: 149 -62 (2015) Ma, X; Lv, X; Qiu, N; Yang, B; He, Q; Hu, Y Discovery of novel quinoline-based mTOR inhibitors via introducing intra-molecular hydrogen bonding scaffold (iMHBS): The design, synthesis and biological evaluation. Bioorg Med Chem 23: 7585 -96 (2015) Tropak, MB; Kornhaber, GJ; Rigat, BA; Maegawa, GH; Buttner, JD; Blanchard, JE; Murphy, C; Tuske, SJ; Coales, SJ; Hamuro, Y; Brown, ED; Mahuran, DJ Identification of pharmacological chaperones for Gaucher disease and characterization of their effects on beta-glucocerebrosidase by hydrogen/deuterium exchange mass spectrometry. Chembiochem 9: 2650 -62 (2008) Peng, YH; Ueng, SH; Tseng, CT; Hung, MS; Song, JS; Wu, JS; Liao, FY; Fan, YS; Wu, MH; Hsiao, WC; Hsueh, CC; Lin, SY; Cheng, CY; Tu, CH; Lee, LC; Cheng, MF; Shia, KS; Shih, C; Wu, SY Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives with IDO1. J Med Chem 59: 282 -93 (2016) Wu, B; Barrios Sosa, AC; Boschelli, DH; Boschelli, F; Honores, EE; Golas, JM; Powell, DW; Wang, YD 7-(aryl/heteroaryl-2-ylethynyl)-4-phenylamino-3-quinolinecarbonitriles as new Src kinase inhibitors: addition of water solubilizing groups. Bioorg Med Chem Lett 16: 3993 -7 (2006) Andreev, S; Pantsar, T; Tesch, R; Kahlke, N; El-Gokha, A; Ansideri, F; Grätz, L; Romasco, J; Sita, G; Geibel, C; Lämmerhofer, M; Tarozzi, A; Knapp, S; Laufer, SA; Koch, P Addressing a Trapped High-Energy Water: Design and Synthesis of Highly Potent Pyrimidoindole-Based Glycogen Synthase Kinase-3β Inhibitors. J Med Chem 65: 1283 -1301 (2022) Cao, X; Sun, Z; Cao, Y; Wang, R; Cai, T; Chu, W; Hu, W; Yang, Y Design, synthesis, and structure-activity relationship studies of novel fused heterocycles-linked triazoles with good activity and water solubility. J Med Chem 57: 3687 -706 (2014) O'Dowd, H; Shannon, DE; Chandupatla, KR; Dixit, V; Engtrakul, JJ; Ye, Z; Jones, SM; O'Brien, CF; Nicolau, DP; Tessier, PR; Crandon, JL; Song, B; Macikenas, D; Hanzelka, BL; Le Tiran, A; Bennani, YL; Charifson, PS; Grillot, AL Discovery and Characterization of a Water-Soluble Prodrug of a Dual Inhibitor of Bacterial DNA Gyrase and Topoisomerase IV. ACS Med Chem Lett 6: 822 -6 (2015) Gu, H; Zhang, T; Guan, T; Wu, M; Li, S; Li, Y; Guo, M; Zhang, L; Peng, Y; Mi, D; Liu, M; Yi, Z; Chen, Y Discovery of a Highly Potent and Selective MYOF Inhibitor with Improved Water Solubility for the Treatment of Gastric Cancer. J Med Chem 66: 16917 -16938 (2023) Showell, GA; Bourrain, S; Neduvelil, JG; Fletcher, SR; Baker, R; Watt, AP; Fletcher, AE; Freedman, SB; Kemp, JA; Marshall, GR High-affinity and potent, water-soluble 5-amino-1,4-benzodiazepine CCKB/gastrin receptor antagonists containing a cationic solubilizing group. J Med Chem 37: 719 -21 (1994) Bonardi, A; Micheli, L; Di Cesare Mannelli, L; Ghelardini, C; Gratteri, P; Nocentini, A; Supuran, CT Development of Hydrogen Sulfide-Releasing Carbonic Anhydrases IX- and XII-Selective Inhibitors with Enhanced Antihyperalgesic Action in a Rat Model of Arthritis. J Med Chem 65: 13143 -13157 (2022) Aso, K; Kobayashi, K; Mochizuki, M; Kanzaki, N; Sako, Y; Yano, T Discovery of pyrrolo[2,3-d]pyrimidin-4-ones as corticotropin-releasing factor 1 receptor antagonists with a carbonyl-based hydrogen bonding acceptor. Bioorg Med Chem Lett 21: 2365 -71 (2011) Uchida, T; Dojun, N; Sekine, Y; Ishimori, K Heme Proximal Hydrogen Bonding between His170 and Asp132 Plays an Essential Role in the Heme Degradation Reaction of HutZ from Vibrio cholerae. Biochemistry 56: 2723 -2734 (2017) Lee, D; Lee, S; Choi, J; Song, YK; Kim, MJ; Shin, DS; Bae, MA; Kim, YC; Park, CJ; Lee, KR; Choi, JH; Seo, J Interplay among Conformation, Intramolecular Hydrogen Bonds, and Chameleonicity in the Membrane Permeability and Cyclophilin A Binding of Macrocyclic Peptide Cyclosporin O Derivatives. J Med Chem 64: 8272 -8286 (2021) Killday, KB; Wright, AE; Jackson, RH; Sills, MA Bis(sulfato)-cyclosiphonodictyol A, a new disulfated sesquiterpene-hydroquinone from a deep water collection of the Marine sponge Siphonodictyon coralliphagum. J Nat Prod 58: 958 -60 (1995) de Graaf, C; Oostenbrink, C; Keizers, PH; van der Wijst, T; Jongejan, A; Vermeulen, NP Catalytic site prediction and virtual screening of cytochrome P450 2D6 substrates by consideration of water and rescoring in automated docking. J Med Chem 49: 2417 -30 (2006) Takaya, D; Inaka, K; Omura, A; Takenuki, K; Kawanishi, M; Yabuki, Y; Nakagawa, Y; Tsuganezawa, K; Ogawa, N; Watanabe, C; Honma, T; Aritake, K; Urade, Y; Shirouzu, M; Tanaka, A Characterization of crystal water molecules in a high-affinity inhibitor and hematopoietic prostaglandin D synthase complex by interaction energy studies. Bioorg Med Chem 26: 4726 -4734 (2018) Navidpour, L; Amini, M; Shafaroodi, H; Abdi, K; Ghahremani, MH; Dehpour, AR; Shafiee, A Design and synthesis of new water-soluble tetrazolide derivatives of celecoxib and rofecoxib as selective cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem Lett 16: 4483 -7 (2006) Ge, J; Normant, E; Porter, JR; Ali, JA; Dembski, MS; Gao, Y; Georges, AT; Grenier, L; Pak, RH; Patterson, J; Sydor, JR; Tibbitts, TT; Tong, JK; Adams, J; Palombella, VJ Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90. J Med Chem 49: 4606 -15 (2006) Inoue, Y; Omodani, T; Shiratake, R; Okazaki, H; Kuromiya, A; Kubo, T; Sato, F Development of a highly water-soluble peptide-based human neutrophil elastase inhibitor; AE-3763 for treatment of acute organ injury. Bioorg Med Chem 17: 7477 -86 (2009) Masuda, T; Nakayama, Y Development of a water-soluble matrix metalloproteinase inhibitor as an intra-arterial infusion drug for prevention of restenosis after angioplasty. J Med Chem 46: 3497 -501 (2003) Danil de Namor, AF; Traboulssi, R; Lewis, DF Host properties of cyclodextrins towards anion constituents of antigenic determinants. A thermodynamic study in water and in N,N-dimethylformamide J Am Chem Soc 112: 8442 -7 (1990) Slee, DH; Zhang, X; Moorjani, M; Lin, E; Lanier, MC; Chen, Y; Rueter, JK; Lechner, SM; Markison, S; Malany, S; Joswig, T; Santos, M; Gross, RS; Williams, JP; Castro-Palomino, JC; Crespo, MI; Prat, M; Gual, S; Díaz, JL; Wen, J; O'Brien, Z; Saunders, J Identification of novel, water-soluble, 2-amino-N-pyrimidin-4-yl acetamides as A2A receptor antagonists with in vivo efficacy. J Med Chem 51: 400 -6 (2008) Chen, C; Wilcoxen, KM; Huang, CQ; McCarthy, JR; Chen, T; Grigoriadis, DE Optimization of 3-phenylpyrazolo[1,5-a]pyrimidines as potent corticotropin-releasing factor-1 antagonists with adequate lipophilicity and water solubility. Bioorg Med Chem Lett 14: 3669 -73 (2004) Fujimoto, K; Yoshida, S; Tadano, G; Asada, N; Fuchino, K; Suzuki, S; Matsuoka, E; Yamamoto, T; Yamamoto, S; Ando, S; Kanegawa, N; Tonomura, Y; Ito, H; Moechars, D; Rombouts, FJR; Gijsen, HJM; Kusakabe, KI Structure-Based Approaches to Improving Selectivity through Utilizing Explicit Water Molecules: Discovery of Selective β-Secretase (BACE1) Inhibitors over BACE2. J Med Chem 64: 3075 -3085 (2021) Nickell, JR; Culver, JP; Janganati, V; Zheng, G; Dwoskin, LP; Crooks, PA Synthesis and in vitro evaluation of water-soluble 1,4-diphenethylpiperazine analogs as novel inhibitors of the vesicular monoamine transporter-2. Bioorg Med Chem Lett 26: 4441 -4445 (2016) Larroque, AL; Peori, B; Williams, C; Fang, YQ; Qiu, Q; Rachid, Z; Jean-Claude, BJ Synthesis of water soluble bis-triazenoquinazolines: an unusual predicted mode of binding to the epidermal growth factor receptor tyrosine kinase. Chem Biol Drug Des 71: 374 -9 (2008) Rai, R; Katzenellenbogen, JA Effect of conformational mobility and hydrogen-bonding interactions on the selectivity of some guanidinoaryl-substituted mechanism-based inhibitors of trypsin-like serine proteases. J Med Chem 35: 4297 -305 (1992) Muley, L; Baum, B; Smolinski, M; Freindorf, M; Heine, A; Klebe, G; Hangauer, DG Enhancement of hydrophobic interactions and hydrogen bond strength by cooperativity: synthesis, modeling, and molecular dynamics simulations of a congeneric series of thrombin inhibitors. J Med Chem 53: 2126 -35 (2010) Oh, J; Quan, KT; Lee, JS; Park, I; Kim, CS; Ferreira, D; Thuong, PT; Kim, YH; Na, M NMR-Based Investigation of Hydrogen Bonding in a Dihydroanthracen-1(4 H)one from Rubia philippinensis and Its Soluble Epoxide Hydrolase Inhibitory Potential. J Nat Prod 81: 2429 -2435 (2018) Nakagawa, Y; Irie, K; Nakamura, Y; Ohigashi, H The amide hydrogen of (-)-indolactam-V and benzolactam-V8's plays a critical role in protein kinase C binding and tumor-promoting activities. Bioorg Med Chem Lett 11: 723 -8 (2001) Scozzafava, A; Briganti, F; Mincione, G; Menabuoni, L; Mincione, F; Supuran, CT Carbonic anhydrase inhibitors: synthesis of water-soluble, aminoacyl/dipeptidyl sulfonamides possessing long-lasting intraocular pressure-lowering properties via the topical route. J Med Chem 42: 3690 -700 (1999) Kim, IH; Morisseau, C; Watanabe, T; Hammock, BD Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J Med Chem 47: 2110 -22 (2004) Richard, DJ; Verheijen, JC; Curran, K; Kaplan, J; Toral-Barza, L; Hollander, I; Lucas, J; Yu, K; Zask, A Incorporation of water-solubilizing groups in pyrazolopyrimidine mTOR inhibitors: discovery of highly potent and selective analogs with improved human microsomal stability. Bioorg Med Chem Lett 19: 6830 -5 (2009) Xie, R; Li, AH; Ji, XD; Melman, N; Olah, ME; Stiles, GL; Jacobson, KA Selective A(3) adenosine receptor antagonists: water-soluble 3, 5-diacyl-1,2,4-trialkylpyridinium salts and their oxidative generation from dihydropyridine precursors. J Med Chem 42: 4232 -8 (1999) Maconi, A; Pastorin, G; Da Ros, T; Spalluto, G; Gao, ZG; Jacobson, KA; Baraldi, PG; Cacciari, B; Varani, K; Moro, S; Borea, PA Synthesis, biological properties, and molecular modeling investigation of the first potent, selective, and water-soluble human A(3) adenosine receptor antagonist. J Med Chem 45: 3579 -82 (2002) Swaminathan, CP; Nandi, A; Visweswariah, SS; Surolia, A Thermodynamic analyses reveal role of water release in epitope recognition by a monoclonal antibody against the human guanylyl cyclase C receptor. J Biol Chem 274: 31272 -8 (1999) Nasief, NN; Said, AM; Hangauer, D Modulating hydrogen-bond basicity within the context of protein-ligand binding: A case study with thrombin inhibitors that reveals a dominating role for desolvation. Eur J Med Chem 125: 975 -991 (2017) Abbate, F; Supuran, CT; Scozzafava, A; Orioli, P; Stubbs, MT; Klebe, G Nonaromatic sulfonamide group as an ideal anchor for potent human carbonic anhydrase inhibitors: role of hydrogen-bonding networks in ligand binding and drug design. J Med Chem 45: 3583 -7 (2002) Thaher, BA; Koch, P; Schattel, V; Laufer, S Role of the hydrogen bonding heteroatom-Lys53 interaction between the p38alpha mitogen-activated protein (MAP) kinase and pyridinyl-substituted 5-membered heterocyclic ring inhibitors. J Med Chem 52: 2613 -7 (2009) Pisani, L; Farina, R; Catto, M; Iacobazzi, RM; Nicolotti, O; Cellamare, S; Mangiatordi, GF; Denora, N; Soto-Otero, R; Siragusa, L; Altomare, CD; Carotti, A Exploring Basic Tail Modifications of Coumarin-Based Dual Acetylcholinesterase-Monoamine Oxidase B Inhibitors: Identification of Water-Soluble, Brain-Permeant Neuroprotective Multitarget Agents. J Med Chem 59: 6791 -806 (2016) Müller, CE; Thorand, M; Qurishi, R; Diekmann, M; Jacobson, KA; Padgett, WL; Daly, JW Imidazo[2,1-i]purin-5-ones and related tricyclic water-soluble purine derivatives: potent A(2A)- and A(3)-adenosine receptor antagonists. J Med Chem 45: 3440 -50 (2002) Kasuga, J; Ishikawa, M; Yonehara, M; Makishima, M; Hashimoto, Y; Miyachi, H Improvement of water-solubility of biarylcarboxylic acid peroxisome proliferator-activated receptor (PPAR)d-selective partial agonists by disruption of molecular planarity/symmetry. Bioorg Med Chem 18: 7164 -73 (2010) Frédérick, R; Charlier, C; Robert, S; Wouters, J; Masereel, B; Pochet, L Investigation of mechanism-based thrombin inhibitors: Implications of a highly conserved water molecule for the binding of coumarins within the S pocket. Bioorg Med Chem Lett 16: 2017 -21 (2006) Poon, SF; Stock, N; Payne, JE; McGuire, AR; Stearns, B; Yang, X; Chen, W; Munoz, B; Smith, ND Novel approach to pro-drugs of lactones: water soluble imidate and ortho-ester derivatives of a furanone-based COX-2 selective inhibitor. Bioorg Med Chem Lett 15: 2259 -63 (2005) Jacobson, KA; Nikodijevic, O; Ji, XD; Berkich, DA; Eveleth, D; Dean, RL; Hiramatsu, K; Kassell, NF; van Galen, PJ; Lee, KS Synthesis and biological activity of N6-(p-sulfophenyl)alkyl and N6-sulfoalkyl derivatives of adenosine: water-soluble and peripherally selective adenosine agonists. J Med Chem 35: 4143 -9 (1992) Parker, EN; Odutola, SO; Wang, Y; Strecker, TE; Mukherjee, R; Shi, Z; Chaplin, DJ; Trawick, ML; Pinney, KG Synthesis and biological evaluation of a water-soluble phosphate prodrug salt and structural analogues of KGP94, a lead inhibitor of cathepsin L. Bioorg Med Chem Lett 27: 1304 -1310 (2017) Verner, E; Katz, BA; Spencer, JR; Allen, D; Hataye, J; Hruzewicz, W; Hui, HC; Kolesnikov, A; Li, Y; Luong, C; Martelli, A; Radika, K; Rai, R; She, M; Shrader, W; Sprengeler, PA; Trapp, S; Wang, J; Young, WB; Mackman, RL Development of serine protease inhibitors displaying a multicentered short (<2.3 A) hydrogen bond binding mode: inhibitors of urokinase-type plasminogen activator and factor Xa. J Med Chem 44: 2753 -71 (2001) Moon, HR; Lee, HJ; Kim, KR; Lee, KM; Lee, SK; Kim, HO; Chun, MW; Jeong, LS Synthesis of 5'-substituted fluoro-neplanocin A analogues: importance of a hydrogen bonding donor at 5'-position for the inhibitory activity of S-adenosylhomocysteine hydrolase. Bioorg Med Chem Lett 14: 5641 -4 (2004) Ozawa, T; Tsuji, E; Ozawa, M; Handa, C; Mukaiyama, H; Nishimura, T; Kobayashi, S; Okazaki, K The importance of CH/pi hydrogen bonds in rational drug design: An ab initio fragment molecular orbital study to leukocyte-specific protein tyrosine (LCK) kinase. Bioorg Med Chem 16: 10311 -8 (2008) Kamaura, M; Kubo, O; Sugimoto, H; Noguchi, N; Miyashita, H; Abe, S; Matsuda, K; Tsujihata, Y; Odani, T; Iwasaki, S; Murata, T; Sato, K Discovery of a novel series of indolinylpyrimidine-based GPR119 agonists: Elimination of ether-a-go-go-related gene liability using a hydrogen bond acceptor-focused approach. Bioorg Med Chem 34: (2021) Pastor, M; Cruciani, G; Watson, KA A strategy for the incorporation of water molecules present in a ligand binding site into a three-dimensional quantitative structure--activity relationship analysis. J Med Chem 40: 4089 -102 (1998) Stierle, DB; Stierle, AA; Patacini, B; McIntyre, K; Girtsman, T; Bolstad, E Berkeleyones and related meroterpenes from a deep water acid mine waste fungus that inhibit the production of interleukin 1-ß from induced inflammasomes. J Nat Prod 74: 2273 -7 (2011) Devkota, L; Lin, CM; Strecker, TE; Wang, Y; Tidmore, JK; Chen, Z; Guddneppanavar, R; Jelinek, CJ; Lopez, R; Liu, L; Hamel, E; Mason, RP; Chaplin, DJ; Trawick, ML; Pinney, KG Design, synthesis, and biological evaluation of water-soluble amino acid prodrug conjugates derived from combretastatin, dihydronaphthalene, and benzosuberene-based parent vascular disrupting agents. Bioorg Med Chem 24: 938 -56 (2016) S El Salamouni, N; Buckley, BJ; Jiang, L; Huang, M; Ranson, M; Kelso, MJ; Yu, H Disruption of Water Networks is the Cause of Human/Mouse Species Selectivity in Urokinase Plasminogen Activator (uPA) Inhibitors Derived from Hexamethylene Amiloride (HMA). J Med Chem 65: 1933 -1945 (2022) Sliskovic, DR; Krause, BR; Picard, JA; Anderson, M; Bousley, RF; Hamelehle, KL; Homan, R; Julian, TN; Rashidbaigi, ZA; Stanfield, RL Inhibitors of acyl-CoA: cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid-regulating activity. J Med Chem 37: 560 -2 (1994) Davies, NG; Browne, H; Davis, B; Drysdale, MJ; Foloppe, N; Geoffrey, S; Gibbons, B; Hart, T; Hubbard, R; Jensen, MR; Mansell, H; Massey, A; Matassova, N; Moore, JD; Murray, J; Pratt, R; Ray, S; Robertson, A; Roughley, SD; Schoepfer, J; Scriven, K; Simmonite, H; Stokes, S; Surgenor, A; Webb, P; Wood, M; Wright, L; Brough, P Targeting conserved water molecules: design of 4-aryl-5-cyanopyrrolo[2,3-d]pyrimidine Hsp90 inhibitors using fragment-based screening and structure-based optimization. Bioorg Med Chem 20: 6770 -89 (2012) Yamamoto, T; Niwa, S; Ohno, S; Tokumasu, M; Masuzawa, Y; Nakanishi, C; Nakajo, A; Onishi, T; Koganei, H; Fujita, S; Takeda, T; Kito, M; Ono, Y; Saitou, Y; Takahara, A; Iwata, S; Shoji, M The structure-activity relationship study on 2-, 5-, and 6-position of the water soluble 1,4-dihydropyridine derivatives blocking N-type calcium channels. Bioorg Med Chem Lett 18: 4813 -6 (2008) Wang, T; Lee, HJ; Tosh, DK; Kim, HO; Pal, S; Choi, S; Lee, Y; Moon, HR; Zhao, LX; Lee, KM; Jeong, LS Design, synthesis, and molecular modeling studies of 5'-deoxy-5'-ureidoadenosine: 5'-ureido group as multiple hydrogen bonding donor in the active site of S-adenosylhomocysteine hydrolase. Bioorg Med Chem Lett 17: 4456 -9 (2007) Discovery of Novel Inhibitors of BRD4 for Treating Prostate Cancer: A Comprehensive Case Study for Considering Water Networks in Virtual Screening and Drug Design. Morimoto, H; Ohashi, N; Shimadzu, H; Kushiyama, E; Kawanishi, H; Hosaka, T; Kawase, Y; Yasuda, K; Kikkawa, K; Yamauchi-Kohno, R; Yamada, K Potent and selective ET-A antagonists. 2. Discovery and evaluation of potent and water soluble N-(6-(2-(aryloxy)ethoxy)-4-pyrimidinyl)sulfonamide derivatives. J Med Chem 44: 3369 -77 (2001) Forsch, RA; Queener, SF; Rosowsky, A Preliminary in vitro studies on two potent, water-soluble trimethoprim analogues with exceptional species selectivity against dihydrofolate reductase from Pneumocystis carinii and Mycobacterium avium. Bioorg Med Chem Lett 14: 1811 -5 (2004) Mikol, V; Papageorgiou, C; Borer, X The role of water molecules in the structure-based design of (5-hydroxynorvaline)-2-cyclosporin: synthesis, biological activity, and crystallographic analysis with cyclophilin A. J Med Chem 38: 3361 -7 (1995) Fu, W; Zhang, M; Liao, J; Tang, Q; Lei, Y; Gong, Z; Shan, L; Duan, M; Chai, X; Pang, J; Tang, C; Wang, X; Xu, X; Li, D; Sheng, R; Hou, T Discovery of a Novel Androgen Receptor Antagonist Manifesting Evidence to Disrupt the Dimerization of the Ligand-Binding Domain via Attenuating the Hydrogen-Bonding Network Between the Two Monomers. J Med Chem 64: 17221 -17238 (2021) Suzuki, F; Shimada, J; Nonaka, H; Ishii, A; Shiozaki, S; Ichikawa, S; Ono, E 7,8-Dihydro-8-ethyl-2-(3-noradamantyl)-4-propyl-1H-imidazo[2,1-i]purin-5(4H)-one: a potent and water-soluble adenosine A1 antagonist. J Med Chem 35: 3578 -81 (1992) Cheng, X; Merz, KH; Vatter, S; Zeller, J; Muehlbeyer, S; Thommet, A; Christ, J; Wölfl, S; Eisenbrand, G Identification of a Water-Soluble Indirubin Derivative as Potent Inhibitor of Insulin-like Growth Factor 1 Receptor through Structural Modification of the Parent Natural Molecule. J Med Chem 60: 4949 -4962 (2017) Zhang, X; Raghavan, S; Ihnat, M; Thorpe, JE; Disch, BC; Bastian, A; Bailey-Downs, LC; Dybdal-Hargreaves, NF; Rohena, CC; Hamel, E; Mooberry, SL; Gangjee, A The design and discovery of water soluble 4-substituted-2,6-dimethylfuro[2,3-d]pyrimidines as multitargeted receptor tyrosine kinase inhibitors and microtubule targeting antitumor agents. Bioorg Med Chem 22: 3753 -72 (2014) Sakamoto, T; Koga, Y; Hikota, M; Matsuki, K; Murakami, M; Kikkawa, K; Fujishige, K; Kotera, J; Omori, K; Morimoto, H; Yamada, K Design and synthesis of novel 5-(3,4,5-trimethoxybenzoyl)-4-aminopyrimidine derivatives as potent and selective phosphodiesterase 5 inhibitors: scaffold hopping using a pseudo-ring by intramolecular hydrogen bond formation. Bioorg Med Chem Lett 24: 5175 -80 (2014) Temperini, C; Cecchi, A; Scozzafava, A; Supuran, CT Carbonic anhydrase inhibitors. Comparison of chlorthalidone, indapamide, trichloromethiazide, and furosemide X-ray crystal structures in adducts with isozyme II, when several water molecules make the difference. Bioorg Med Chem 17: 1214 -21 (2009) Federico, S; Paoletta, S; Cheong, SL; Pastorin, G; Cacciari, B; Stragliotto, S; Klotz, KN; Siegel, J; Gao, ZG; Jacobson, KA; Moro, S; Spalluto, G Synthesis and Biological Evaluation of a New Series of 1,2,4-Triazolo[1,5-a]-1,3,5-triazines as Human A(2A) Adenosine Receptor Antagonists with Improved Water Solubility. J Med Chem 54: 877 -89 (2011) Eibl, C; Tomassoli, I; Munoz, L; Stokes, C; Papke, RL; Gündisch, D The 3,7-diazabicyclo[3.3.1]nonane scaffold for subtype selective nicotinic acetylcholine receptor (nAChR) ligands. Part 1: the influence of different hydrogen bond acceptor systems on alkyl and (hetero)aryl substituents. Bioorg Med Chem 21: 7283 -308 (2013) Meanwell, NA; Hewawasam, P; Thomas, JA; Wright, JJ; Russell, JW; Gamberdella, M; Goldenberg, HJ; Seiler, SM; Zavoico, GB Inhibitors of blood platelet cAMP phosphodiesterase. 4. Structural variation of the side-chain terminus of water-soluble 1,3-dihydro-2H-imidazo[4,5-b]quinolin-2-one derivatives. J Med Chem 36: 3251 -64 (1993) Cox, CD; Torrent, M; Breslin, MJ; Mariano, BJ; Whitman, DB; Coleman, PJ; Buser, CA; Walsh, ES; Hamilton, K; Schaber, MD; Lobell, RB; Tao, W; South, VJ; Kohl, NE; Yan, Y; Kuo, LC; Prueksaritanont, T; Slaughter, DE; Li, C; Mahan, E; Lu, B; Hartman, GD Kinesin spindle protein (KSP) inhibitors. Part 4: Structure-based design of 5-alkylamino-3,5-diaryl-4,5-dihydropyrazoles as potent, water-soluble inhibitors of the mitotic kinesin KSP. Bioorg Med Chem Lett 16: 3175 -9 (2006) Seo, J; Igarashi, J; Li, H; Martasek, P; Roman, LJ; Poulos, TL; Silverman, RB Structure-based design and synthesis of N(omega)-nitro-L-arginine-containing peptidomimetics as selective inhibitors of neuronal nitric oxide synthase. Displacement of the heme structural water. J Med Chem 50: 2089 -99 (2007) Grunewald, GL; Dahanukar, VH; Caldwell, TM; Criscione, KR Examination of the role of the acidic hydrogen in imparting selectivity of 7-(aminosulfonyl)-1,2,3,4-tetrahydroisoquinoline (SK&F 29661) toward inhibition of phenylethanolamine N-methyltransferase vs the alpha 2-adrenoceptor. J Med Chem 40: 3997 -4005 (1998) Alonso, JA; Andrés, M; Bravo, M; Buil, MA; Calbet, M; Castro, J; Eastwood, PR; Esteve, C; Ferrer, M; Forns, P; Gómez, E; González, J; Lozoya, E; Mir, M; Moreno, I; Petit, S; Roberts, RS; Sevilla, S; Vidal, B; Vidal, L; Vilaseca, P Structure-activity relationships (SAR) and structure-kinetic relationships (SKR) of bicyclic heteroaromatic acetic acids as potent CRTh2 antagonists III: the role of a hydrogen-bond acceptor in long receptor residence times. Bioorg Med Chem Lett 24: 5127 -33 (2014) Scozzafava, A; Menabuoni, L; Mincione, F; Briganti, F; Mincione, G; Supuran, CT Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem 42: 2641 -50 (1999) Woon, EC; Sunderland, PT; Paine, HA; Lloyd, MD; Thompson, AS; Threadgill, MD One-pot tandem Hurtley-retro-Claisen-cyclisation reactions in the synthesis of 3-substituted analogues of 5-aminoisoquinolin-1-one (5-AIQ), a water-soluble inhibitor of PARPs. Bioorg Med Chem 21: 5218 -27 (2013) Kawada, H; Ebiike, H; Tsukazaki, M; Yamamoto, S; Koyama, K; Nakamura, M; Morikami, K; Yoshinari, K; Yoshida, M; Ogawa, K; Shimma, N; Tsukuda, T; Ohwada, J Optimization of the phenylurea moiety in a phosphoinositide 3-kinase (PI3K) inhibitor to improve water solubility and the PK profile by introducing a solubilizing group and ortho substituents. Bioorg Med Chem 24: 2897 -2906 (2016) Bohacek, RS; McMartin, C Definition and display of steric, hydrophobic, and hydrogen-bonding properties of ligand binding sites in proteins using Lee and Richards accessible surface: validation of a high-resolution graphical tool for drug design. J Med Chem 35: 1671 -84 (1992) Huber, JD; Bentzien, J; Boyer, SJ; Burke, J; De Lombaert, S; Eickmeier, C; Guo, X; Haist, JV; Hickey, ER; Kaplita, P; Karmazyn, M; Kemper, R; Kennedy, CA; Kirrane, T; Madwed, JB; Mainolfi, E; Nagaraja, N; Soleymanzadeh, F; Swinamer, A; Eldrup, AB Identification of a potent sodium hydrogen exchanger isoform 1 (NHE1) inhibitor with a suitable profile for chronic dosing and demonstrated cardioprotective effects in a preclinical model of myocardial infarction in the rat. J Med Chem 55: 7114 -40 (2012) Scozzafava, A; Menabuoni, L; Mincione, F; Supuran, CT Carbonic anhydrase inhibitors. A general approach for the preparation of water-soluble sulfonamides incorporating polyamino-polycarboxylate tails and of their metal complexes possessing long-lasting, topical intraocular pressure-lowering properties. J Med Chem 45: 1466 -76 (2002) Bookser, BC; Ugarkar, BG; Matelich, MC; Lemus, RH; Allan, M; Tsuchiya, M; Nakane, M; Nagahisa, A; Wiesner, JB; Erion, MD Adenosine kinase inhibitors. 6. Synthesis, water solubility, and antinociceptive activity of 5-phenyl-7-(5-deoxy-beta-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidines substituted at C4 with glycinamides and related compounds. J Med Chem 48: 7808 -20 (2005) Grdadolnik, SG; Mierke, DF; Byk, G; Zeltser, I; Gilon, C; Kessler, H Comparison of the conformation of active and nonactive backbone cyclic analogs of substance P as a tool to elucidate features of the bioactive conformation: NMR and molecular dynamics in DMSO and water. J Med Chem 37: 2145 -52 (1994) Cox, CD; Breslin, MJ; Whitman, DB; Coleman, PJ; Garbaccio, RM; Fraley, ME; Zrada, MM; Buser, CA; Walsh, ES; Hamilton, K; Lobell, RB; Tao, W; Abrams, MT; South, VJ; Huber, HE; Kohl, NE; Hartman, GD Kinesin spindle protein (KSP) inhibitors. Part V: discovery of 2-propylamino-2,4-diaryl-2,5-dihydropyrroles as potent, water-soluble KSP inhibitors, and modulation of their basicity by beta-fluorination to overcome cellular efflux by P-glycoprotein. Bioorg Med Chem Lett 17: 2697 -702 (2007) Nasief, NN; Tan, H; Kong, J; Hangauer, D Water mediated ligand functional group cooperativity: the contribution of a methyl group to binding affinity is enhanced by a COO(-) group through changes in the structure and thermodynamics of the hydration waters of ligand-thermolysin complexes. J Med Chem 55: 8283 -302 (2012) Kyriakis, E; Karra, AG; Papaioannou, O; Solovou, T; Skamnaki, VT; Liggri, PGV; Zographos, SE; Szennyes, E; Bokor, É; Kun, S; Psarra, AG; Somsák, L; Leonidas, DD The architecture of hydrogen and sulfur σ-hole interactions explain differences in the inhibitory potency of C-β-d-glucopyranosyl thiazoles, imidazoles and an N-β-d glucopyranosyl tetrazole for human liver glycogen phosphorylase and offer new insights to structure-based design. Bioorg Med Chem 28: (2020) Kotsikorou, E; Navas, F; Roche, MJ; Gilliam, AF; Thomas, BF; Seltzman, HH; Kumar, P; Song, ZH; Hurst, DP; Lynch, DL; Reggio, PH The importance of hydrogen bonding and aromatic stacking to the affinity and efficacy of cannabinoid receptor CB2 antagonist, 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide (SR144528). J Med Chem 56: 6593 -612 (2013) Huang, B; Chen, W; Zhao, T; Li, Z; Jiang, X; Ginex, T; Vílchez, D; Luque, FJ; Kang, D; Gao, P; Zhang, J; Tian, Y; Daelemans, D; De Clercq, E; Pannecouque, C; Zhan, P; Liu, X Exploiting the Tolerant Region I of the Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI) Binding Pocket: Discovery of Potent Diarylpyrimidine-Typed HIV-1 NNRTIs against Wild-Type and E138K Mutant Virus with Significantly Improved Water Solubility and Favorable Safety Profiles. J Med Chem 62: 2083 -2098 (2019) Zhao, J; Mao, Q; Lin, F; Zhang, B; Sun, M; Zhang, T; Wang, S Intramolecular hydrogen bond interruption and scaffold hopping of TMC-5 led to 2-(4-alkoxy-3-cyanophenyl)pyrimidine-4/5-carboxylic acids and 6-(4-alkoxy-3-cyanophenyl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-ones as potent pyrimidine-based xanthine oxidase inhibitors. Eur J Med Chem 229: (2022) Girotra, NN; Biftu, T; Ponpipom, MM; Acton, JJ; Alberts, AW; Bach, TN; Ball, RG; Bugianesi, RL; Parsons, WH; Chabala, JC Development, synthesis, and biological evaluation of (-)-trans-(2S,5S)-2-[3-[(2-oxopropyl)sulfonyl]-4-n-propoxy-5-(3- hydroxypropoxy)-phenyl]-5-(3,4,5-trimethoxyphenyl)tetrahydrofuran, a potent orally active platelet-activating factor (PAF) antagonist and its water-soluble prodrug phosphate ester. J Med Chem 35: 3474 -82 (1992)
Park, CH; Chung, BY; Lee, SS; Bai, HW; Cho, JY; Jo, C; Kim, TH Bioorg Med Chem Lett 23: 1099 -103 (2013) Bonhaus, DW; Bach, C; DeSouza, A; Salazar, FH; Matsuoka, BD; Zuppan, P; Chan, HW; Eglen, RM Br J Pharmacol 115: 622 -8 (1995) Kang, JH; Ting, Z; Moon, MR; Sim, JS; Lee, JM; Doh, KE; Hong, S; Cui, M; Choi, S; Chang, HW; Park Choo, HY; Yim, M Bioorg Med Chem 23: 7069 -78 (2015) Zhang, Y; Wu, X; Xue, X; Li, C; Wang, J; Wang, R; Zhang, C; Wang, C; Shi, Y; Zou, L; Li, Q; Huang, Z; Hao, X; Loomes, K; Wu, D; Chen, HW; Xu, J; Xu, Y J Med Chem 62: 4716 -4730 (2019) Lee, G; Lim, CS; Cho, HW; Seo, J; Gyorkos, AC; Cho, SY; Choi, EK; Kim, CS; Hwang, JJ US Patent US8664230 (2014) Jang, SJ; Choi, HW; Choi, DL; Cho, S; Rim, HK; Choi, HE; Kim, KS; Huang, M; Rhim, H; Lee, KT; Lee, JY Bioorg Med Chem Lett 23: 6656 -62 (2013) Chung HW Bryant, HU; Nelson, DL; Button, D; Cole, HW; Baez, MB; Lucaites, VL; Wainscott, DB; Whitesitt, C; Reel, J; Simon, R; Koppel, GA Life Sci 59: 1259 -68 (1996) Zou, XQ; Peng, SM; Hu, CP; Tan, LF; Yuan, Q; Deng, HW; Li, YJ Bioorg Med Chem 18: 3020 -5 (2010) Ding, HW; Wang, S; Qin, XC; Wang, J; Song, HR; Zhao, QC; Song, SJ Bioorg Med Chem 27: 2729 -2740 (2019) Hagmann, WK; Dorn, CP; Frankshun, RA; O'Grady, LA; Bailey, PJ; Rackham, A; Dougherty, HW J Med Chem 29: 1436 -41 (1986) Zhang, Z; Sun, S; Kodumuru, V; Hou, D; Liu, S; Chakka, N; Sviridov, S; Chowdhury, S; McLaren, DG; Ratkay, LG; Khakh, K; Cheng, X; Gschwend, HW; Kamboj, R; Fu, J; Winther, MD J Med Chem 56: 568 -83 (2013) Steinbaugh, BA; Hamilton, HW; Patt, WC; Rapundalo, ST; Batley, BL; Lunney, EA; Ryan, MJ; Hicks, GW Bioorg Med Chem Lett 4: 2029 -2034 (1994) Sun, WX; Han, HW; Yang, MK; Wen, ZL; Wang, YS; Fu, JY; Lu, YT; Wang, MY; Bao, JX; Lu, GH; Qi, JL; Wang, XM; Lin, HY; Yang, YH Bioorg Med Chem 27: (2019) He, HW; Yuan, JL; Peng, H; Chen, T; Shen, P; Wan, SQ; Li, Y; Tan, HL; He, YH; He, JB; Li, Y J Agric Food Chem 59: 4801 -13 (2011) Campiani, G; Fattorusso, C; Butini, S; Gaeta, A; Agnusdei, M; Gemma, S; Persico, M; Catalanotti, B; Savini, L; Nacci, V; Novellino, E; Holloway, HW; Greig, NH; Belinskaya, T; Fedorko, JM; Saxena, A J Med Chem 48: 1919 -29 (2005) JOHNSON HW YOON, SH; JOO, HW; SEO, BK; LEE, EJ; JUNG, JY; YOON, SY; CHO, WY US Patent US20230322706 (2023) More, KN; Jang, HW; Hong, VS; Lee, J Bioorg Med Chem Lett 24: 2424 -8 (2014) Jeong, HW; Kim, MR; Son, KH; Han, MY; Ha, JH; Garnier, M; Meijer, L; Kwon, BM Bioorg Med Chem Lett 10: 1819 -22 (2000) Li, YS; Tian, H; Zhao, DS; Hu, DK; Liu, XY; Jin, HW; Song, GP; Cui, ZN Bioorg Med Chem Lett 26: 3632 -5 (2016) Ibrahim, MA; Johnson, HW; Jeong, JW; Lewis, GL; Shi, X; Noguchi, RT; Williams, M; Leahy, JW; Nuss, JM; Woolfrey, J; Banica, M; Bentzien, F; Chou, YC; Gibson, A; Heald, N; Lamb, P; Mattheakis, L; Matthews, D; Shipway, A; Wu, X; Zhang, W; Zhou, S; Shankar, G J Med Chem 55: 1368 -81 (2012) Kim, YK; Park, SY; Joo, HW; Choi, ES; Paek, SY; Kang, SW; Kim, BG; Lee, CS; Kim, SW; Lee, SD US Patent US11261186 (2022) Anh, DT; Hai, PT; Dung, DTM; Dung, PTP; Huong, LT; Park, EJ; Jun, HW; Kang, JS; Kwon, JH; Tung, TT; Han, SB; Nam, NH Bioorg Med Chem Lett 30: (2020) Nhoek, P; Ahn, S; Pel, P; Kim, YM; Huh, J; Kim, HW; Noh, M; Chin, YW J Nat Prod 86: 138 -148 (2023) Peukert, S; Brendel, J; Pirard, B; Brüggemann, A; Below, P; Kleemann, HW; Hemmerle, H; Schmidt, W J Med Chem 46: 486 -98 (2003) Archer, S; Seyed-Mozaffari, A; Ward, S; Kosterlitz, HW; Paterson, SJ; McKnight, AT; Corbett, AD J Med Chem 28: 974 -6 (1985) Rawson, TE; Rüth, M; Blackwood, E; Burdick, D; Corson, L; Dotson, J; Drummond, J; Fields, C; Georges, GJ; Goller, B; Halladay, J; Hunsaker, T; Kleinheinz, T; Krell, HW; Li, J; Liang, J; Limberg, A; McNutt, A; Moffat, J; Phillips, G; Ran, Y; Safina, B; Ultsch, M; Walker, L; Wiesmann, C; Zhang, B; Zhou, A; Zhu, BY; Rüger, P; Cochran, AG J Med Chem 51: 4465 -75 (2008) Yoon, JS; Kim, G; Jarhad, DB; Kim, HR; Shin, YS; Qu, S; Sahu, PK; Kim, HO; Lee, HW; Wang, SB; Kong, YJ; Chang, TS; Ogando, NS; Kovacikova, K; Snijder, EJ; Posthuma, CC; van Hemert, MJ; Jeong, LS J Med Chem 62: 6346 -6362 (2019) Li, J; Gu, BB; Sun, F; Xu, JR; Jiao, WH; Yu, HB; Han, BN; Yang, F; Zhang, XC; Lin, HW J Nat Prod 80: 1436 -1445 (2017) Jadhav, PK; Woerner, FJ; Man, HW Bioorg Med Chem Lett 7: 2145 -2148 (1997) Walsh, DA; Moran, HW; Shamblee, DA; Welstead, WJ; Nolan, JC; Sancilio, LF; Graff, G J Med Chem 33: 2296 -304 (1990) Ważyńska, MA; Butera, R; Requesens, M; Plat, A; Zarganes-Tzitzikas, T; Neochoritis, CG; Plewka, J; Skalniak, L; Kocik-Krol, J; Musielak, B; Magiera-Mularz, K; Rodriguez, I; Blok, SN; de Bruyn, M; Nijman, HW; Elsinga, PH; Holak, TA; Dömling, A J Med Chem 66: 9577 -9591 (2023) Bedford, CD; Harris, RN; Howd, RA; Miller, A; Nolen, HW; Kenley, RA J Med Chem 27: 1431 -8 (1984) Ong HW Jeong, P; Moon, Y; Lee, JH; Lee, SD; Park, J; Lee, J; Kim, J; Lee, HJ; Kim, NY; Choi, J; Heo, JD; Shin, JE; Park, HW; Kim, YG; Han, SY; Kim, YC Eur J Med Chem 195: (2020) Becker, MR; Ewing, WR; Davis, RS; Pauls, HW; Ly, C; Li, A; Mason, HJ; Choi-Sledeski, YM; Spada, AP; Chu, V; Brown, KD; Colussi, DJ; Leadley, RJ; Bentley, R; Bostwick, J; Kasiewski, C; Morgan, S Bioorg Med Chem Lett 9: 2753 -8 (1999) Ries, UJ; Priepke, HW; Hauel, NH; Handschuh, S; Mihm, G; Stassen, JM; Wienen, W; Nar, H Bioorg Med Chem Lett 13: 2297 -302 (2003) Kim, JY; Lee, JW; Kim, YS; Lee, Y; Ryu, YB; Kim, S; Ryu, HW; Curtis-Long, MJ; Lee, KW; Lee, WS; Park, KH Chembiochem 11: 2125 -31 (2010) Saha, P; Hödl, C; Strauss, WS; Steiner, R; Goessler, W; Kunert, O; Leitner, A; Haslinger, E; Schramm, HW Bioorg Med Chem 18: 1891 -8 (2010) Lee, JH; Bok, JH; Park, SB; Pagire, HS; Na, YJ; Rim, E; Jung, WH; Song, JS; Kang, NS; Seo, HW; Jung, KY; Lee, BH; Kim, KY; Ahn, JH Bioorg Med Chem Lett 30: (2020) Maeng, CY; Jang, YK; Cha, SB; Shin, HW; Joung, CM; Yi, EJ US Patent US10456385 (2019) Kubas, H; Meyer, U; Hechenberger, M; Klein, KU; Plitt, P; Zemribo, R; Spexgoor, HW; van Assema, SG; Abel, U Bioorg Med Chem Lett 23: 6370 -6 (2013) Morley, RM; Tse, HW; Feng, B; Miller, JC; Monaghan, DT; Jane, DE J Med Chem 48: 2627 -37 (2005) Jagtap, AD; Chang, PT; Liu, JR; Wang, HC; Kondekar, NB; Shen, LJ; Tseng, HW; Chen, GS; Chern, JW Eur J Med Chem 85: 268 -88 (2014) Zhang, XR; Wang, HW; Tang, WL; Zhang, Y; Yang, H; Hu, DX; Ravji, A; Marchand, C; Kiselev, E; Ofori-Atta, K; Agama, K; Pommier, Y; An, LK J Med Chem 61: 9908 -9930 (2018) Williams, HW; Eichler, E; Randall, WC; Rooney, CS; Cragoe, EJ; Streeter, KB; Schwam, H; Michelson, SR; Patchett, AA; Taub, D J Med Chem 26: 1196 -200 (1983) Liu, GZ; Xu, HW; Wang, P; Lin, ZT; Duan, YC; Zheng, JX; Liu, HM Eur J Med Chem 65: 323 -36 (2013) Lin, XD; Yang, HW; Ma, S; Li, WW; Zhang, CH; Wang, WJ; Xiang, R; Li, LL; Yang, SY Bioorg Med Chem Lett 25: 4534 -8 (2015) Lin, TS; Liw, YW; Song, JS; Hsieh, TC; Yeh, HW; Hsu, LC; Lin, CJ; Wu, SH; Liang, PH Bioorg Med Chem 21: 6282 -91 (2013) Imbriglio, JE; Shen, DM; Liang, R; Marby, K; You, M; Youm, HW; Feng, Z; London, C; Xiong, Y; Tata, J; Verras, A; Garcia-Calvo, M; Song, X; Addona, GH; McLaren, DG; He, T; Murphy, B; Metzger, DE; Salituro, G; Deckman, D; Chen, Q; Jin, X; Stout, SJ; Wang, SP; Wilsie, L; Palyha, O; Han, S; Hubbard, BK; Previs, SF; Pinto, S; Taggart, A J Med Chem 58: 9345 -53 (2015) Zhang, HW; Detorio, M; Herman, BD; Solomon, S; Bassit, L; Nettles, JH; Obikhod, A; Tao, SJ; Mellors, JW; Sluis-Cremer, N; Coats, SJ; Schinazi, RF Eur J Med Chem 46: 3832 -44 (2011) Liu, FZ; Fang, H; Zhu, HW; Wang, Q; Yang, Y; Xu, WF Bioorg Med Chem 16: 578 -85 (2008) Sokias, R; Werry, EL; Alison Cheng, HW; Lloyd, JH; Sohler, G; Danon, JJ; Montgomery, AP; Du, JJ; Gao, Q; Hibbs, DE; Ittner, LM; Reekie, TA; Kassiou, M Eur J Med Chem 207: (2020) El-Shafey, HW; Gomaa, RM; El-Messery, SM; Goda, FE Bioorg Med Chem Lett 30: (2020) van Galen, PJ; van Vlijmen, HW; IJzerman, AP; Soudijn, W J Med Chem 33: 1708 -13 (1990) Lange, UE; Baucke, D; Hornberger, W; Mack, H; Seitz, W; Höffken, HW Bioorg Med Chem Lett 16: 2648 -53 (2006) Biamonte, MA; Van de Water, R; Arndt, JW; Scannevin, RH; Perret, D; Lee, WC J Med Chem 53: 3 -17 (2010)
ChEMBL_1555051 (CHEMBL3767629) Inhibition of catalase (unknown origin) using hydrogen peroxide as substrate ChEMBL_141046 (CHEMBL746894) Screened in AP1 cells expressing human NHE-1 for sodium hydrogen exchange activity ChEMBL_306146 (CHEMBL831381) Inhibitory concentration against human sodium/hydrogen exchanger (NHE-1) in PS120 variant cells ChEMBL_2271918 Inhibition of AQP1 water channel in human erythrocyte by stopped-flow spectrometry ChEMBL_468962 (CHEMBL952107) Inhibition of aromatase in human SKBR3 cells by tritiated water release assay ChEMBL_141048 (CHEMBL872728) Compound is screened in AP1 cells expressing human NHE-2 for sodium hydrogen exchange activity ChEMBL_141050 (CHEMBL748618) Compound is screened in AP1 cells expressing human NHE-3 for sodium hydrogen exchange activity ChEMBL_141051 (CHEMBL748619) Compound is screened in AP1 cells expressing human NHE-5 for sodium hydrogen exchange activity ChEMBL_428436 (CHEMBL919505) Inhibition of aromatase in SK-BR-3 cells by tritiated water release assay ChEMBL_202731 (CHEMBL809059) Sodium/hydrogen exchanger antiport activity was measured using [22]Na+ uptake inhibition assay in acidified rabbit erythrocytes ChEMBL_1287262 (CHEMBL3110842) Inhibition of human recombinant His-tagged LSD1 (171 to 836) assessed as hydrogen peroxide formation after 5 mins ChEMBL_582869 (CHEMBL1051107) Binding affinity to ecdysone receptor ligand binding domain in Drosophila melanogaster assessed as number of hydrogen bonds formed ChEMBL_607020 (CHEMBL1074648) Inhibition of PARP1 in human HeLa cells assessed as inhibition of hydrogen peroxide-induced poly(ADP-ribosyl)ation ChEMBL_2152147 (CHEMBL5036694) Inhibition of human recombinant HAO1 assessed as fluorescent intensity using hydrogen peroxide as substrate by HRP interference detection assay ChEMBL_2271917 Inhibition of human AQP1 water channel expressed in CHO cells incubated for 10 mins by FLIPR Tetra assay ChEMBL_872732 (CHEMBL2185145) Inhibition of human placental aromatase using [3H]-1beta-androstenedione as substrate after 16 hrs by [3H]-water method ChEMBL_2271916 Agonist activity at human AQP1 water channel expressed in Xenopus laevis oocytes assessed as inhibition of AQP1-mediated osmotic swelling ChEMBL_629407 (CHEMBL1105760) Inhibition of human recombinant MAO-A assessed as inhibition of production of hydrogen peroxide after 15 mins by Amplex Red fluorimetric method ChEMBL_908843 (CHEMBL3067865) Binding affinity to salt water mollusc Aplysia californica AChBP Y55W mutant assessed as [3H]acetamiprid binding by radioligand binding assay ChEMBL_629410 (CHEMBL1105763) Inhibition of human recombinant MAO-B assessed as inhibition of production of hydrogen peroxide after 15 mins by the Amplex Red fluorimetric method ChEMBL_2446254 Inhibition of human leukocyte MPO chlorinating activity using hydrogen peroxide as substrate preincubated for 15 mins followed by substrate addition and measured after 8 mins ChEMBL_663024 (CHEMBL1250927) Inhibition of human recombinant MAOA expressed in baculovirus infected BTI insect cells assessed as hydrogen peroxide production after 15 mins by Amplex red assay ChEMBL_663025 (CHEMBL1250928) Inhibition of human recombinant MAOB expressed in baculovirus infected BTI insect cells assessed as hydrogen peroxide production after 15 mins by Amplex red assay ChEMBL_743261 (CHEMBL1769015) Inhibition of human recombinant MAO-B expressed in baculovirus infected BTI-TN-5B1-4 insect sells assessed as hydrogen peroxide production by fluorimetric method ChEMBL_743262 (CHEMBL1769016) Inhibition of human recombinant MAO-A expressed in baculovirus infected BTI-TN-5B1-4 insect sells assessed as hydrogen peroxide production by fluorimetric method ChEMBL_1473802 (CHEMBL3418527) Inhibition of human recombinant MAO-A expressed in Sf9 cells using 5-hydroxytryptamine substrate assessed as hydrogen peroxide production after 1 hr by fluorescence assay ChEMBL_1473827 (CHEMBL3418726) Inhibition of human recombinant MAO-B expressed in Sf9 cells using 5-phenylacetaldehyde substrate assessed as hydrogen peroxide production after 1 hr by fluorescence assay Thallium Flux Assay FluxOR Kit Components (Invitrogen F10017) FluxOR Reagent (Component A) FluxOR Assay Buffer (Component B)-10x Concentrate PowerLoad Concentrate (Component C)-100x Concentrate Probenecid (Component D)-Lyophilized sample is kept at -20 C. Water soluble, 100x after solubilization in 1 ml water. Store at 4 C. FluxOR Chloride-free Buffer (Component E)-5x Concentrate Potassium sulfate (K2SO4) Concentrate (Component F)-125 mM in water. Store at 4 C. Thallium sulfate (Tl2SO4) Concentrate (Component G)-50 mM in water. Store at 4 C. DMSO (dimethyl sulfoxide, Component H)-1 ml (100%) Reagent Preparation- FluxOR Working Solutions 1000x FluxOR Reagent: Reconstitute a vial of component A in 100 ul DMSO; Mix well; Store 10 ul aliquots at -20 C. 1x FluxOR Assay Buffer: Dilute Component B 10-fold with water; Adjust pH to 7.4 with Hepes/NaOH; Filter and store at 4 C. ChEMBL_2122235 (CHEMBL4831382) Inhibition of Mycobacterium tuberculosis recombinant DprE1 expressed in Escherichia coli assessed as formation of resorufin using FPR as substrate by Amplex Red hydrogen/peroxidase coupled assay ChEMBL_212902 (CHEMBL822095) Inhibition constant (Ki) when mixed with p-nitroanilide against trypsin enzyme for the conversion of water-soluble compound to fluorescent oil-soluble compound ChEMBL_2165791 (CHEMBL5050652) Inhibition of Mycobacterium tuberculosis H37Rv recombinant DprE1 expressed in Escherichia coli assessed as formation of resorufin using FPR as substrate by Amplex Red hydrogen/peroxidase coupled assay ChEMBL_661995 (CHEMBL1251643) Inhibition of human recombinant MAOA expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as hydrogen peroxide production from p-tyramine by amplex red assay ChEMBL_661996 (CHEMBL1251644) Inhibition of human recombinant MAOB expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as hydrogen peroxide production from p-tyramine by amplex red assay ChEMBL_1333130 (CHEMBL3232285) Competitive inhibition of Lactobacillus casei thymidylate synthetase using dTMP as substrate assessed as release of water after 15 mins by double reciprocal plot analysis Inhibition Assay The inhibition of the activity of xanthine oxidase (XO) is tested through a coupled enzymatic reaction of xanthine oxidase, horseradish peroxidase (HRP) and a substrate thereof. First, xanthine oxidase oxidizes hypoxanthine to produce xanthine and hydrogen peroxide, and further oxidizes xanthine to produce uric acid and hydrogen peroxide. Then, hydrogen peroxide reacts with 10-acetyl-3,7-dihydroxyphenoxazine (Ampliflu Red) under catalytic action of horseradish peroxidase so as to produce resorufin, a compound with strong fluorescence. The fluorescence intensity of resorufin is determined by using a fluorescence microplate, which is in direct proportion to the activity of xanthine oxidase. ChEMBL_2055417 (CHEMBL4710418) Inhibition of Mycobacterium tuberculosis DprE1 expressed in Escherichia coli BL21 (DE3) cells assessed as formation of resorufin using FPR as substrate by Amplex Red hydrogen/peroxidase coupled assay ChEMBL_2355736 Inhibition of human NOX1 transfected in CHO cells assessed as PMA-induced hydrogen peroxide pre-incubated for 10 and measured after 15 mins by Amplex-red based fluorescence assay ChEMBL_2520737 Inhibition of human neutrophil elastase using MeOSuc-AAPV-MCA as substrate measured at 60 mins interval for up to several hrs in presence of hydrogen peroxide by fluorescence assay ChEMBL_657398 (CHEMBL1246374) Inhibition of human recombinant MAOA expressed in baculovirus infected insect BTI-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine by amplex red assay ChEMBL_657399 (CHEMBL1246375) Inhibition of human recombinant MAOB expressed in baculovirus infected insect BTI-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine by amplex red assay ChEMBL_512098 (CHEMBL978974) Inhibition of aromatase in human placental microsomes assessed as tritiated water release after 15 mins using [1-beta, 3H]androstenedione as substrate by scintillation counting ChEMBL_2355737 Inhibition of human NOX2 transfected in PLB-985 cells assessed as PMA-induced hydrogen peroxide pre-incubated for 10 and measured after 15 mins by Amplex-red based fluorescence assay ChEMBL_2355741 Inhibition of human DUOX1 transfected in HEK cells assessed as PMA-induced hydrogen peroxide production preincubated for 15 mins and measured after 10 mins by Amplex-red based fluorescence assay ChEMBL_2355742 Inhibition of human DUOX2 transfected in HEK cells assessed as PMA-induced hydrogen peroxide production preincubated for 15 mins and measured after 10 mins by Amplex-red based fluorescence assay ChEMBL_2152146 (CHEMBL5036693) Inhibition of human recombinant HAO1 assessed as detecting hydrogen peroxide using glycolic acid as substrate preincubated for 30 min followed by substrate addition measured after 15 mins by microplate reader ChEMBL_2355740 Inhibition of human NOX5 transfected in HEK cells assessed as PMA-induced hydrogen peroxide production pre-incubated for 10 mins and measured after 15 mins by Amplex-res based fluorescence assay ChEMBL_786455 (CHEMBL1920950) Inhibition of human recombinant MAOA expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as hydrogen peroxide production from p-tyramine after 15 mins by amplex red assay ChEMBL_786456 (CHEMBL1920951) Inhibition of human recombinant MAOB expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as hydrogen peroxide production from p-tyramine after 15 mins by amplex red assay ChEMBL_2271914 Inhibition of human AQP1 water channel expressed in Xenopus laevis oocytes assessed as inhibition of AQP1-mediated osmotic swelling incubated for 1 to 2 hrs by videomicroscopy analysis ChEMBL_2271915 Inhibition of rat AQP4 water channel expressed in Xenopus laevis oocytes assessed as inhibition of AQP1-mediated osmotic swelling incubated for 1 to 2 hrs by videomicroscopy analysis Biological Assay All assays were performed in room temperature with purified recombinantly expressed human SSAO. The enzyme activity was measured with benzylamine as substrate and utilized the production of hydrogen peroxide for detection. ChEMBL_2355769 Inhibition of NOX2 (unknown origin) transfected in HEK293 cells assessed as inhibition of hydrogen peroxide production pe-incubated for 15 mins and measured after 10 mins by Amplex Red based fluorescence analysis ChEMBL_769477 (CHEMBL1831417) Inhibition of human recombinant MAO-A expressed in baculovirus infected BTI-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine after 15 mins by microplate fluorescence assay ChEMBL_769479 (CHEMBL1833115) Inhibition of human recombinant MAO-B expressed in baculovirus infected BTI-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine after 15 mins by microplate fluorescence assay Malachite Green ATPase assay Chemicals are of the highest purity commercially available and all aqueous solutions are made up in AR water. Because of the need to minimise contamination with inorganic phosphate, precautions should be taken with solutions and apparatus used in the assays. Glassware and pH meters are rinsed with double distilled or deionised water before use and, wherever possible, plastic ware should be used. ChEMBL_2355738 Inhibition of human NOX3 transfected in HEK293-T-REx cells assessed as PMA-induced hydrogen peroxide production pre-incubated for 10 mins and measured after 15 mins by Amplex-res based fluorescence assay ChEMBL_2355739 Inhibition of human NOX4 transfected in HEK293-T-REx cells assessed as PMA-induced hydrogen peroxide production pre-incubated for 10 mins and measured after 15 mins by Amplex-res based fluorescence assay ChEMBL_795153 (CHEMBL1936625) Inhibition of human recombinant MAO-A expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as inhibition of hydrogen peroxide production from p-tryptamine after 15 mins by fluorimetric method ChEMBL_795208 (CHEMBL1936776) Inhibition of human recombinant MAO-B expressed in baculovirus infected BTI-TN-5B1-4 insect cells assessed as inhibition of hydrogen peroxide production from p-tryptamine after 15 mins by fluorimetric method ChEMBL_1462109 (CHEMBL3396816) Inhibition of SCD1 in ICR mouse liver microsome using [9,10 3H]- stearoyl-Coenzyme A substrate assessed as decreased production of tritiated water after 20 hrs by scintillation counting analysis ChEMBL_934509 (CHEMBL2321456) Inhibition of SCD1 in ICR mouse liver microsome using [9,10 3H]- stearoyl-Coenzyme A substrate assessed as decreased production of tritiated water after 20 hrs by scintillation counting analysis ChEMBL_772924 (CHEMBL1838812) Inhibition of human recombinant MAO-A expressed in insect BT1-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine up to 15 mins by amplex red-based fluorometric assay ChEMBL_772926 (CHEMBL1838814) Inhibition of human recombinant MAO-B expressed in insect BT1-TN-5B1-4 cells assessed as production of hydrogen peroxide from p-tyramine up to 15 mins by amplex red-based fluorometric assay ChEMBL_946070 (CHEMBL2342563) Inhibition of recombinant human MAO-B expressed in baculovirus infected BT1-TN-5B1-4 cells assessed as inhibition of production of hydrogen peroxide from p-tyramine after 15 mins by Amplex Red assay ChEMBL_946072 (CHEMBL2342565) Inhibition of recombinant human MAO-A expressed in baculovirus infected BT1-TN-5B1-4 cells assessed as inhibition of production of hydrogen peroxide from p-tyramine after 15 mins by Amplex Red assay ChEMBL_1333129 (CHEMBL3232284) Competitive inhibition of Lactobacillus casei thymidylate synthetase using 2'-deoxy[5-3H]uridine-5'-phosphate as substrate assessed as release of water after 15 mins by double reciprocal plot analysis ChEMBL_1515820 (CHEMBL3614582) Inhibition of recombinant human DAAO expressed in HEK cells using D-serine as substrate assessed as formation of alpha-keto acid, ammonia, hydrogen peroxidase after 20 mins by horseradish peroxidase/o-phenylenediamine-based assay ChEMBL_2355732 Inhibition of human NOX2 expressed in HEK cells assessed as inhibition of ionomycin-induced hydrogen peroxide production pre-incubated for 15 mins followed by ionomycin stimulation and measured after 10 mins by Fluorescence polarization assay ChEMBL_2355733 Inhibition of human NOX1 expressed in HEK cells assessed as inhibition of ionomycin-induced hydrogen peroxide production pre-incubated for 15 mins followed by ionomycin stimulation and measured after 10 mins by Fluorescence polarization assay In vitro D-Amino Acid Oxidase Assay D-amino acid oxidase (DAAO) was assayed in a 96-well plate format. D-serine was oxidatively deaminated by porcine D-amino acid oxidase in the presence of molecular oxygen and flavin adenosine dinucleotide (FAD) to yield the corresponding alpha-keto acid, ammonia and hydrogen peroxide. The resulting hydrogen peroxide was quantified using horseradish peroxidase and o-phenylenediamine, which displays a defined yellow absorbance at 411 nm when it becomes oxidized. Aromatase Assay The enzyme activity was assayed by measuring the formation of tritiated water from [1beta, 2beta-3H ]androstenedione in the presence of increasing concentrations of compounds. IC50 values of inhibition were calculated. ChEMBL_2355728 Inhibition of human NOX1 expressed in CHO cells assessed as inhibition of PMA-induced hydrogen peroxide production pretreated for 15 mins followed by PMA stimulation and measured after 10 mins by Amplex Red based fluorescence analysis ChEMBL_2355729 Inhibition of NOX2 in human PLB-985 cells assessed as inhibition of PMA-induced hydrogen peroxide production pretreated for 15 mins followed by PMA stimulation and measured after 10 mins by Amplex Red based fluorescence analysis ChEMBL_2355731 Inhibition of human NOX5 expressed in HEK293 cells assessed as inhibition of ionomycin-induced hydrogen peroxide production pretreated for 15 mins followed by ionomycin stimulation and measured after 10 mins by Amplex Red based fluorescence analysis PDGFRβ HTRF Assay Stock Solutions:Assay buffer stock solution, contains 50 mM Hepes, 10 mM MgCl2, 1 mM EGTA, and 0.01% Brij-35, 0.01% ovalbumin, 2 mM DTT at pH 7.5, in molecular biology grade water. Store at room temperature.DTT, 2 M in molecular biology grade water, store at −20° C. in aliquots.Ovalbumin, 10% or 100 mg/mL, prepare fresh on experimental day.PDGFRβ, 116 μM (PDGFRb_08 Prep 02), produced at Accelagen. Store at −80° C. in aliquots.TK-biotin peptide, 0.5 μM in molecular biology grade water, store at −20° C. in aliquots.ATP, 100 mM in molecular biology grade water, store at −20° C. in aliquots.HTRF KinEASE-TK kit: Allow the contents of the Cisbio kit to warm up to room temperature before use. This kit contains HTRF detection buffer, TK-Antibody labeled with Eu3+-cryptate, TK-substrate biotin and Streptavidin-XL665.TK Substrate-Biotin, reconstitute 500 μg lyopholized with 574 μL molecular biology grade water to prepare a 500 μM stock; After use, aliquot the rest and store at −20° C.TK Antibody-Cryptate, reconstitute lyophilized with 1 mL of molecular biology grade water (100× solution) then add 99 mL detection buffer to prepare a ready to use TK-antibody-cryptate solution; the concentration of the TK-antibody-cryptate reagent is not known. After use, aliquot the rest and store at −20° C.Streptavidin-XL665, reconstitute 3 mg lyophilized with 3 mL molecular biology grade water to prepare a 1 mg/mL or 16.67 μM stock; MW=60 kDa; After use, aliquot the rest and store at −20° C. ChEMBL_2064944 (CHEMBL4720197) Inhibition of human liver microsome CYP1A2 using phenacetin as substrate incubated for 5 mins followed by NADPH addition and further incubated for 10 mins in shaking water bath by LC-MS/MS analysis ChEMBL_2064945 (CHEMBL4720198) Inhibition of human liver microsome CYP3A4 using dextromethorphan as substrate incubated for 5 mins followed by NADPH addition and further incubated for 10 mins in shaking water bath by LC-MS/MS analysis ChEMBL_2064946 (CHEMBL4720199) Inhibition of human liver microsome CYP2C9 using tolbutamide as substrate incubated for 5 mins followed by NADPH addition and further incubated for 10 mins in shaking water bath by LC-MS/MS analysis ChEMBL_2064947 (CHEMBL4720200) Inhibition of human liver microsome CYP2C19 using omeprazole as substrate incubated for 5 mins followed by NADPH addition and further incubated for 10 mins in shaking water bath by LC-MS/MS analysis ChEMBL_2064948 (CHEMBL4720201) Inhibition of human liver microsome CYP2D6 using dextromethorphan as substrate incubated for 5 mins followed by NADPH addition and further incubated for 10 mins in shaking water bath by LC-MS/MS analysis ChEMBL_2064949 (CHEMBL4720202) Inhibition of human liver microsome CYP2E1 using chlorzoxazone as substrate incubated for 5 mins followed by NADPH addition and further incubated for 10 mins in shaking water bath by LC-MS/MS analysis ChEMBL_2355770 Inhibition of NOX1 (unknown origin) expressed in HT 29 cells assessed as inhibition of PMA-induced hydrogen peroxide production pre-incubated for 15 mins and followed by PMA stimulation and measured after 10 mins by Amplex Red based analysis ChEMBL_1292875 (CHEMBL3122614) Inhibition of human recombinant MAO-B expressed in baculovirus infected insect cells assessed as hydrogen peroxide production using p-tyramine as substrate incubated for 30 mins prior to substrate addition measured for 45 mins by Amplex Red monoamine oxidase assay ChEMBL_1292876 (CHEMBL3122615) Inhibition of human recombinant MAO-A expressed in baculovirus infected insect cells assessed as hydrogen peroxide production using p-tyramine as substrate incubated for 30 mins prior to substrate addition measured for 45 mins by Amplex Red monoamine oxidase assay Inhibition Assay MAT2A was incubated with FIDAS agents at RT for 20 min andthen mixed with L-methionine and ATP in 0.5 mL of reaction buffer.Cold deionized water (2 mL) was added to stop the reaction anddilute the samples. KinaseGlo Plus Luminescent Assay A stock solution of 5 % DMSO (BioReagent for molecular biology, Sigma Aldrich) in water was prepared. The CK1 substrate peptide RRKDLHDDEEDEAMSITA (Jena Bioscience, cat. no. PE-206, 2 mM stock solution in water) was diluted in water to a final concentration of 250 μM. CK1δ or CK1Ɛ kinases were diluted with CK1 dilution buffer to 50 ng / µL. In addition, fresh Reaction buffer was prepared containing 250 mM Tris-HCl (pH 7.5), 50 mM MgCl2, 5 mM DTT and 0.5 mg / mL bovine serum albumin (BSA). Compounds were serially diluted in 5 % DMSO solution to the desired concentrations (maximal concentration employed in the assay was 50 µM). Additional samples included blank sample containing Reaction buffer, water and CK1δ / CK1Ɛ kinase and control sample containing Reaction buffer, CK1 substrate peptide and CK1δ / CK1Ɛ kinase. CK1 substrate peptide, Reaction buffer and CK1δ / CK1Ɛ kinase stock solutions were mixed in 1:1:1 ratio. 6 μL of this mixtur ChEMBL_2355730 Inhibition of human NOX4 expressed in HEK293T cells assessed as inhibition of teracycline-induced hydrogen peroxide production pretreated for 18 hrs with tetracycline and treated with compound for 15 mins and measured at 10 mins post-compound treatment by Amplex Red based fluorescence analysis Enzyme Activity Assay The formation of ADP from ATP was quantified using a coupled enzyme assay (DiscoverX) in with a fluorescent resorufin dye is generated from the interaction of ADP with hydrogen peroxide and 10-acetyl-3,7-dihydroxy-phenoxazine (excitation and emission wavelenghts of 540 and 590 nm). Aromatase Assay The enzyme activity was assayed by measuring the 3H-labeled H2O formed from [1beta-3H] Androstenedione during aromatization. After incubation, the reaction was terminated by CHCl3 extraction and centrifugation. Aliquots of the water phase were assayed for the presence of 3H2O by counting using a liquid scintillation spectrometer. Inhibition Mass Spectrometer LRRKtide Detection settings: Q1 mass 644.8 Da, Q3 mass 638.8, declustering potential 76 volts, collision energy 37 volts, CXP 34 volts Phospho-LRRKtide Detection settings: Q1 mass 671.4 Da, Q3 mass 638.8, Declustering potential 76 volts, Collision energy 37 volts, CXP 34 volts. A C4 cartridge was used and running buffers were: A (aqueous) 0.1% formic acid in water B (organic) 0.1% formic acid, 80% acetonitrile, 20% water 6. Data was analysed using ActivityBase software (IDBS). A percent conversion from LRRKtide to Phospho-LRRKtide was calculated using the following formula: % conversion=(Phospho-LRRKtide product peak area/(Phospho-LRRKtide product peak area+LRRKtide substrate peak area))*100 3) Recombinant Cellular LRRK2 AlphaScreen Assay Inhibitory Activity Assay 1. Materials, Kits and EquipmentsSodium L-ascorbate (Cat: A4034-100G, SIGMA)4-(dimethylamino)benzaldehyde (Cat: 156477-25g, SIGMA)Trichloroacetic acid (Cat: T0699-100ML, SIGMA)L-Tryptophan (Cat: T8941-25G, SIGMA)Methylene blue (Cat: M9140-25G, SIGMA)Potassium dihydrogen phosphate (Cat: 10017618, Sinopharm Chemical Reagent)Disodium hydrogen phosphate (Cat: 20040618, Sinopharm Chemical Reagent)Constant temperature water tank (Cat: DK-8D, Shanghai Jinghong Experimental Equipment)Multifunctional microplate reader (Cat: M5, Molecular Devices)96-well reaction plate (Cat: 3590, costar)IDO1 protease (commercially available)Desktop Microplate Reader: SpectraMax M5 Microplate Reader (Molecular Devices)Test compounds: self-madePositive control agent: INCB024360 (commercially available)2. Reagent Preparation100 mM PBS:100 mM disodium hydrogen phosphate and 100 mM potassium dihydrogen phosphate mixed in a ratio of 3:5, pH 6.5IDO1 assay buffer:100 mM PBS containing 400 μM L-tryptophan, 20 mM ascorbate, 20 μM methylene blue and 1000 U/ml catalase, pH 6.530% trichloroacetic acidddH2 O solution of 30% trichloroacetic acidEhrlich reagent1% (w/v) diluted solution of 4-(dimethylamino) benzaldehyde compoundAll compounds were dissolved with DMSO. During the assay, each compound was diluted to a concentration as needed. The compound of each concentration was added to multi-wells, and the final concentration of DMSO was controlled at 1%.3. Test Methoda.) the reaction mixture was prepared by adding 50 nM IDO1 and the desired concentration of the test compound to 100 μL of IDO1 assay buffer. IDO1 and assay buffer need to be preheated to 37° C.b.) The mixture was reacted in a constant temperature water tank at 37° C. for 30 minutes.c.) 50 μL of 30% trichloroacetic acid was added.d.) The above mixture was reacted in a constant temperature water tank at 52° C. for 30 minutes.e.) The reaction mixture was centrifuged at 12000 g for 10 minutes at room temperature.f.) 100 μL of the obtained supernatant and 100 μL of Ehrlich reagent were mixed.g.) the absorbance at 480 nm was measured using an M5 microplate reader.4. Data AnalysisInhibition rate=(ODpostive−ODsample)/(ODpositive−ODnegative)*100%5. Results and Discussion Myeloperoxidase Assay Assays were performed at 22 °C with 2 nM MPO and 10 μM hydrogen peroxide (H2O2) in 20 mM NaH2PO4 buffer, pH 6.5 containing 140 mM NaCl, 10 mM taurine, and 1 mM L-tyrosine. Inhibitor compounds were preincubated with MPO for 15 min prior to the addition of H2O2, and the accumulation of taurine chloramine was determined after 1 min. CYP Enzyme Inhibitory Activity IC50 Assay The CYP enzyme probe substrates used in the experiment were: Phenacetin (1A2), Bupropion (2B6), Amodiaquine (2C8), Mephenytoin (2C19), Diclofenac (2C9), Dextromethorphan (2D6) and Testosterone (3A4/5). The final concentration of microsomes in the experimental system was 0.1 mg/mL. PBS Buffer was 50 mM K2HPO4 buffer. The concentrations of the compound to be tested were 50 μM, 12.5 μM, 3.125 μM, 0.781 μM, 0.195 μM, and 0.0488 μM, respectively. The corresponding probe substrates and microsomes were added into PBS, mixed well and dispensed into each reaction system, then control compound/compound to be tested/DMSO solution was added into the corresponding reaction systems respectively. The reaction system was mixed well, pre-incubated in a water bath at 37° C. for 5 minutes, added with 10 mM NADPH solution and mixed well, and reacted in a water bath at 37° C. for 10 minutes. After the reaction was completed, an internal standard acetonitrile solution was added to terminate the reaction. Centrifugation was performed at 4000 rpm, and the supernatant solution was taken and mixed well with an equal volume of pure water. ITK Enzyme Assay 1.0 M HEPES Buffer pH 7.5 solution was prepared as follows: 238.3 g HEPES free acid (Sigma) and 800 mL of water were combined, and the mixture was stirred until complete dissolution. The pH was adjusted to 7.5 via titration with 5N NaOH and the volume adjusted to 1000 mL. The solution was filtered and sterilized.ITK assay buffer was prepared as follows: 50 mL of HPLC-grade water was treated with 2 mL of 1.0 M HEPES Buffer, 500 μL of 2% Gelatin (Sigma), 1.0 mL of aqueous MgCl2 solution (1.0 M), and 1.0 mL of aqueous glutathione solution (0.5 M), and the solution was mixed. The solution was brought to 99 mL in a graduated cylinder by addition of water and sterilized through a 0.2 μm filter. 0.1 mL of Brij-35 Surfact-Amps Detergent Solution (10% w/v aqueous solution, ThermoFisher) and 1.0 mL of ATP (Teknova,100 mM) were added and the solution was mixed.Preparation of 1.33 ITK enzyme solution was as follows: 49.99 mL of ITK assay buffer was treated with 4.1 μL of ITK enzyme (ITK FL (N-Flag and C-His tagged, −72 kDa) Lake Pharma, 0.25 mg/ml in a buffer containing 25 mM Tris pH 7.8, 150 mM NaCl, 10% glycerol and 2 mM TCEP) and the mixture was gently agitated. The resulting solution was stored on ice. 30 Minutes prior to use, the enzyme solution was removed from ice and equilibrated to RT by incubation in a RT water bath. CYP2C9, CYP2D6 and CYP3A4 Enzymatic Activity Assay 1. Preparation of 100 mM phosphate buffered saline (PBS): 7.098 g of Na2HPO4 was weighed, 500 mL of pure water was added and subjected to ultrasonic dissolution to obtain solution A. 3.400 g of KH2PO4 was weighed, 250 mL of pure water was added and subjected to ultrasonic dissolution to obtain solution B. Solution A was placed on a stirrer and solution B was slowly added until the pH value reached 7.4 to prepare 100 mM PBS buffer.2. Preparation of 10 mM NADPH solution with 100 mM PBS buffer. The 10 mM stock solution of the compound of the present disclosure was diluted with DMSO to obtain a 200× concentration of compound working solution (6000, 2000, 600, 200, 60, 20, 0 μM). The stock solution of positive inhibitors was diluted with DMSO to obtain a 200× concentration of positive inhibitor working solution (sulfaphenazole, 1000, 300, 100, 30, 10, 3, 0 μM; quinidine/ketoconazole, 100, 30, 10, 3, 1, 0.3, 0 μM). A 200× concentration of substrate working solution (120 μM diclofenac, 400 μM dextromethorphan, and 200 μM midazolam) was prepared with water, acetonitrile, or acetonitrile/methanol.3. 2 μL of 20 mg/ml liver microsome solution, 1 μL of substrate working solution, 1 μL of compound working solution and 176 μL of PBS buffer were taken, mixed uniformly, and pre-incubated in a 37° C. water bath for 15 min. To the positive control group was added 1 μL of sulfaphenazole, quinidine or ketoconazole working solution instead of compound working solution. A 10 mM NADPH solution was also pre-incubated in the 37° C. water bath for 15 min. After 15 min, 20 μL of NADPH was taken and added to each well to initiate the reaction, and the reaction was incubated at 37° C. for 5 min (CYP2C9), 20 min (CYP2D6) or 5 min (CYP3A4). Double samples were set for all incubation samples. After the corresponding time of incubation, 400 μL of glacial methanol containing internal standard was added to all samples to stop the reaction. The mixture was mixed uniformly by vortex and centrifuged at 3220 g for 40 min at 4° C. After the completion of the centrifugation, 100 μL of the supernatant was transferred to a loading plate, and 100 μL of ultrapure water was added and mixed uniformly for LC-MS/MS analysis. Cholinesterase Inhibition Assay The IC50 values were determined using the spectrophotometric Ellman's method. All of the tested compounds were dissolved in 0.01 M DMSO and then diluted in demineralised water to 0.001 M and 0.0001 M. Acetylcholinesterase was obtained from electric eel (Electrophorus electricus L.) and butyrylcholinesterase was fromequine serum. Rivastigmine and galantamine were involved as reference drugs. Biochemical Assay In brief, this Rpn11 bioassay employs a fluorescent polarization readout based on the ability of the 26S proteasome to cleave the protein substrate including four tandem ubiquitin proteins fused to a peptide having a unique cysteine labeled with a fluorophore. Cleavage of this substrate by Rpn11 at the junction between the fourth ubiquitin and the peptide, releases the low molecular weight fluorescent peptide. Accordingly, inhibition of fluorescence correlates with inhibition of Rpn11. Inhibition is reported as the half maximal inhibitory concentration (IC50) for the candidate compound.The catalytic JAB1/MPN/Mov34 metalloenzyme (JAMM) motif of Rpn11 is found in 7 different human proteins including the Csn5 subunit of the COP9 signalosome, AMSH, AMSH-LP, the BRCC36 subunit of BRISC, MPND, and MYSM1. All of these enzymes cleave the isopeptide linkage that joins ubiquitin (or the ubiquitin-like protein Nedd8 in the case of Csn5) to a second molecule of ubiquitin or to a substrate. The conserved JAMM domain has the consensus sequence EXnHS/THX7SXXD, in which the histidine (His) and aspartic acid (Asp) residues bind the Zn2+ ion and the fourth coordination site is occupied by a water molecule that is engaged in hydrogen bonding with a conserved glutamic acid (Glu). The Zn2+ acts as a Lewis acid and increases the nucleophilic character of the bound water enough to allow hydrolytic cleavage of the isopeptide bond. CRBN/DDB1 Protein Activity Microplate reader (BMG PHERAstar FSX), ECHO (LABCYTE Echo 665), microplate thermostatic oscillator (Hangzhou Ruicheng Instrument Co., Ltd.), disodium hydrogen phosphate (Sigma-Aldrich (Shanghai) Trading Co., Ltd.), sodium dihydrogen phosphate (Sigma-Aldrich (Shanghai) Trading Co., Ltd.), bovine serum albumin (Sigma-Aldrich (Shanghai) Trading Co., Ltd.), Anti-6His-Tb crypate Gold (Cisbio Bioassays Company CISBIO), CRBN/DDB1 protein (HitGen Inc.), 384-well plate (Grenier Bio-one (Shanghai) Co., Ltd.). Alphascreen Assays A PI3K Alphascreen assay (PerkinElmer, Waltham, Mass.) was used to measure the activity of a panel of four phosphoinositide 3-kinases: PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ. Enzyme reaction buffer was prepared using sterile water (Baxter, Deerfield, Ill.) and 50 mM Tris HCl pH 7, 14 mM MgCl2, 2 mM sodium cholate, and 100 mM NaCl. 2 mM DTT was added fresh the day of the experiment. The Alphascreen buffer was made using sterile water and 10 mM Tris HCl pH 7.5, 150 mM NaCl, 0.10% Tween 20, and 30 mM EDTA. 1 mM DTT was added fresh the day of the experiment. Compound source plates used for this assay were 384-well Greiner clear polypropylene plates containing test compounds at 5 mM and diluted 1:2 over 22 concentrations. In Vitro Assay A reliable 96-well plate D-amino acid oxidase (DAAO) assay was developed based on previously published reports (J. Biol. Chem. 277: 27782 (2002)). Briefly, D-serine (5 mM) was oxidatively deaminated by human recombinant D-amino acid oxidase in the presence of molecular oxygen and flavin adenosine dinucleotide (FAD, 1 μM), to yield the corresponding α-keto acid, ammonia and hydrogen peroxide. The resulting hydrogen peroxide was quantified using horseradish peroxidase (0.01 mg/mL) and o-phenylenediamine (180 μg/mL), which displays a defined yellow absorbance at 411 nm when it becomes oxidized. All reactions were carried out for 20 min at room temperature in a 100-μL volume in Tris buffer (50 mM, pH 8.5). Additionally, stock solutions and serial dilutions of potential DAO inhibitors were made in 10:90 DMSO:buffer with a final assay DMSO concentration of 1%. ICL Enzyme Assay Isocitrate lyase activity was determined at 37°C by measuringthe formation of glyoxylate phenylhydrazone at 324 nm [Bai et al., Drug Dev. Res., 67:818-23]. The reaction mixture contained 100 μL of 0.5 mM potassium phosphate buffer, 1.2 μL of 1 mM magnesium chloride, 24 μL of 100 mM 2-mercaptoethanol, 7 μL of 4 mM phenylhydrazine hydrochloride, 6 μL of 50 mM trisodium isocitric acid, and ICL enzyme (usually 3-6 μL). This mixture was made up to 200 μL with MilliQ water (water that has been purified and deionized to a high degree by a water purification system manufactured by Millipore Corporation). At the end of the 10th minute this reaction mixture was made up to 1 mL and ultraviolet (UV) absorbance was measured at 324 nM, which served as a control. For the test compounds, 3 μL of 100 mM 3-nitropropionic acid (3-NPA) was used, and in the case of the candidate molecules, 10 μL of 10 mM concentration was added to the abovementioned reaction mixture. At the end of the 10th minute this reaction mixture was made up to 1 mL and UV absorbance was measured at 324 nM, which served as the test. Enzyme Assay The functional activity of compounds inhibiting the DAAO enzyme was determined by utilizing the co-product of the catalysis of D-Serine, H2O2 which can be quantitatively measured using the Amplex Red (Invitrogen) detection. Amplex Red reagent is a colorless substrate that reacts with hydrogen peroxide (H2O2) with a 1:1 stoichiometry in the presence of hydrogen peroxide to produce highly fluorescent resorufin (excitation/emission maxima=570/585 nm). The changes in fluorescence were monitored by a fluorescence plate reader, Envision (Perkin Elmer) and increases in DAAO activity were readily detected upon addition of D-Serine and suppression of this response observed with the application of test compounds.Human DAAO enzyme was supplied by the Takeda Pharmaceutical Company (Osaka) and each batch was tested and used at concentrations giving comparable levels of activity. The Km of D-Serine was measured for each enzyme batch to maintain consistency; this Km was used in subsequent assays. Inhibition Assay NSC-87877 ranked among top 10% (175th) of the compounds with the best GLIDE scores for the docking to the human Shp2 PTP domain in our virtual screening of 2368 3D structures derived from the NCI Diversity Set. Computer docking of NSC-87877 (FIG. 2) suggested that the B-ring sulfonic acid group forms hydrogen bond with the backbone NH group of Arg-465. Arg-465 is a conserved residue in the PTP signature motif (motif 9) VHCSXGXGR[T/S]G located at the base of the PTP catalytic cleft (Andersen et al., 2001). The A-ring sulfonic acid forms hydrogen bonds with the side-chain NH3 group of Lys-280 and the side-chain NH2 group of Asn-281. Lys-280/Asn-281 are non-conserved PTP residues located adjacent to the phosphotyrosine recognition loop (motif 1) (Andersen et al., 2001). The interaction between aromatic rings of the compound and the protein contributes to the binding through hydrophobic stabilization. PAR Assay Cellular activity of PARP-1 inhibitors was assessed by measuring the inhibition of the hydrogen peroxide induced PAR formation in HeLa cells (ECACC). Cellular PAR levels were measured by immunocytochemistry, and quantified using an ArrayScan vTi instrument (Cellomics Thermo Scientific). Studies were performed as follows: 6000 cells/well were seeded in 96 well plates (Perkin Elmer) in MEM/10% FCS and incubated for 24 hours at 37 C., 5% carbon dioxide. Test compounds were then added at the required concentration for 30 minutes. DNA damage was then induced adding hydrogen peroxide at the concentration of 0.1 mM for 15 minutes. Concentration curves were prepared in MEM/10% FCS from compound stocks in DMSO, and final DMSO concentration was 0.002% (v/v). Duplicate wells for each concentration point were prepared with a typical highest compound concentration of 20 uM and serial dilution 1:3. Plates were dried and fixed adding cold methanol-acetone (70:30) solution for 15 minutes at room temperature. CD73 Biochemical IC50 Assay Compound serial dilutions were pre-spotted into Thermo Nunc assay plate. 50 μL CD73 enzyme buffer (CD73 purchased from R&D system=0.6 nM, 25 mM Tris, pH 7.4, 5 mM MgCl2) was added into the assay plate and incubated for 15 mins. 50 μL AMP buffer (AMP=30 uM in 25 mM Tris, pH 7.4, 5 mM MgCl2, final AMP=15 uM, 2×Km, final CD73=0.3 nM) was added into assay plate. After incubation for 60 mins, the supernatant 20 uL was transferred into 384-well NUNC plate pre-filled with 60 ul Quench buffer (80% organic and 20% water+0.1% FA) with internal standard. The plate was spun down at 4500 rpm for 20 mins, then 200 of supernatant was transferred to another Nunc plate prefilled with 800 of water. The samples were run using Rapid fire. CD73 Biochemical IC50 Assay Compound serial dilutions were pre-spotted into Thermo Nunc assay plate. 50 uL CD73 enzyme buffer (CD73 purchased from R&D system=0.6 nM, 25 mM Tris, pH 7.4, 5 mM MgCl2, 1 mM NaH2PO4) was added into the assay plate and incubate for 15 mins. 50 uL AMP buffer (AMP=30 uM in 25 mM Tris, pH 7.4, 5 mM MgCl2, final AMP=15 uM, 2×Km, final CD73=0.3 nM) was added into assay plate. After incubation for 60 mins, the supernatant (20 uL) was transferred into 384-well NUNC plate pre-filled with 60 uL Quench buffer (80% organic and 20% water+0.1% FA) with internal standard. The plate was spun down at 4500 rpm for 20 mins, then 20 uL of supernatant was transferred to another Nunc plate prefilled with 80 uL of water. The samples were analyzed using Rapid fire. Cholinesterase Inhibition Assay Test compounds were prepared in DMSO (maximum concentration used 1% v/v), and 10 μL of each (0.001-25 μm final concentration range) was incubated for 5 min at room temperature with 160 μL of 1.5 mm DTNB, 50 μL 0.22 U/mL AChE [in 50 mm Tris-HCl, pH 8.0, 0.1% w/v bovine serum albumin (BSA)], or 50 μL 0.12 U/mL BuChE (in 50 mm Tris-HCl, pH 8.0, 0.1% w/v BSA). After the incubation period, 30 μLof ATChI (15 mm, prepared in ultrapure water) or BuTChI (15 mm, prepared in ultrapure water) was added to 96-well plates. Test compound inhibition of ChE was measured via UV absorbance (412 nm wavelength) using a microplate reader (SpectraMax M5) at various time intervals (t = 0, 1, 2, 3, 4, and 5 min). Enzymatic Activity Assay I. Test Materials and Instruments1. Human liver microsomes (Corning 452117)2. NADPH (Solarbio 705Y021)3. Positive substrates diclofenac (Sigma SLBV3438), dextromethorphan (TRC 3-EDO-175-1) and midazolam (Cerilliant FE01161704)4. Positive inhibitors sulfaphenazole (D. Ehrenstorfer GmbH 109012), quinidine (TCI WEODL-RE) and ketoconazole (Sigma 100M1091V)5. AB Sciex Triple Quad 5500 liquid chromatography-mass spectrometryII. Test Steps: 1. Preparation of 100 mM phosphate buffered saline (PBS): 7.098 g of Na2HPO4 was weighed, 500 mL of pure water was added and subjected to ultrasonic dissolution to obtain solution A. 3.400 g of KH2PO4 was weighed, 250 mL of pure water was added and subjected to ultrasonic dissolution to obtain solution B. Solution A was placed on a stirrer and solution B was slowly added until pH reaches 7.4 to prepare 100 mM PBS buffer. 2. Preparation of 10 mM NADPH solution with 100 mM PBS buffer. 10 mM stock solution of compounds of the invention was diluted with DMSO to obtain a 200× concentration of compound working solution (6000, 2000, 600, 200, 60, 20, 0 µM). The stock solution of positive inhibitors was diluted with DMSO to obtain a 200× concentration of positive inhibitor working solution (sulfaphenazole, 1000, 300, 100, 30, 10, 3, 0 µM; quinidine/ketoconazole, 100, 30, 10, 3, 1, 0.3, 0 µM). A 200 × concentration of substrate working solution (120 µM of diclofenac, 400 µM of dextromethorphan, and 200 µM of midazolam) was prepared in water, acetonitrile, or acetonitrile/methanol. 3. 2 µl of 20 mg/ml liver microsome solution, 1 µl of substrate working solution, 1 µl of compound working solution and 176 µl of PBS buffer were taken, mixed uniformly, and pre-incubated in a 37° C. water bath for 15 minutes. To the positive control group was added 1 µl of diclofenac, dextromethorphan or midazolam working solution to replace the compound working solution. Simultaneously, 10 mM of NADPH solution was pre-incubated together in a 37° C. water bath for 15 minutes. After 15 minutes, 20 µl of NADPH was added to each well to initiate the reaction, and the reaction was incubated at 37° C. for 5 minutes (CYP2C9) or 20 minutes (CYP2D6). Double samples were set for all incubation samples. After incubation for corresponding time, the reaction was terminated by adding 400 ul of ice methanol containing an internal standard to all samples. The mixture was mixed uniformly by vortex and centrifuged at 3220 g at 4° C. for 40 minutes. After centrifugation, 100 µL of the supernatant was transferred to a loading plate, and 100 µL of ultrapure water was added and mixed uniformly for LC-MS/MS analysis. Protease Inhibition Assay A peptide cleavage assay was performed using the icosapeptide H-Arg-Arg-Ser-Asn-Gln-Val-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln-Asn-Ile-Gln-Gly-Arg-Arg-OH, and the activity was monitored by HPLC. The cleavage products were analyzed after elution with a water/acetonitrile gradient, and detection at 215 nm. Ro 31-895919 served as a reference compound in this assay with an IC50 of 6.3 nM. Z-LYTE biochemical assay To the assay kit, 2.5 µL of different concentrate of the test compounds, Pazopanib or water (control) were added and incubated for 1 h at room temperature followed by the addition of 5 µL development agent. The reaction was stopped by the addition of 5 µL stop reagent. The fluorescence intensity at 445 and 520 nm were monitored and the standard inhibitory reference compound was Staurosporine. NAAA Assay Recombinant NAAA or native rat lung NAAA was incubated at 37 °C. for 30 min in 0.2 ml of sodium hydrogen phosphate buffer (50 mM, pH 5.0) containing 0.1% Triton X-100, 3 mM dithiothreitol (DTT) and 50 mM heptadecenoylethanolamide as substrate. The reaction was terminated by the addition of 0.2 ml cold methanol containing 1 mmol of heptadecanoic acid (HDA, NuChek Prep, Elysian, Minn.). Samples were analyzed by LC/MS (liquid chromatography/mass spectrometry). Heptadecanoic acid was eluted on an XDB Eclipse C18 column isocratically at 2.2 ml/min for 1 min with a solvent mixture of 95% methanol and 5% water, both containing 0.25% acetic acid and 5 mM ammonium acetate. The column temperature was 50 °C. ESI was in the negative mode, capillary voltage was 4 kV, and fragmentor voltage was 100 V. N2 was used as drying gas at a flow rate of 13 liters/min and a temperature of 350 °C. Nebulizer pressure was set at 60 psi. β-Arrestin Assay Assay procedures: OCC culture solution was preheated in water tank at 37° C. Cells were taken out from liquor nitrogen tank, left on the dry-ice, and dissolved in water tank at 37° C. 0.5 ml OCC culture solution was added and blended slowly. The cells were diluted in 11.5 mL OCC culture solution, and 100 μL cell suspension (containing 8333 cells) was added to each well of the 96-well plate. The 96-well plate was incubated for 8 hours under 5% CO2 at 37° C., and 10 μL test compound dissolved in DMSO was added in; 10 μL DMSO was used as negative control. After the plate was incubated for 90 min at 37° C., 55 μL detection solution was added to each well. Then the results were read out by EnVision after the cell plate was incubated for 90 min at room temperature. CDK9 Biochemical Assay To each well of a 96-well plate was added 5× kinase assay buffer with 10 mM DTT (6 μL), 500 μM ATP (1 μL), 5×CDK substrate (10 μL), and water (8 μL). 5 μL of compound were added to the test and positive control groups, while 5 μL of solvent was added to the blank groups. 100 ng of CDK9/CyclinT in 20 μL water was added to the test and positive control groups, while 20 μL of 1× kinase assay buffer was added to the blank groups. The reaction mixtures were incubated at 30° C. for 45 minutes, and 50 μL of Kinase-Glo Max was added to each well and the plates were shielded from light and incubated for 15 minutes at room temperature. Luminescence was measured on a microplate reader and IC50values were calculated using Prism 9 software. Enzymatic Activity Assay A protocol for determination of the carbonic anhydrase enzymatic activity at rt using the pH indicator method is described below:1 μL inhibitor (50 mM stock solution in DMSO) is diluted to a final test concentration ranging from 100 μM down to 1 nM (or 1 μL water in controls) and incubated for 2 min with 0.5 to 2 EU human Carboanhydrase I (180 U/mg) in 400 μL water and 200 μL phenol red indicator solution (20 mg/L). An enzymactic unit (EU) is defined as an amount which doubles the non catalyzied rate. The hydration reaction is initiated by adding 100 μL 0.5M bicarbonate buffer (0.3M Na2CO3; 0.2M NaHCO3) and subsequent dumping of CO2 through a needle (0.7×30 mm; 22 G×1.25) into the assay solution at a rate of 10 mL gas/minute. The time to colour change (pH 7.2) is determined with a microchronometer or stop watch. Enzymatic Assay 0.1 nM of FLAG-tagged Vanin-1 (AA 22-493, T26I, produced internally) and test compounds are incubated at room temperature for 20 minutes in assay buffer (1 mM DTT, 0.0025% Brij-35, 50 mM HEPES, pH7.5). D-Pantethine (Sigma, Cat #P2125-5G) in assay buffer is added (final concentration 3 μM) and incubated for additional 30 minutes at room temperature. Total assay volume typically is 40 μl but might be adjusted according to needs. Reaction is stopped by adding equal volume of stop solution as the reaction mixture to reach 100 nM HD-pantothenic acid (as an internal standard) and 1% TFA. Assay plates are centrifuged for 2 minutes and the formation of pantothenic acid is detected by RapidFire Mass Spectrometry (mobile phase A: 0.1% formic acid and 0.01% trifluoroacetic acid in water; mobile phase B: 47.5% acetonitrile, 47.5% methanol, 0.1% formic acid and 0.01% trifluoroacetic acid in water) using a C18, 12 μL cartridge (Agilent Cat #G9205A). Filter-paper Assay This assay relies on Whatman P-81 filter paper, which binds peptides but not SAM. Protein Methyl Transferases (PMTs) transfer 3H-Me of [3H-Me]-SAM to peptide substrates and the resultant 3H-methylated, filter-paper-bound peptide is quantified with a scintillation counter. Briefly, 6 μL of the methylation reaction was spotted onto Whatman P-81 phosphocellulose filter paper (1.2×1.2 cm2) to immobilize the 3H-labeled peptide. After drying in air for 20 min, the filter paper was immersed into 20 mL of 50 mM Na2CO3/NaHCO3 buffer (pH=9.2), and washed 5 times for 10 min each time. The washed filter paper was then transferred to a 20 mL scintillation vial containing 1 mL of distilled water and 10 mL of Ultima Gold scintillation cocktail or 7 mL scintillation vial containing 0.5 mL od distilled water and 5 mL of scintillation cocktail (PerkinElmer). The radioactivity was quantified by a Beckman LS6000IC liquid scintillation counter. Fluorescence Quench Assay The assay was performed in the presence of OptiMEM (supernatant collected over 24 h and cleared from cellular debris by centrifugation) containing the ectodomain of BACE1, 25 μl water containing the desired 2-fold concentration of test compound and 2% DMSO, 1 μM substrate peptide, 20 mM NaOAc, pH 4.4, and 0.04% Triton-X100 in a total assay volume of 50 μl in a 384 well plate. In general, 25 μl of compound dilution were added to the plate followed by the addition of 10 μl of BACE1 containing OptiMEM diluted 1:10 in water with 0.2% Triton X-100. The reaction was started with the addition of 15 μl substrate in NaOAc buffer. The reaction was incubated at rt (dark) in an Envision multilabel reader (Perkin Elmer) and the cleavage of the substrate was recorded as kinetic for 60 min at ex: 485 nm, em: 538 nm. Blank wells containing no enzyme were included on each plate. Inhibition Assay Solutions of each test compound were separately prepared at concentrations of 20000, 6000, 2000, 600, 200, and 60 μM by serial dilution with DMSO:MeCN (50:50 v/v). The individual test compound solutions were then diluted 20-fold with DMSO:MeCN:deionized water (5:5:180 v/v/v) to concentrations of 1000, 300, 100, 30, 10, and 3 μM. Mixtures of isozyme inhibitors (sulfaphenazole, tranylcypromine, and ketoconazole as specific inhibitors of isozymes 2C9, 2C19, and 3A4, respectively) were prepared containing each inhibitor at concentrations of 6000, 2000, 600, 200, 60, 20, 6, and 2 μM by serial dilution with DMSO:ACN (50:50 v/v). The mixed inhibitor solutions were then diluted 20-fold with DMSO:MeCN:deionized water (5:5:180 v/v/v) to concentrations of 300, 100, 30, 10, 3, 1, 0.3, and 0.1 μM. The percent of organic solvent attributable to the test compound or inhibitor mixture in the final reaction mixture was 2% v/v. Inhibition Assay Solutions of each test compound were separately prepared at concentrations of 20000, 6000, 2000, 600, 200, and 60 uM by serial dilution with DMSO:MeCN (50:50 v/v). The individual test compound solutions were then diluted 20-fold with DMSO:MeCN:deionized water (5:5:180 v/v/v) to concentrations of 1000, 300, 100, 30, 10, and 3 uM. Mixtures of isozyme inhibitors (sulfaphenazole, tranylcypromine, and ketoconazole as specific inhibitors of isozymes 2C9, 2C19, and 3A4, respectively) were prepared containing each inhibitor at concentrations of 6000, 2000, 600, 200, 60, 20, 6, and 2 uM by serial dilution with DMSO:ACN (50:50 v/v). The mixed inhibitor solutions were then diluted 20-fold with DMSO:MeCN:deionized water (5:5:180 v/v/v) to concentrations of 300, 100, 30, 10, 3, 1, 0.3, and 0.1 uM. The percent of organic solvent attributable to the test compound or inhibitor mixture in the final reaction mixture was 2% v/v.Pooled human liver microsome suspension (20 mg/mL) was determined. Enzyme Activity Inhibition Assay The scintillation Proximity Assay (SPA) method was used to determine the inhibitory effect of compounds on the activity of PDE4D catalytic domain. Human PDE4D catalytic domain protein was obtained by expression and purification in E. coli. The positive compound Apremilast was purchased from Topscience Biochemical, Microplate Scintillation Counter (MicroBeta2, Perkin Elmer), constant temperature water bath (DK420, Shanghai Medical Device Factory), Micro-vibrator (XW-80A, Shanghai Jingke Industrial Co., Ltd.) are a public instrument in the radioactive laboratory, and the step-by-step pipette (Multipette Plus, Eppendorf) and supporting tips were purchased from Ebende Biotechnology Company, 3.5. [3H]-cAMP, scintillation beads (RPNQO150, Perkin Elmer), and 96-well scintillation microplates (Isoplate-96, Perkin Elmer) were purchased from Perkin Elmer Company. 10× SPA buffer was prepared in the laboratory (500 mM Tris pH7.5, 83 mM MgCl2, 17 mM EGTA).In the experiment, 60 μl water and 10 μl reaction solution were added into 100 μl total volume reaction to achieve the final concentration of each component being 50 mM Tris-HCl, pH7.5, 8.3 mM MgCl2, 1.7 mM EGTA, 10 μl compound and 10 μl enzyme (0.1 ng/ul). Finally, 10 μl [3H]-cAMP (0.005 μCi/μl) was added and incubated for 30 min at 30° C. in a water bath. 50 μl SPA beads was added to quench the reaction, shaked appropriately, and stood for 20 minutes. A microplate scintillation counter was used Enzymatic Activity of CYP2C9, CYP2D6, and CYP3A4 I. Experimental materials and instruments1. Human liver microsome (Corning 452117)2. NADPH (Solarbio 705Y021)3. Positive substrates diclofenac (Sigma SLBV3438), dextromethorphan (TRC 3-EDO-175-1), and midazolam (Cerilliant FE01161704)4. Positive inhibitors sulfaphenazole (D. Ehrenstorfer GmbH 109012), quinidine (TCI WEODL-RE), and ketoconazole (Sigma 100M1091V)5. AB Sciex Triple Quad 5500 liquid chromatography-mass spectrometry systemII. Procedures1. Preparation of 100 mM phosphate-buffered saline (PBS): 7.098 g Na2HPO4 was weighed. 500 mL pure water was added. The mixture was dissolved by sonication to give solution A. 3.400 g KH2PO4 was weighed. 250 mL pure water was added. The mixture was dissolved by sonication to give solution B. The solution A was placed in a stirrer, and the solution B was slowly added until the pH reached 7.4, so that the 100 mM PBS buffer was prepared.2. A 10 mM NADPH solution was prepared with a 100 mM PBS buffer. A 10 mM stock solution of the compound of the present application was diluted with DMSO to give a compound working solution at a concentration of 200×(6000, 2000, 600, 200, 60, 20, and 0 μM). The positive inhibitor stock solution was diluted with DMSO to give a positive inhibitor working solution at a concentration of 200×(sulfaphenazole, 1000, 300, 100, 30, 10, 3, and 0 μM; quinidine/ketoconazole, 100, 30, 10, 3, 1, 0.3, and 0 μM). Substrate working solutions (120 μM diclofenac, 400 μM dextromethorphan, and 200 μM midazolam) at a concentration of 200× were prepared with water, acetonitrile, or acetonitrile/methanol.3. 2 μL of 20 mg/mL liver microsome solution, 1 μL of substrate working solution, 1 μL of compound working solution, and 176 μL of PBS buffer were taken, mixed well, and placed in a 37° C. water bath for pre-incubation for 15 min. 1 μL of sulfaphenazole, quinidine, or ketoconazole working solution was added to the positive control group instead of the compound working solution. At the same time, 10 mM NADPH solution was placed together in the 37° C. water bath for pre-incubation for 15 min. After 15 min, 20 μL of NADPH was added to each well to initiate the reaction. The mixture was incubated at 37° C. for 5 min (CYP2C9), 20 min (CYP2D6), or 5 min (CYP3A4). All incubated samples were in duplicate. After incubation for the corresponding period of time, 400 μL of icy methanol containing internal standard was added to all samples to stop the reaction. The mixture was vortexed, mixed well, and centrifuged for 40 min at 4° C. at 3220 g. 100 μL of the supernatant was transferred to a feeding plate after the centrifugation was completed, and 100 μL ultrapure water was added. The mixture was well mixed for LC-MS/MS analysis. Inhibitory Effect of Compound of the Present Invention on Enzymatic Activity of CYP2C9 and CYP2D6 I. Experimental Materials and Instruments1. Human liver microsome (Corning 452117)2. NADPH (Solarbio 705Y021)3. Positive substrates diclofenac (Sigma SLBV3438) and dextromethorphan (TRC 3-EDO-175-1)4. Positive inhibitors sulfaphenazole (D. Ehrenstorfer GmbH 109012) and quinidine (TCI WEODL-RE)5. AB Sciex Triple Quad 5500 liquid chromatography-mass spectrometry systemII. Procedures1. Preparation of 100 mM phosphate-buffered saline (PBS): 7.098 g Na2HPO4 was weighed. 500 mL pure water was added. The mixture was dissolved by sonication to give solution A. 3.400 g KH2PO4 was weighed. 250 mL pure water was added. The mixture was dissolved by sonication to give solution B. The solution A was placed in a stirrer, and the solution B was slowly added until the pH reached 7.4, so that the 100 mM PBS buffer was prepared.2. A 10 mM NADPH solution was prepared with a 100 mM PBS buffer. A 10 mM stock solution of the compound of the present invention was diluted with DMSO to give a compound working solution at a concentration of 200×(6000, 2000, 600, 200, 60, 20, and 0 μM). The positive inhibitor stock solution was diluted with DMSO to give a positive inhibitor working solution at a concentration of 200×(sulfaphenazole, 1000, 300, 100, 30, 10, 3, and 0 μM; quinidine, 100, 30, 10, 3, 1, 0.3, and 0 μM). Substrate working solutions (120 μM diclofenac and 400 μM dextromethorphan) at a concentration of 200× were prepared with water, acetonitrile, or acetonitrile/methanol.3.2 μL of 20 mg/mL liver microsome solution, 1 μL of substrate working solution, 1 μL of compound working solution, and 176 μL of PBS buffer were taken, mixed well, and placed in a 37° C. water bath for pre-incubation for 15 min. 1 μL of sulfaphenazole or quinidine working solution was added to the positive control group instead of the compound working solution. At the same time, 10 mM NADPH solution was placed together in the 37° C. water bath for pre-incubation for 15 min. After 15 min, 20 μL of NADPH was added to each well to initiate the reaction. The mixture was incubated at 37° C. for 5 min (CYP2C9) or 20 min (CYP2D6). All incubated samples were in duplicate. After incubation for the corresponding period of time, 400 μL of icy methanol containing internal standard was added to all samples to stop the reaction. The mixture was vortexed, mixed well, and centrifuged for 40 min at 4° C. at 3220 g. 100 μL of the supernatant was transferred to a feeding plate after the centrifugation was completed, and 100 μL ultrapure water was added. The mixture was well mixed for LC-MS/MS analysis. Luciferase reporter assay 10 μL of supernatant from the assay was transferred to white 384-plate with flat bottom and squared wells. One pouch of QUANTI-Luc Plus was dissolved in 25 mL of water. 100 μL of QLC Stabilizer per 25 mL of QUANTI-Luc Plus solution was added. 50 μL of QUANTI-Luc Plus/QLC solution per well was then added. Luminescence was measured on a Platereader (e.g., Spectramax I3X (Molecular Devices GF3637001)). CYP1A2 Inhibition Assay CYP1A2 Inhibition in Human Liver Microsomes. Pooled human liver microsomes were incubated with individual CYP, CYP1A2, isozyme-specific marker substrate (Phenacetin) in the presence of test compound at various concentrations (0.05, 0.15, 0.5, 1.5, 5, 15, 50 uM). The specific marker metabolites are measured with LC/MS/MS. The remaining enzymatic activities and inhibitory potency IC50 are determined. Procedure: Microsomes are removed out of the −80° C. freezer to thaw on ice and 20 μL of the substrates solution added to the corresponding wells. Then 20 μL PB was added to blank wells and 2 μL of the test compounds and positive control working solution added to the corresponding wells. 2 μL of solvent was added to No Inhibitor wells and Blank wells. Then 158 μL of the HLM working solution was added to all wells of the incubation plate. The plate was pre-warmed for about 10 min using a 37° C. water bath. Then 20 μL of the NADPH cofactor solution was added to all incubation wells, mixed and incubated for 10 minutes at 37° C. water bath. The reaction is terminated by adding 400 μL cold stop solution (200 ng/mL Tolbutamide and 200 ng/mL Labetalol in ACN). The samples were centrifuged at 4000 rpm for 20 minutes to precipitate protein. Finally 200 μL of the supernatant was transferred to 100 μL HPLC water, shaken for 10 min and analyzed by LC/MS/MS. Inhibition Assay Test compound is dissolved in DMSO 500× the highest final concentration desired in the IC50 assay. 30 ml of deionized water is pre-warmed to 37° C. and all kit components are placed on ice. For each well of column 1, 149.4 μL of NADPH-Cofactor Mix, (187.5 μl of Cofactors, 150 μl of G6PDH, 100 μl of Control Protein and 14.56 ml of 37° C. water) are added. In each well from Column 2 to 12, 100 μl of Cofactor/DMSO mix (40 μL DMSO in 9.96 ml of NADPH-Cofactor Mix) are added. To each well of column 1, 0.6 μl of test compound or positive control are added. 50 μl from each well of column 1 are serially diluted to column 8. The extra 50 μl from column 8 are discarded. The plate is covered and pre-incubated at 37° C. for 10 minutes. Preparation of enzyme/substrate mix: 7.92 ml of pre-warmed deionized water, 75 μl of enzyme, 3 μl of 10 mM AMMC and 2 ml of pre-warmed buffer are mixed. After the pre-incubation time (10'), 100 μl of enzyme/substrate mix to each well from column 1 to 10 are added. The plate is incubated at 37° C. for 30 minutes. After this time, 75 μl of Stop Reagent to each well are added. For blank controls, 100 μl of enzyme/substrate mix are added to columns 11 and 12. Study with Mouse and Human Intestinal Tissue Homogenates The aim of this study was to evaluate TK-112690 in vivo as an inhibitor of uridine phosphorylase (UPase) enzyme activity. The range of TK-112690 doses studied for ability to prevent metabolic breakdown of uridine, through the in vitro inhibition of mouse and human small intestinal UPase enzyme, was 0, 0.1, 0.5, 1, 5, 10, 50, 100, 500, 1000, 5000 and 10000 μM). Detection of UPase activity was determined by HPLC analysis using UV detection of uracil concentration (UPase catabolizes uridine into uracil and ribose-1-phosphate).The UPase enzyme material was prepared from homogenized mouse and human being small intestinal tissue. TK-112690 was dissolved in water (50 mg/ml) and analyzed for UPase inhibition in aqueous solution containing 5 mM uridine, 0.01 M Tris, 0.01 M phosphate, 1 mM EDTA, and 1 mM DTT. Reactions were performed at 37° C. at pH of 7.3.TK-11260 inhibition of mouse and human UPase was analyzed by reverse phase HPLC using UV detection. HPLC analysis was performed at ambient temperature with a Water 2695 Alliance system equipped with a C18 ECONOSIL 5 U ALLtech column. 20 μl of UPase reaction samples were auto-injected onto column. Mobile phase consisted of water eluting for first 2.5 ml and acetonitrile gradient to 100% in 12.5 ml (flow rate 0.5 ml/min). The outlet fluent was monitored by UV absorption in the range of 240-320 nm. UPase enzymatic activity was based on the AUC of the uracil peaks. Alphascreen Assay A PI3K Alphascreen assay (PerkinElmer, Waltham, Mass.) was used to measure the activity of a panel of four phosphoinositide 3-kinases: PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ. Enzyme reaction buffer was prepared using sterile water (Baxter, Deerfield, Ill.) and 50 mM Tris HCl pH 7, 14 mM MgCl2, 2 mM sodium cholate, and 100 mM NaCl. 2 mM DTT was added fresh the day of the experiment. The Alphascreen buffer was made using sterile water and 10 mM Tris HCl pH 7.5, 150 mM NaCl, 0.10% Tween 20, and 30 mM EDTA. 1 mM DTT was added fresh the day of the experiment. Compound source plates used for this assay were 384-well Greiner clear polypropylene plates containing test compounds at 5 mM and diluted 1:2 over 22 concentrations. Columns 23 and 24 contained only DMSO as these wells comprised the positive and negative controls, respectively. Source plates were replicated by transferring 0.5 uL per well into 384-well Optiplates (PerkinElmer, Waltham, Mass.). Autotaxin LPC Cascade Assay The efficacy of the Examples described herein, as inhibitors of ATX are demonstrated and confirmed by pharmacological in vitro assays. The following assays and their respective methods are carried out with the compounds according to the present invention. Activity possessed by the compounds may be demonstrated in vivo. Those skilled in the art will appreciate that a variety of assay formats may be used to determine the activity of the compounds of this invention.Autotaxin LPC Cascade Assay:Assay Buffer:100 mM Tris-HCl, pH=9500 mM NaCl5 mM MgCl25 mM CaCl20.05% Triton X 100Reagents:Enzyme Source Stock conc Working conc. Final conc. Autotaxin X-Chem 4.6 μM 20 nM 5 nM LPC Sigma L5254 8 mM in assay buffer 400 μM 100 μM Choline Oxidase Sigma C5896 50 U/mL in water 0.8 U/mL 0.1 U/mL HRP Sigma P8375 1000 U/mL in water 8 U/mL 1 U/mL Ampliflu Red Sigma 90101 100 mM in DMSO 400 μM 50 μM. CA Inhibition Assay An SX.18MV-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic/inhibition of various CA isozymes. Phenol Red (0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, following the CA-catalyzed CO2 hydration reaction for a period of 5-10 s. Saturated CO2 solutions in water at 25 deg C were used as substrate. Stock solutions of inhibitors were prepared at a concentration of 10 mM (in DMSO-water 1:1, v/v) and dilutions up to 0.01 nM. At least seven different inhibitor concentrations have been used for measuring the inhibition constant. Inhibitor and enzyme solutions were preincubated together for 10 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. Triplicate experiments were done for each inhibitor concentration, and the values reported throughout the paper are the mean of such results. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, and represent the mean from at least three different determinations. Fluorescence Quench Assay The inhibitory activity of compounds was assessed by a fluorescence quench assay of BACE1 activity using commercially available substrate HiLyte Fluor 488-Glu-Val-Asn-Leu-Asp-Ala-Glu-Phe-Lys-(QXLT 520)-OH (SEQ ID NO:1) AnaSpec, San Jose, Calif.) and truncated human beta-secretase, BACE1 (amino acids 1-454) fused to a myc-his tag and secreted from HEK293/BACEect. cells into OptiMEM (Invitrogen). The substrate was dissolved at 1 mg/ml in DMSO.The assay was performed in the presence of OptiMEM (supernatant collected over 24 h and cleared from cellular debris by centrifugation) containing the ectodomain of BACE1, 25 ul water containing the desired 2-fold concentration of test compound and 2% DMSO, 1 uM substrate peptide, 20 mM NaOAc, pH 4.4, and 0.04% Triton-X100 in a total assay volume of 50 ul in a 384 well plate. In general, 25 ul of compound dilution were given to the plate followed by the addition of 10 ul of BACE1 containing OptiMEM diluted 1:10 in water. Functional Assay To measure D2 and D3 stimulation of mitogenesis (agonist assay) or D2 and D3 inhibition of quinpirole stimulation of mitogenesis (antagonist assay), CHOp-cells (human receptor) were seeded in a 96-well plate at a concentration of 5,000 cells/well. The cells were incubated at 37C. in alpha -MEM with 10% FBS, 0.05% penicillin-streptomycin, and 200 ug/mL of G418. After 48 hours, the cells were rinsed twice with serum-free alpha -MEM and incubated for 24 hours at 37 C. In the functional assay for agonism, the medium was removed and replaced with 90 ul of serum-free alpha -MEM and 10 ul of test compound in sterile water; in the antagonist assay, the test compound was diluted in sterile water plus 30 nM quinpirole. After another 24-hour incubation at 37 C., 0.25 uCi of [3H]thymidine was added to each well and the plates were further incubated for 2 hours at 37 C. The cells were then trypsinized, and the plates were filtered and counted as usual in the art. High ATP Caliper Endpoint Assay JAK1 enzyme (Invitrogen) stock solution is made up at 5.2 uM in sterile water. JAK1 enzyme stock is diluted to 20 nM in assay buffer (10 mM HEPES free acid pH 7.5, 10 mM HEPES free base pH 7.5, 10 mM MgCL2, 0.0005% Tween-20, 0.01% BSA) containing 2 mM DTT (all supplied by Sigma) with the addition of one protease tablet per 25 mls buffer (Roche). ATP is made up at 10 mM stock in sterile water and diluted to 5 mM in assay buffer. Peptide H236 (Caliper Life Sciences) is made up at 1.5 mM in 100% DMSO and diluted to 3 uM in assay buffer. Stop buffer comprises 140 mM HEPES, 22.5 mM EDTA (Sigma) and 0.15% coating reagent (Caliper Life Sciences).Assays are performed in Greiner polypropylene 384 well plates. Following compound preparation within the plate 10 ul of enzyme in assay buffer containing DTT is added using a Multidrop Micro. Final assay concentration of enzyme is 10 nM. Compound and enzyme are pre-incubated for 30 minutes at room temperature. High ATP Caliper Endpoint Assay JAK3 enzyme (Invitrogen) stock solution is made up at 4.1 uM in sterile water. JAK3 enzyme stock is diluted to 2 nM in assay buffer (10 mM HEPES free acid pH 7.5, 10 mM HEPES free base pH 7.5, 10 mM MgCL2, 0.0005% Tween-20, 0.01% BSA) containing 2 mM DTT (all supplied by Sigma). ATP is made up at 10 mM stock in sterile water and diluted to 800 uM in assay buffer. Peptide (American peptide company) is made up at 30 mM in 100% DMSO and diluted to 3 uM in assay buffer. Stop buffer comprises 140 mM HEPES, 22.5 mM EDTA (Sigma) and 0.15% coating reagent (Caliper Life Sciences).Assays are performed in Greiner polypropylene 384 well plates. Following compound preparation within the plate 10 ul of enzyme in assay buffer containing DTT is added using a Multidrop Micro. Final assay concentration of enzyme is 1 nM. Compound and enzyme are pre-incubated for 60 minutes at room temperature using low evaporation lids before addition of 10 ul ATP/peptide mixture in assay buffer. In Vitro Enzyme Assay The proteolytic enzyme, Dipeptidyle Peptidase IV (DppIV) was monitored in vitro by the substrate Gly-Pro 4-methoxy-β-naphthylamide while in the presence of compound 7. 20 ul of 1.3 mM DppIV was pipetted into twenty-four micro centrifuge tubes containing various concentrations of compound 7 in a total volume of 100 uL. The samples were thoroughly vortexed and centrifuged at 5,000 RPM for 30 seconds. The micro centrifuge tube were then placed in a 37° C. water bath and incubated for 30 minutes. After incubation, 0.625 uM of substrate was added and the volume was adjusted to 130 uM with incubation buffer. Samples were thoroughly mixed and centrifuged at 5,000 RPM for 30 seconds before placed in a 37° C. water bath for an additional 30 minutes. The reaction was terminated by adding 1 ml of 100 mM Citrate buffer pH 4.0 and vortexing thoroughly for 1 minute. The excitation and emission spectrum of each sample was measured at 340 nm and 425 nm respectfully on a Fluoromax-II Fluorometer. In-vitro Plasma Kallikrein Inhibition Enzymatic reactions were conducted in assay buffer , comprising 50 mM Hepes/NaOH at pH 7.8, 150 mM NaCl, 1 mM EDTA and 0.05% (w/v) CHAPS. For the determination of IC50 values, the assays were performed at room temperature in 384-well plates with a total assay volume of 25.25 ul per well. The test compound was dissolved in 90% (v/v) DMSO/water. For the assays, 250 nL of the 90% (v/v) DMSO/water solution or compound solution were added per well, followed by the addition of 12.5 ul protease solution (protease in assay buffer). The final assay concentration of the human plasma kallikrein was nominally 25 pM, the 11 compound concentrations in the dilution series were in the range form 1 nM to 100 uM. After 1 hour of pre-incubation at room temperature, the reactions were started by the addition of 12.5 ul substrate solution (in assay buffer, final assay concentration was 0.5 uM). After the addition of the substrate solution, the final DMSO concentration in the assay was 0.9% (v/v). Inhibition Assay By a method similar to the measurement of PDE9-inhibiting activity, PDE5-inhibiting activity of each of the test compounds was measured, percent inhibition was calculated and IC50 value against PDE5 was determined. To 150 uL of buffer B (70 mmol/L Tris-HCl, pH7.5, 16.7 mmol/L MgCl2, 33.3 nmol/L [3H]-cGMP) solution containing [3H]-cGMP (specific activity=244.2 GBq/mmol) at a concentration of 33.3 nmol/L, 50 uL of a solution of the compound to be evaluated (formed by dissolving the compound in DMSO and diluting it with distilled water to DMSO concentration of 5%) and 50 uL of the PDE9 protein solution as prepared in the above, as diluted with buffer C (40 mmol/L Tris-HCl, pH7.5, 15 mmol/L benzamidine, 15 mmol/L 2-mercaptoethanol, 1 ug/mL Pepstatin A, 1 ug/mL Leupeptin) by 1,500x, were added under cooling with ice. This mixed solution was incubated at 30 C. for 30 minutes and the enzymatic reaction of PDE9 was terminated by heating the system in boiling water. Inhibition Assay Master solutions were prepared containing human liver microsomes (Gibco, 0.2 mg/mL) and MgCl2 (5 mM) in potassium phosphate buffer (10 mM). To aliquots (169 μL) of the microsome solution was added test compound in acetonitrile (1 μL) and DMSO (1 μL) to provide final test compound concentrations of 0, 0.005, 0.05, 0.25, 1, 5, 10, and 25 μM.NADPH (10 mM) in ultra-pure water (20 μL) was added, and this mixture was incubated at 37° C. for 30 minutes. The enzyme reaction then was initiated by the addition of enzyme substrate (dextromethorphan) dissolved in 1 μL of acetonitrile and 9 μL of ultra-pure water. The final substrate concentration was 10 μM.After 20 minutes, the incubation mixture was diluted with 3 volumes of cold methanol containing imipramine (200 nM), labetalol (200 nM), and ketoprofen (2 μM) as internal standards. Samples were centrifuged at 16,000 g for 10 minutes, then an aliquot of the supernatant (200 μL) was removed and a Inhibition Assay Solutions of each test compound were separately prepared at concentrations of 20000, 6000, 2000, 600, 200, and 60 uM by serial dilution with DMSO:MeCN (50:50 v/v). The individual test compound solutions were then diluted 20-fold with DMSO:MeCN:deionized water (5:5:180 v/v/v) to concentrations of 1000, 300, 100, 30, 10, and 3 M. Mixtures of isozyme inhibitors (sulfaphenazole, tranylcypromine, and ketoconazole as specific inhibitors of isozymes 2C9, 2C19, and 3A4, respectively) were prepared containing each inhibitor at concentrations of 6000, 2000, 600, 200, 60, 20, 6, and 2 uM by serial dilution with DMSO:CH3CN (50:50 v/v). The mixed inhibitor solutions were then diluted 20-fold with DMSO: CH3CN:deionized water (5:5:180 v/v/v) to concentrations of 300, 100, 30, 10, 3, 1, 0.3, and 0.1 uM. The percent of organic solvent attributable to the test compound or inhibitor mixture in the final reaction mixture was 2% v/v. Pooled human liver microsome suspension (20 mg/mL) was diluted with phosphate buffer to obtain a 5 mg/mL suspension. Inhibition Assay To 150 uL of buffer B (70 mmol/L Tris-HCl, pH7.5, 16.7 mmol/L MgCl2, 33.3 nmol/L [3H]-cGMP) solution containing [3H]-cGMP (specific activity=244.2 GBq/mmol) at a concentration of 33.3 nmol/L, 50 uL of a solution of the compound to be evaluated (formed by dissolving the compound in DMSO and diluting it with distilled water to DMSO concentration of 5%) and 50 uL of the PDE9 protein solution as prepared in the above, as diluted with buffer C (40 mmol/L Tris-HCl, pH7.5, 15 mmol/L benzamidine, 15 mmol/L 2-mercaptoethanol, 1 ug/mL Pepstatin A, 1 ug/mL Leupeptin) by 1,500x, were added under cooling with ice. This mixed solution was incubated at 30 C. for 30 minutes and the enzymatic reaction of PDE9 was terminated by heating the system in boiling water for 90 seconds. Returning the system to room temperature, 50 uL of Snake venom (SIGMA: 1 mg/mL) was added, followed by 10 minutes' incubation at 30 C. Enzymatic Activity Assay The inhibition of purified recombinant human GAC by varying concentrations of inhibitors is assessed via a dual-coupled enzymatic assay. The glutamate produced by the glutaminase reaction is used by glutamate oxidase to produce α-ketoglutarate, ammonia, and hydrogen peroxide, with this hydrogen peroxide subsequently being used by horseradish peroxidase to produce resorufin in the presence of Amplex UltraRed. The assay buffer consisted of 50 mM Hepes (pH 7.4), 0.25 mM EDTA and 0.1 mM Triton X-100. GAC was incubated with potassium phosphate (10 minutes at room temperature) prior to incubation with inhibitor (10 minutes at room temperature). The final reaction conditions were as follows: 2 nM GAC, 50 mM potassium phosphate, 100 mU/mL glutamate oxidase (Sigma), 1 mM glutamine (Sigma), 100 mU/mL horseradish peroxidase (Sigma), 75 μM Amplex UltraRed (Life Technologies), and 1% (v/v) DMSO. The production of resorufin was monitored on a Perkin Elmer Envision plate reader (excitation 530 nm, emission 590 nm) either in a kinetics or endpoint mode (at 20 minutes). IC50 values were calculated using a four-parameter logistic curve fit. Biological Assays of the SSAO Enzyme Inhibitors Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≤2%.Hydrogen Peroxide Detection:In a horseradish peroxidase (HRP) coupled reaction, hydrogen peroxide oxidation of 10-acetyl-3,7-dihydroxyphenoxazine produced resorufin, which is a highly fluorescent compound (Zhout and Panchuk-Voloshina. Analytical Biochemistry 253 (1997) 169-174; Amplex Red Hydrogen Peroxide/peroxidase Assay kit, Invitrogen A22188). Enzyme and compounds in 50 mM sodium phosphate, pH 7.4 were set to pre-incubate in flat-bottomed microtiter plates for approximately 15 min before initiating the reaction by addition of a mixture of HRP, benzylamine and Amplex reagent. Benzylamine concentration was fixed at a concentration corresponding to the Michaelis constant, determined using standard procedures. Fluorescence intensity was then measured at several time points during 1-2 h, exciting at 544 nm and reading the emission at 590 nm. For the human SSAO assay final concentrations of the reagents in the assay wells were: SSAO enzyme 1 ug/mL, benzylamine 100 uM, Amplex reagent 20 uM, HRP 0.1 U/mL and varying concentrations of test compound. The inhibition was measured as % decrease of the signal compared to a control without inhibitor (only diluted DMSO). The background signal from a sample containing no SSAO enzyme was subtracted from all data points. Data was fitted to a four parameter logistic model and IC50 values were calculated using the GraphPad Prism 4 or XLfit 4 programs. Cyclooxygenase activity assay Reaction mixture containing 100 mM Tris-HCl buffer (pH 8) and COX-1 (ovine) or COX-2 (human recombinant) was preincubated for 10 min in a water bath at 37 °C. The reaction was initiated by the addition of 10 µL arachidonic acid (final concentration 100 µM). After 2 min, the reaction was terminated by adding 1 M HCl. Test compounds were dissolved in DMSO and diluted to the desired concentration with potassium phosphate buffer of pH 7.4. Cytochrome P450 Isoenzyme Inhibitory Activity Assay Test compounds, standard inhibitors (at 100× final concentration), and mixed substrate working solutions were prepared; the microsomes (purchased from Corning Inc.) stored at −80° C. were taken out and thawed. To the corresponding wells were added 20 μL of the test compound and standard inhibitor solutions. Meanwhile, 20 μL of the respective solvent was added to the No Inhibitor Control (NIC) and blank control wells. Next, 20 μL of mixed substrate solution was added to the corresponding wells, except the blank wells where 20 μL of phosphate buffer (PB) was added. A human liver microsome solution was prepared (immediately returned to the fridge after the date of use was marked). Then, 158 μL of this solution was added to all the wells. The sample plate was placed in a 37° C. water bath for pre-incubation. A co-enzyme factor (NADPH) solution was then promptly prepared. After 10 minutes, 20 μL of NADPH solution was added to all wells. The sample plate was shaken to mix and placed back into a 37° C. water bath for an additional 10 minutes of incubation. At the respective time points, the reaction was terminated by adding 400 μL of cold acetonitrile solution (internal standard at 200 ng/ml of tolbutamide and labetalol). The sample plate was mixed thoroughly and was then centrifuged at 4000 rpm for 20 minutes to precipitate proteins. 200 μL of the supernatant was taken and mixed with 100 μL of water, after which it was sent for LC/MS/MS analysis. Ca2+-Calmodulin Dependent PDE Enzyme Assays PDE1B, PDE1A, and PDE1C are cloned and purified following standard protein generation procedures. The assay buffer is prepared to give a final concentration in the assay of 50 mM Tris-HCl, 50 mM MgCl2, 4 mM CaCl2, 0.1% Bovine serum albumin and 6 U/ml Calmodulin in water, at pH 7.5. The final enzyme concentration is 0.25, 0.074 and 0.0012 nM, for PDE1A, PDE1B and PDE1C respectively. The reactions are started by addition of the substrate, [3H]cAMP, to give a final concentration of 47 nM. Enzyme Inhibition Assay The enzyme reaction was started by the addition of enzyme to the reaction mixture containing substrate and test compounds. The reaction was stopped by spotting an aliquot onto a silica gel plate that had been prespotted with thymine and thymidine. The plate was developed in ethyl acetate-water-formic acid (60:35:5), and the spots were visualized under UV light (254 nm) and cut out for radioactivity determination in toluene-based scintillation cocktail. IC50 values were determined from dose response curves, and were means of minimum three experiments. Trypanosoma brucei Brucei Lysis Assay Briefly, the AlamarBlue active compound, the resazurin, a blue, water soluble, non-toxic and cell permeable molecule, which can be followed by absorbance, is reduced by various metabolic pathways into resorufin, a red compound which can be followed by either absorbance or fluorescence. The assay allows the calculation of the percent viability (percent of living Trypanosomes remaining in each well) at the end of a lysis relative to the untreated condition by interpolation of fluorescent values (FLU) on a standard curve with a known amount of seeded trypanosome/well. [35S]GTPγS Coupling Assay For antagonist experiments, protein was preincubated with test compounds for 15 min prior to the addition of 100 nM U69,593 and [35S]GTPγS. Reactions were terminated by rapid filtration using a 96-well plate Brandel cell harvester (Brandel, Gaithersburg, MD) followed by washes with ice cold water. Microscint-20 (PerkinElmer Life Sciences) was added to the plates after drying, and radioactivity was read with a TopCount NXT HTS microplate scintillation and luminescence counter (PerkinElmer Life Sciences). All compounds were run in parallel assays in duplicate for comparison. Inhibition of DAO (Diamine Oxidase) Test purpose: The following method is used to determine the selective inhibitory activity of the compound of the present invention on DAO.Test Materials:Human recombinant DAO (Recombinant Human ABP-1/DAO) was purchased from R&D, Cat. No. 8298-AO;Amplex® Red Hydrogen PeroxidePeroxidase Assay Kit was purchased from Invitrogen, Cat. No. A22188;1,4-Diaminobutane dihydrochloride was purchased from Aladdin, Cat. No. D106194-25G.Test Method:The test compound was dissolved in DMSO and diluted 5 times to a total of 6 concentrations. In a 384-well plate, 24 μL of human recombinant DAO (1 μg/mL) was added into each well. 1 μL of test compounds at different concentrations were added to each well containing human recombinant DAO, and the plate was incubated at 37° C. for 30 min. After incubating for 30 min, 25 μL of Amplex® Red Hydrogen Peroxide Peroxidase Assay Kit (containing 100 μM Amplex® Red and 0.2 U/ml HRP) containing 1 M 1,4-butanediamine dihydrochloride was added into the corresponding wells, and the plate was incubated in the dark at 37° C. for 30 min. After 30 min, PHERAstar FSX microplate reader of BMG LABTECH was used to read the fluorescence value (RFU) under excitation at 540 nm and emission at 580 nm. CA Inhibition Assay An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity [Khalifah et al., J. Biol. Chem., 246:2561-2573]. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10-100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to0.01 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. Inhibition Assay 5 uM ATP: Test compounds dissolved and diluted in DMSO (2 μl) were added to a reaction mixture comprising 10 μl of 5× Reaction Buffer (40 mM MOPS pH 7.0, 5 mM EDTA), 10 μl of recombinant human PIM2 solution (4 ng PIM-2 dissolved in dilution buffer (20 mM MOPS pH 7.0; EDTA 1 mM; 5% Glycerol; 0.01% Brij 35; 0.1%; 0.1% 2-mercaptoethanol; 1 mg/ml BSA)) and 8 ul of water. Reactions were initiated by the addition of 10 ul of ATP Solution (49% (15 mM MgCl2; 5 uM ATP) 1% ([γ-33P]ATP: Stock 1 mCi/100 μl; 3000 Ci/mmol (Perkin Elmer)) and 10 ul of substrate peptide solution (RSRSSYPAGT (SEQ ID NO.: 6), dissolved in water at a concentration of 1 mM), Reactions were maintained for 10 min at 30° C. The reactions were quenched with 100 ul of 0.75% Phosphoric acid, then transferred to and filtered through a Phosphocellulose filter plate (Millipore, MSPH-N6B-50). After washing each well 4 times with 0.75% Phosphoric acid, scintillation fluid (20 uL) was added to each well and Inhibition Assay Test compounds dissolved and diluted in DMSO (2 μl) were added to a reaction mixture comprising 10 μl of 5× Reaction Buffer (40 mM MOPS pH 7.0, 5 mM EDTA), 10 μl of recombinant human PIM2 solution (4 ng PIM-2 dissolved in dilution buffer (20 mM MOPS pH 7.0; EDTA 1 mM; 5% Glycerol; 0.01% Brij 35; 0.1%; 0.1% 2-mercaptoethanol; 1 mg/ml BSA)) and 8 ul of water. Reactions were initiated by the addition of 10 ul of ATP Solution (49% (15 mM MgCl2; 75 uM ATP) 1% ([γ-33P]ATP: Stock 1 mCi/100 μl; 3000 Ci/mmol (Perkin Elmer)) and 10 ul of substrate peptide solution (RSRSSYPAGT (SEQ ID NO.: 6), dissolved in water at a concentration of 1 mM), Reactions were maintained for 10 min at 30° C. The reactions were quenched with 100 ul of 0.75% Phosphoric acid, then transferred to and filtered through a Phosphocellulose filter plate (Millipore, MSPH-N6B-50). After washing each well 4 times with 0.75% Phosphoric acid, scintillation fluid (20 uL) was added to each well and the resid Measurement of Dissociation Constants (Kd) by SPR SPR experiments were performed using the P4SPR from Affinité Instruments using Ni-NTA immobilization chips and his-tagged protein. Ni-NTA coated surfaces allow the immobilization of his-tagged proteins by chelation of histidine residues to the nickel ion. The sensor chip was inserted into a quad inlet model P4SPR (with 4 independent channels). Once the instrument was turned on, the baseline was stabilized by deionized (DI) water, followed by signal stabilization by the running buffer. His-tagged protein at 10 μg/mL was injected into all 4 channels of the P4SPR and was left to react for 20 min. The sensor chip was then washed with DI water. The lowest concentration of the ligand was injected into the channels of the P4SPR and was left to react for 10 min. The SPR shift was saved. Then, a higher concentration of the ligand was injected, and the sample injection steps were repeated until all 5 concentrations have been added. The KD of the binding interaction between the ligand and protein was determined by using the affinity curve fitting function in the P4SPR Control software. SOD Assay (Fluorimetric Analysis) SOD activity of the purified samples was determined by the SOD assay kit using (2-(4-iodophenyl)-3-(4nitrophenyl)-5-(2,4-disulphenyl)-2H-tetrazolium, monosodium salt) [WST] which is a colorimetric assay that monitors the rate of inhibition of WST to a water soluble formazan dye. Purified SOD from bovine brain (20 μl) was incubated (37°C, 20 min) with WST working solution (200 μl) and distilled water (20 μl) according to the Sigma Information Bulletin, 19160 after which SOD activity was determined from the absorbance at 450 nm. Spectrofluorimetry was used to analyse the interaction of amyloid peptides [Aβ25-37, Aβ29-33 and Aβ1-40] with SOD. The excitation wavelength was fixed at 295 nm, the wavelength at which tryptophan absorbs, and the emission wavelength was at 482 nm. The change in fluorescence of the solution was monitored as increasing concentrations of amyloid peptides (0-40 μM) were added to a reaction mixture of SOD (5.0 μl) in Tris-HCl buffer (pH 8.0, 50 mM) in a final volume of 300 μl. Cytochrome P450 Isoenzyme Inhibitory Activity Test The inhibitory activities of test compound against different isoforms of human cytochrome P450 isoenzymes were determined. Experimental Operation. The test compound, standard inhibitor (100×final concentration) and mixed substrate working solution were prepared; the microsome frozen in −80° C. refrigerator was taken out and thawed. 2 μL of the compound to be tested and standard inhibitor solution were added to the corresponding wells, and at the same time, 2 μL of the corresponding solvent was added to the non-inhibitor control wells (NIC) and the blank control wells; secondly, 20 μL of mixed substrate solution was added to the corresponding wells except the blank wells (20 μL of Pb was added to the blank wells); human liver microsome solution was prepared (the solution was put back in the refrigerator immediately after using and marking the date), and then 158 μL of human liver microsome solution was added to all wells; the sample plate was put in a 37° C. water bath for pre-incubation, and then a coenzyme factor (NADPH) solution was prepared; after 10 minutes, 20 μL of NADPH solution was added to all wells, the sample plate was shaken well, and incubated in a 37° C. water bath for 10 minutes; at the corresponding time point, 400 μL of cold acetonitrile solution (internal standard is 200 ng/mL tolbutamide and labetalol) was added to terminate the reaction; after the plates were evenly mixed, the mixture was centrifuged at 4000 rpm for 20 minutes to precipitate protein; 200 μL of supernatant was added into 100 μL of water, shaken well and detected by LC/MS/MS. In vitro kinase assay In vitro kinase assays described below can be used to determine the activity of a test compound for inhibiting the activity of JAK2 kinase. The test compounds were dissolved in dimethyl sulfoxide and diluted with water to a serial concentration gradient as required in the experiment. JAK2 substrates (Cell Signaling Technology, Catalog Number: 1305s) and ATP (2 mM) solution were diluted with water to obtain a final concentration of 20 μM ATP and 1.2 μM substrate solution. The appropriate amount of JAK2 kinase (Invitrogen, Catalog Number: pv4210) was mixed with 4× buffer (prepared by user, and comprising 50 mM HEPES, pH 7.3, 125 mM NaCl, 24 mM MgCl2, 1.25 mM DTT) to a final concentration of 8 ng/μL. To each well of a microplate [DELFIA Streptavidin-coated clear plate (Perkin Elmer, Catalog Number: AAAND-0005)]17.5 μL of ATP/substrate mixture, 5 μL of aqueous solution of a test compound (5 μL of pure water only were added to control and blank), and 7.5 μL of the kinase solution prepared above (4× buffer only was added to control) were added. Each well was mixed sufficiently, then incubated at room temperature (27° C.) for 50 minutes, washed with wash buffer, and dried three times, then HRP conjugated antibody [Phospho-Tyrosine Mouse mAb (P-Tyr-100) (HRP Conjugate, Cell signaling Technology, Catalog Number: 5465)] was added, and incubated for 1 hour. The microplate was washed with wash buffer and dried three times, and then TMB (Sigma, Catalog Number: T4444) was added and incubated for 5 to 15 minutes to allow for color change. Stop solution (1 N sulfuric acid solution) was added to stop the reaction. Absorbance was measured on a Novostar microplate reader at a wavelength of 450 nm. In vitro kinase assay JAK1: In vitro kinase assays described below can be used to determine the activity of a test compound for inhibiting the activity of JAK1 kinase. The test compounds were dissolved in dimethyl sulfoxide and diluted with water to a serial concentration gradient as required in the experiment. JAK1 substrates (Cell Signaling Technology, Catalog Number: 1305s) and ATP (2 mM) solution were diluted with water to obtain a final concentration of 20 μM ATP and 1.2 μM substrate solution. The appropriate amount of JAK1 kinase (Invitrogen, Catalog Number: pv4774) was mixed with 4× buffer (prepared by user, and comprising 50 mM HEPES, pH 7.3, 125 mM NaCl, 24 mM MgCl2, 1.25 mM DTT) to a final concentration of 8 ng/μL. To each well of a microplate [DELFIA Streptavidin-coated clear plate (Perkin Elmer, Catalog Number: AAAND-0005)] 17.5 μL of ATP/substrate mixture, 5 μL of an aqueous solution of a test compound (5 μL of pure water only were added to the control and blank), and 7.5 μL of the kinase solution prepared above (4× buffer only was added to the control) were added. Each well was mixed sufficiently, then incubated at room temperature (27° C.) for 50 minutes, washed with wash buffer, and dried three times, then HRP conjugated antibody was added [Phospho-Tyrosine Mouse mAb (P-Tyr-100) (HRP Conjugate, Cell Signaling Technology, Catalog Number: 5465)], and incubated for 1 hour. The microplate was washed with wash buffer and dried three times, and then TMB (Sigma, Catalog Number: T4444) was added, and incubated for 5 to 15 minutes to allow for color change. Stop solution (1 N sulfuric acid solution) was added to stop the reaction. Absorbance was measured on a Novostar microplate reader at a wavelength of 450 nm. In vitro kinase assay TBDIn vitro kinase assays described below can be used to determine the activity of a test compound for inhibiting the activity of JAK3 kinase. The test compounds were dissolved in dimethyl sulfoxide and diluted with water to a serial concentration gradient as required by the experiment. JAK3 substrates (Cell Signaling Technology, Catalog Number: 1305s) and ATP (2 mM) solution were diluted with water to obtain a final concentration of 20 M ATP and 1.2 M substrate solution. The appropriate amount of JAK3 kinase (Invitrogen, Catalog Number: pv3 855) was mixed with 4× buffer (prepared by user, and comprising 50 mM HEPES, pH 7.3, 125 mM NaCl, 24 mM MgCl2, 1.25 mM DTT) to a final concentration of 8 ng/μL. To each well of a microplate [DELFIA Streptavidin-coated clear plate (Perkin Elmer, Item: AAAND-0005)]17.5 μL of ATP/substrate mixture, 5 μL of aqueous solution of a test compound (L of pure water only were added to the control and blank), and 7.5 μL of the kinase solution prepared above (4× buffer only was added to the control) were added. Each well was mixed sufficiently, then incubated at room temperature (27° C.) for 50 minutes, washed with wash buffer, and dried three times, then HRP conjugated antibody [Phospho-Tyrosine Mouse mAb (P-Tyr-100) (HRP Conjugate, Cell signaling Technology, Catalog Number: 5465)] was added, and incubated for 1 hour. The microplate was washed with wash buffer and dried three times, and then TMB (Sigma, Catalog Number: T4444) was added and incubated for 5 to 15 minutes to allow for color change. Stop solution (1 N sulfuric acid solution) was added to stop the reaction. Absorbance was measured on a Novostar microplate reader at a wavelength of 450 nm. Inhibition Against Cytochrome P450 Isoenzymes Table 9: The inhibition of the test compound against different subtypes of the human cytochrome P450 isoenzyme was determined. The test compound, a standard inhibitor (100×final concentration) and a mixed substrate working solution were prepared; the microsomes frozen in a refrigerator at −80° C. were taken out and thawed. 2 μL of a solution of the test compound and the standard inhibitor was added to corresponding wells, and 2 μL of a corresponding solvent was added to a non-inhibitor control (NIC) well and a blank control (Blank) well; then, 20 μL of a solution of mixed substrate was added to corresponding wells except for the Blank well (adding 20 μL of PB to the Blank well); a human liver microsome solution (labeled with the date after use and immediately putting back to a refrigerator) was prepared and then added to all wells at 158 μL per well; the sample plate was put into a 37° C. water bath for pre-incubation, and then a coenzyme factor (NADPH) solution was prepared; after 10 min, the NADPH solution was added to all the wells at 20 μL per well, and the sample plate was shaken to mix well and then incubated in a 37° C. water bath for 10 min; at corresponding time points, 400 μL of cold acetonitrile solution (internal standard: 200 ng/mL tolbutamide and labetalol) was added to stop the reaction; after being mixed well, the mixture in the sample plate was centrifuged at 4,000 rpm for 20 min, and proteins were precipitated; 200 μL of supernatant was collected and added into 100 μL of water, and the mixture was mixed well and then analyzed by LC/MS/MS. JAK1 Kinase Assay TBDIn vitro kinase assays described below can be used to determine the activity of a test compound for inhibiting the activity of JAK1 kinase. The test compounds were dissolved in dimethyl sulfoxide and diluted with water to a serial concentration gradient as required in the experiment. JAK1 substrates (Cell Signaling Technology, Catalog Number: 1305s) and ATP (2 mM) solution were diluted with water to obtain a final concentration of 20 μM ATP and 1.2 μM substrate solution. The appropriate amount of JAK1 kinase (Invitrogen, Catalog Number: pv4774) was mixed with 4× buffer (prepared by user, and comprising 50 mM HEPES, pH 7.3, 125 mM NaCl, 24 mM MgCl2, 1.25 mM DTT) to a final concentration of 8 ng/μL. To each well of a microplate [DELFIA Streptavidin-coated clear plate (Perkin Elmer, Catalog Number: AAAND-0005)]17.5 μL of ATP/substrate mixture, 5 μL of an aqueous solution of a test compound (5 μL of pure water only were added to the control and blank), and 7.5 μL of the kinase solution prepared above (4× buffer only was added to the control) were added. Each well was mixed sufficiently, then incubated at room temperature (27° C.) for 50 minutes, washed with wash buffer, and dried three times, then HRP conjugated antibody was added [Phospho-Tyrosine Mouse mAb (P-Tyr-100) (HRP Conjugate, Cell Signaling Technology, Catalog Number: 5465)], and incubated for 1 hour. The microplate was washed with wash buffer and dried three times, and then TMB (Sigma, Catalog Number: T4444) was added, and incubated for 5 to 15 minutes to allow for color change. Stop solution (1 N sulfuric acid solution) was added to stop the reaction. ITK Enzyme Assay 1.0M HEPES Buffer pH 7.5 solution was prepared as follows: 238.3 g HEPES free acid (Sigma) and 800 mL of water were combined, and the mixture was stirred until complete dissolution. The pH was adjusted to 7.5 via titration with 5N NaOH and the volume adjusted to 1000 mL. The solution was filtered and sterilized. ITK assay buffer was prepared as follows: 50 mL of HPLC-grade water was treated with 2 mL of 1.0M HEPES Buffer, 500 μL of 2% Gelatin (Sigma), 1.0 mL of aqueous MgCl2 solution (1.0M), and 1.0 mL of aqueous glutathione solution (0.5M), and the solution was mixed. The solution was brought to 99 mL in a graduated cylinder by addition of water and sterilized through a 0.2 μm filter. 0.1 mL of Brij-35 Surfact-Amps Detergent Solution (10% w/v aqueous solution, ThermoFisher) and 1.0 mL of ATP (Teknova,100 mM) were added and the solution was mixed. Preparation of 1.33× ITK enzyme solution was as follows: 49.99 mL of ITK assay buffer was treated with 4.1 μL of ITK enzyme (ITK FL (N-Flag and C-His tagged, 72 kDa) Lake Pharma, 0.25 mg/ml in a buffer containing 25 mM Tris pH 7.8, 150 mM NaCl, 10% glycerol and 2 mM TCEP) and the mixture was gently agitated. The resulting solution was stored on ice. 30 Minutes prior to use, the enzyme solution was removed from ice and equilibrated to RT by incubation in a RT water bath. Preparation of 4× ITK substrate solution was as follows: 50 mL of ITK assay buffer was treated with 100 μL of BTK peptide (China Peptide Company, 2 mM stock solution in DMSO). The tube was capped, mixed by gently inverting the tube, and then stored on ice. 30 Minutes prior to use, the substrate solution was removed from ice and equilibrated to RT by incubation in a RT water bath.At the time of assay, 7.5 μL of the 1.33× ITK enzyme solution was added to plate wells containing 0.1 μL of varying concentrations of test compound in DMSO. The plate was incubated 30 min at RT. The plate wells were each treated with 2.5 uL of the 4× ITK substrate solution and the plate was sealed (TopSeal , Perkin Elmer). The plate was spun at 1000 rpm for 30 sec and then incubated for 60 min at RT. The seal was removed, and each well was treated with 10 μL of Stop/Detect Buffer (20 mM HEPES pH 7.5, 0.01% gelatin, 1 nM LANCE PT66 (Perkin Elmer), 16.5 μg/ml Surelight APC (Perkin Elmer), 10 mM EDTA, 250 mM NaCl). The plate was again covered and was spun at 1000 rpm for seconds. The plate was allowed to incubate overnight at RT and in a closed carrier to reduce dehydration. Biological Assay of the SSAO Enzyme Inhibitors All assays were performed in room temperature with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 2006, 46, 321-331). The enzyme activity was measured with benzylamine as substrate and utilized the production of hydrogen peroxide for detection. In a horseradish peroxidise (HRP) coupled reaction, hydrogen peroxide oxidation of 10-acetyl-3,7-dihydroxyphenoxazine produced resorufin, which is a highly fluorescent compound (Zhout and Panchuk-Voloshina. Analytical Biochemistry 1997, 253, 169-174; Amplex Red Hydrogen Peroxide/peroxidise Assay kit, Invitrogen A22188).Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer (50 mM sodium phosphate, pH 7.4) yielded a final DMSO concentration ≤2%. Enzyme and compounds were set to pre-incubate in flat-bottomed microtiter plates for approximately 60 minutes before initiating the reaction by addition of a mixture of HRP, benzylamine and Amplex reagent. Fluorescence intensity was then measured at several time points (15 minutes, 20 minutes and 30 minutes) exciting at 544 nm and reading the emission at 590 nm). Final concentrations of the reagents in the assay wells were: SSAO enzyme 2 μg/ml, benzylamine 100 μM, Amplex reagent 20 μM, HRP 0.1 U/mL and varying concentrations of test compound. The inhibition was measured as % decrease of the signal compared to a control without inhibitor (only diluted DMSO). The background signal from a sample containing no SSAO enzyme was subtracted from all data points. Data was fitted to a four parameter logistic model and IC50 values were calculated using the GraphPad Prism 4 or XLfit 4 programs. MAO Enzyme Inhibition Assay The effects of the test compounds on hMAO isoform enzymatic activity were evaluated by measuring their effects on the production of hydrogen peroxide (and therefore, of resorufin) from p-tyramine, using the Amplex Red MAO assay kit (Molecular Probes, Eugene, Oregon, USA) and microsomal MAO isoforms prepared from insect cells infected with recombinant baculovirus containing cDNA inserts for human MAO-A or MAO-B. The production of H2O2 was quantified in a multidetection microplate fluorescence reader (FLX800, Bio-Tek Instruments, Inc., Winooski, VT) based on the fluorescence generated (excitation, 545 nm; emission, 590 nm) over a 15 min period, in which the fluorescence increased linearly. Carbonic Anhydrase II Kinetic Assay Kinetic studies were performed by using different concentration of inhibitors over different concentrations of substrate (4-NPA) such as 0.175, 0.35, 0.70 and 1.40 mM. The enzyme 0.2 mg/mL concentration for each well was used after dissolving in de-ionized water. HEPES-tris ammonia was used as buffer at pH of 7.4. The change in absorbance was measured by keeping the 96-well flat bottom plate in ELISA reader (Spectra max 384, Molecular Devices USA) after addition of substrate for a period of 30 min at 25 °C with 1 min interval. Cholinesterase Activity Assay The eight synthesized dialkyl-3-cyanopropylphosphates (3a-h) were dissolved in0.01 M dimethyl sulphoxide and then diluted in demineralized water to 1 mM and 0.1 mM. Pursuing the procedure described in [Carletti et al., Biochem. J., 421:97-106], the activities were determined as follows: the reaction mixture containing the phosphate buffer, AChE or BChE and the chosen dialkyl-3-cyanopropylphosphate, was prepared and intensively stirred. DTNB and ATCh were then added to the sample and mixed, and the absorbance was then measured at 405 nm using a Thermo Scientific Multiskan FC Microplate Photometer. Inhibitory Activity Assay The reaction mixture contained 160 uL of reaction buffer [50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5 ug/mL bovine serum albumin (BSA), 5 uM CaCl2, and 10 mM anilinonaphthalene sulfonate (ANS)], 20 uL of substrate stock solution (2 mM DMPC), and 2 mM of sodium deoxycholate in water. After 5 min of sonication, 10 uL of inhibitor stock solution (in DMSO) were incubated at 28 uC for 10 min. The reactions were started by adding 10 uL of hnps-PLA2 stock solution (0.1 mg/mL) and monitored by excitation at 377 nm and emission at 470 nm. ROS Production Measurement on hNOX1 Membranes All solutions were placed on ice and protected from light. The final concentration in the 1x hNOX1 membrane fluorescent assay buffer was PBS pH7, 6 μM FAD, 15 μM PA, 1 mM MgCl2, 12.5 μM AR, 0.02 u/ml, 125 ng membranes, 1.5 μg of cofactors and 30 μM NADPH.The NADPH was dissolved in water at a concentration of 12 mM and was transferred in a metal transfer plate kept at 4° C. The NADPH was added to the assay plate to initiate the reaction just before the measurement. Nox4 Assay Cells: HEK (CJ Nox4) stably expressing Nox4 was purchased from Redoxis AB (Lund). The adherent cells were cultivated in RPMI 1640 with L-Glutamine were supplemented with FBS (10%), penicillin (10 U/ml) streptomycin (100 μg/ml) and neomycin (200 μg/ml) at 37° C. in air with 5% CO2.Hydrogen peroxide produced by Nox4 was measured (fluorescence emission: 590 and excitation: 544) using Amplex red (Molecular Probes) in Fluorescan Ascent plate reader Type 374. Cells were collected from cultures by Trypsin mediated detachment of adherent cells. Cells were seeded in 96-well plates at a density of 50 000 cells in 200 l assay volume. Inhibitors were added for 30 min (37° C.) and then reagents was added to give a final concentration of Amplex Red 35 mM and 0.17 U/ml horseradish peroxidase. Nox4 activity was measured up to 100 min with readings every minute. Inhibition curves of different Nox4 inhibitors were evaluated at 50% inhibition (IC50) in comparison to cell control without inhibitor present. Y-axes: turnover of hydrogen peroxide; x-axes: concentration of inhibitor. Inhibitors were diluted in a compound plate in DMSO (100%) then transferred to Hanks buffer solution and in assay plate DMSO were 2% in all the wells.Compounds (Nox inhibitors) were diluted at 3× working concentration and titrated from 200 μM to 0.003 μM in 11 steps. ITK Enzyme Assay 1.0 M HEPES Buffer pH 7.5 solution was prepared as follows: 238.3 g HEPES free acid (Sigma) and 800 mL of water were combined, and the mixture was stirred until complete dissolution. The pH was adjusted to 7.5 via titration with 5N NaOH and the volume adjusted to 1000 mL. The solution was filtered and sterilized.ITK assay buffer was prepared as follows: 50 mL of HPLC-grade water was treated with 2 mL of 1.0 M HEPES Buffer, 500 μL of 2% Gelatin (Sigma), 1.0 mL of aqueous MgCl2 solution (1.0 M), and 1.0 mL of aqueous glutathione solution (0.5 M), and the solution was mixed. The solution was brought to 99 mL in a graduated cylinder by addition of water and sterilized through a 0.2 μm filter. 0.1 mL of Brij-35 Surfact-AmpS Detergent Solution (10% w/v aqueous solution, ThermoFisher) and 1.0 mL of ATP (Teknova, 100 mM) were added and the solution was mixed.Preparation of 1.33×ITK enzyme solution was as follows: 49.99 mL of ITK assay buffer was treated with 4.1 μL of ITK enzyme (ITK FL (N-Flag and C-His tagged, 72 kDa) Lake Pharma, 0.25 mg/ml in a buffer containing 25 mM Tris pH 7.8, 150 mM NaCl, 10% glycerol and 2 mM TCEP) and the mixture was gently agitated. The resulting solution was stored on ice. 30 Minutes prior to use, the enzyme solution was removed from ice and equilibrated to RT by incubation in a RT water bath.Preparation of 4×ITK substrate solution was as follows: 50 mL of ITK assay buffer was treated with 100 μL of BTK peptide (China Peptide Company, 2 mM stock solution in DMSO). The tube was capped, mixed by gently inverting the tube, and then stored on ice. 30 Minutes prior to use, the substrate solution was removed from ice and equilibrated to RT by incubation in a RT water bath.At the time of assay, 7.5 μL of the 1.33×ITK enzyme solution was added to plate wells containing 0.1 μL of varying concentrations of test compound in DMSO. The plate was incubated 30 min at RT. The plate wells were each treated with 2.5 uL of the 4×ITK substrate solution and the plate was sealed (TopSeal , Perkin Elmer). The plate was spun at 1000 rpm for 30 sec and then incubated for 60 min at RT. The seal was removed, and each well was treated with 10 μL of Stop/Detect Buffer (20 mM HEPES pH 7.5, 0.01% gelatin, 1 nM LANCE PT66 (Perkin Elmer), 16.5 μg/ml Surelight APC (Perkin Elmer), 10 mM EDTA, 250 mM NaCl). The plate was again covered and was spun at 1000 rpm for 30 seconds. The plate was allowed to incubate overnight at RT and in a closed carrier to reduce dehydration. The seal was removed, and the fluorescence was read with a plate reader with an excitation wavelength of 665 nm and an emission wavelength of 615 nm. BPGM Phosphatase Assay The following materials were used in this assay: Tris buffer (50 mM Tris, 0.01% Tween 20, pH 7.4); cyclohexylammonium salt of 2,3-BPG (stock solution of 500 mM in ultrapure distilled water; human Bisphosphoglycerate mutase (hBPGM; stock solution of 110 μM in Standard Buffer); 3-Phosphoglyceric acid (3-PG; stock solution of 100 mM in ultrapure distilled water); 384-Well Polystyrene Plates (Fisherbrand , Catalog No.: 12-566-625); BIOMOL GREEN REAGENT (Enzo life sciences, BML-AK111-0250); and 1 mM DMSO stock of exemplary compounds. A 3-PG enzyme mix solution (final enzyme concentration 62.5 nM, 3-PG concentration at 50 M, 14.5 uL) was added to onto a 384-well clear plate. Solutions of the test compounds were then added to the plate and the DMSO content was normalized across the plate. The plate was incubated at RT for 15 min. Next, the substrate 2,3-BPG (5 μL) was added to all the wells, and the plate was sealed and incubated for 24 hr at RT. After incubation, BIOMOL green dye (40 μL) was added to all the wells, the plate was shaken to mix, and the plate was incubated at RT for 20-30 min. Absorbance measurements were recorded at 620 nm. CA Inhibition Assay An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity. Phenol red (at a concentration of 0.2 mM) has been used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10-100 s. The CO2 concentrations ranged from 1.7 to 17 mM for thedetermination of the kinetic parameters and inhibition constants19. For each inhibitor at least six traces of the initial 5-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutionsup to 0.01 nM were done thereafter with distilled deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min-72 h at room temperature(15 min) or 4 °C (all other incubation times) prior to assay, in order to allow for the formation of the E-I complex or for the eventual active site mediated hydrolysis of the inhibitor. CA Inhibition Assay An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity. Phenol red (at a concentration of 0.2mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction [Khalifah et al., J. Biol. Chem., 246:2561-73] for a period of 10-100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled deionized water and dilutions up to 0.01nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex (GTX3 was incubated also for longer periods, of 1-24 h, but no differences of activity have been detected). CA Inhibition Assay An applied photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activity. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20mMHepes (pH 7.5) as buffer, and 20mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10-100 s. The CO2 concentrations ranged from 1.7 to 17mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1mM) were prepared in distilled-deionized water and dilutions up to 0.01nM were done thereafter with distilled deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min to 72 h at room temperature (15 min) or 4 °C (all other incubation times) prior to assay, in order to allow for the formation of the E-I complex or for the eventual active site mediated hydrolysis of the inhibitor. CA Inhibition Assay Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Tris-HCl (pH 7.5) as buffer, 0.1 M Na2SO4 (for maintaining constant the ionic strength), at 25°C, following the CA-catalyzed CO2-hydration reaction for a period of 10-80 s (the uncatalyzedreaction needs around 60-100 s in the assay conditions, whereas the catalyzed ones are of around 6-10 s). The CO2 concentrations ranged from 1.7 to 17 mM for thedetermination of kinetic parameters. For each inhibitor, tested in the concentration range between 0.01 and 100 μM, at least six traces of the initial 5-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (1 mM) were prepared in distilled-deionized water with 10-20% (v/v) dimethyl sulfoxide (which is not inhibitory at these concentrations), and dilutions up to 0.001 μM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay in order to allow for the formation of the E-I complex. Carbonic Anhydrase I, II, IV and XII Assay An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity [Khalifah et al., J. Biol. Chem., 246:2561-2573]. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10-20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10-100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionised water and dilutions up to 0.01 nM were done thereafter with distilled-deionised water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. In Vitro Inhibition Assay Method The in vitro inhibition assay method was adapted from Svenson and Jaffrey, 2016. All reactions were performed in a 96-well plate with 200 μL assay buffer (50 mM HEPES pH 6, 300 μM 2-oxoglutarate, 300 μM (NH4)2Fe(SO4)2 6H2O, 2 mM ascorbic acid in RNase-free water) with 7.5 μm6A7-Broccoli RNA and 0.250 μM FTO. Inhibitors were added in concentrations ranging from 0.008-40 μM; all inhibitors were dissolved in DMSO and added to a final concentration of 0.2% DMSO. Prior to incubation, 40 μL read buffer (250 mM HEPES pH 9.0, 1 M KCl, 40 mM MgCl2, 2.2 μM DFHBI-1T in RNase-free water) was added to bring the final well volume to 200 μL. After incubation at room temperature for 2 hours, the plates were left at 4 C. overnight (16 hours) to allow DFHBI-1T to bind to A7-Broccoli RNA. Specificity assays were performed by the same method with 0.250 μM ALKBH5. Fluorescence intensity was measured with a BioTek Synergy plate reader with FITC filters (excitation 485 nm, emission 510 nm). Sigmoidal dose-response curves were fitted in GraphPad Prism 6. Measurement of Human SPR Inhibitory Activity Assay Human SPR inhibitory activity was measured by using a 384-well low adsorption clear plate (Greiner) with buffer D containing 100 mM Tris-HCl (pH 7.5) (Thermo Fisher Scientific) and 0.01% bovine serum albumin (Sigma). A compound was diluted with DMSO to a concentration of 1 mM, and a 1 μL aliquot of the preparation was diluted with 250 μL of buffer D. The thus-diluted compound was dispensed into the 384-well plate at L/well. Thereafter, human SPR was diluted with buffer D to 200 ng/mL and then dispensed into the 384-well plate at 20 μL/well. Subsequently, 40 μL of ultrapure water containing 120 μM Sepiapterin (WuXi AppTec) and 120 μM NADPH (Nacalai) was added to each well, and mixed with a vortex mixer, to thereby initiate the reaction. The reaction mixture was incubated at room temperature for 240 minutes, and the reaction was terminated by addition of 10 μL of ultrapure water containing 2% formic acid (Nacalai). In order to determine the amount of sepiapterin after termination of the reaction, the absorbance at 420 nm was measured by applying the 384-well plate to EnSpire multimode plate reader (Perkin Elmer). In Vitro Kinase Assay In vitro kinase assays described below can be used to determine the activity of a test compound for inhibiting the activity of JAK1 kinase. The test compounds were dissolved in dimethyl sulfoxide and diluted with water to a serial concentration gradient as required in the experiment. JAK1 substrates (Cell Signaling Technology, Catalog Number: 1305s) and ATP (2 mM) solution were diluted with water to obtain a final concentration of 20 uM ATP and 1.2 uM substrate solution. The appropriate amount of JAK1 kinase (Invitrogen, Catalog Number: pv4774) was mixed with 4x buffer (prepared by user, and comprising 50 mM HEPES, pH 7.3, 125 mM NaCl, 24 mM MgCl2, 1.25 mM DTT) to a final concentration of 8 ng/uL. To each well of a microplate [DELFIA Streptavidin-coated clear plate (Perkin Elmer, Catalog Number: AAAND-0005)]17.5 uL of ATP/substrate mixture, 5 uL of an aqueous solution of a test compound (5 uL of pure water only were added to the control and blank), and 7.5 uL of the kinase solution prepared above (4x buffer only was added to the control) were added. Each well was mixed sufficiently, then incubated at room temperature (27° C.) for 50 minutes, washed with wash buffer, and dried three times, then HRP conjugated antibody was added [Phospho-Tyrosine Mouse mAb (P-Tyr-100) (HRP Conjugate, Cell Signaling Technology, Catalog Number: 5465)], and incubated for 1 hour. The microplate was washed with wash buffer and dried three times, and then TMB (Sigma, Catalog Number: T4444) was added, and incubated for 5 to 15 minutes to allow for color change. Stop solution (1 N sulfuric acid solution) was added to stop the reaction. Absorbance was measured on a Novostar microplate reader at a wavelength of 450 nm. IC50 values of test compounds was calculated from the data of the test compounds in inhibiting the activity of JAK1 kinase at different concentrations. In Vitro Kinase Assay In vitro kinase assays described below can be used to determine the activity of a test compound for inhibiting the activity of JAK2 kinase. The test compounds were dissolved in dimethyl sulfoxide and diluted with water to a serial concentration gradient as required in the experiment. JAK2 substrates (Cell Signaling Technology, Catalog Number: 1305s) and ATP (2 mM) solution were diluted with water to obtain a final concentration of 20 uM ATP and 1.2 uM substrate solution. The appropriate amount of JAK2 kinase (Invitrogen, Catalog Number: pv4210) was mixed with 4x buffer (prepared by user, and comprising 50 mM HEPES, pH 7.3, 125 mM NaCl, 24 mM MgCl2, 1.25 mM DTT) to a final concentration of 8 ng/uL. To each well of a microplate [DELFIA Streptavidin-coated clear plate (Perkin Elmer, Catalog Number: AAAND-0005)]17.5 uL of ATP/substrate mixture, 5 uL of aqueous solution of a test compound (5 uL of pure water only were added to control and blank), and 7.5 uL of the kinase solution prepared above (4x buffer only was added to control) were added. Each well was mixed sufficiently, then incubated at room temperature (27° C.) for 50 minutes, washed with wash buffer, and dried three times, then HRP conjugated antibody [Phospho-Tyrosine Mouse mAb (P-Tyr-100) (HRP Conjugate, Cell signaling Technology, Catalog Number: 5465)] was added, and incubated for 1 hour. The microplate was washed with wash buffer and dried three times, and then TMB (Sigma, Catalog Number: T4444) was added and incubated for 5 to 15 minutes to allow for color change. Stop solution (1 N sulfuric acid solution) was added to stop the reaction. Absorbance was measured on a Novostar microplate reader at a wavelength of 450 nm. IC50 values of test compounds were calculated from the data of the test compounds for inhibiting the activity of JAK2 kinase at different concentrations. In Vitro Kinase Assay In vitro kinase assays described below can be used to determine the activity of a test compound for inhibiting the activity of JAK3 kinase. The test compounds were dissolved in dimethyl sulfoxide and diluted with water to a serial concentration gradient as required by the experiment. JAK3 substrates (Cell Signaling Technology, Catalog Number: 1305s) and ATP (2 mM) solution were diluted with water to obtain a final concentration of 20 M ATP and 1.2 M substrate solution. The appropriate amount of JAK3 kinase (Invitrogen, Catalog Number: pv3 855) was mixed with 4x buffer (prepared by user, and comprising 50 mM HEPES, pH 7.3, 125 mM NaCl, 24 mM MgCl2, 1.25 mM DTT) to a final concentration of 8 ng/uL. To each well of a microplate [DELFIA Streptavidin-coated clear plate (Perkin Elmer, Item: AAAND-0005)]17.5 uL of ATP/substrate mixture, 5 uL of aqueous solution of a test compound (L of pure water only were added to the control and blank), and 7.5 uL of the kinase solution prepared above (4x buffer only was added to the control) were added. Each well was mixed sufficiently, then incubated at room temperature (27° C.) for 50 minutes, washed with wash buffer, and dried three times, then HRP conjugated antibody [Phospho-Tyrosine Mouse mAb (P-Tyr-100) (HRP Conjugate, Cell signaling Technology, Catalog Number: 5465)] was added, and incubated for 1 hour. The microplate was washed with wash buffer and dried three times, and then TMB (Sigma, Catalog Number: T4444) was added and incubated for 5 to 15 minutes to allow for color change. Stop solution (1 N sulfuric acid solution) was added to stop the reaction. Absorbance was measured on a Novostar microplate reader at a wavelength of 450 nm. IC50 values of test compounds were calculated from the data of the test compounds for inhibiting the activity of JAK3 kinase at different concentrations. In vitro kinase assay JAK2: In vitro kinase assays described below can be used to determine the activity of a test compound for inhibiting the activity of JAK2 kinase. The test compounds were dissolved in dimethyl sulfoxide and diluted with water to a serial concentration gradient as required in the experiment. JAK2 substrates (Cell Signaling Technology, Catalog Number: 1305s) and ATP (2 mM) solution were diluted with water to obtain a final concentration of 20 μM ATP and 1.2 μM substrate solution. The appropriate amount of JAK2 kinase (Invitrogen, Catalog Number: pv4210) was mixed with 4× buffer (prepared by user, and comprising 50 mM HEPES, pH 7.3, 125 mM NaCl, 24 mM MgCl2, 1.25 mM DTT) to a final concentration of 8 ng/μL. To each well of a microplate [DELFIA Streptavidin-coated clear plate (Perkin Elmer, Catalog Number: AAAND-0005)] 17.5 μL of ATP/substrate mixture, 5 μL of aqueous solution of a test compound (5 μL of pure water only were added to control and blank), and 7.5 μL of the kinase solution prepared above (4× buffer only was added to control) were added. Each well was mixed sufficiently, then incubated at room temperature (27° C.) for 50 minutes, washed with wash buffer, and dried three times, then HRP conjugated antibody [Phospho-Tyrosine Mouse mAb (P-Tyr-100) (HRP Conjugate, Cell signaling Technology, Catalog Number: 5465)] was added, and incubated for 1 hour. The microplate was washed with wash buffer and dried three times, and then TMB (Sigma, Catalog Number: T4444) was added and incubated for 5 to 15 minutes to allow for color change. Stop solution (1 N sulfuric acid solution) was added to stop the reaction. Absorbance was measured on a Novostar microplate reader at a wavelength of 450 nm. IC50 values of test compounds were calculated from the data of the test compounds for inhibiting the activity of JAK2 kinase at different concentrations. In vitro kinase assay JAK3: In vitro kinase assays described below can be used to determine the activity of a test compound for inhibiting the activity of JAK3 kinase. The test compounds were dissolved in dimethyl sulfoxide and diluted with water to a serial concentration gradient as required by the experiment. JAK3 substrates (Cell Signaling Technology, Catalog Number: 1305s) and ATP (2 mM) solution were diluted with water to obtain a final concentration of 20 μM ATP and 1.2 μM substrate solution. The appropriate amount of JAK3 kinase (Invitrogen, Catalog Number: pv3855) was mixed with 4× buffer (prepared by user, and comprising 50 mM HEPES, pH 7.3, 125 mM NaCl, 24 mM MgCl2, 1.25 mM DTT) to a final concentration of 8 ng/μL. To each well of a microplate [DELFIA Streptavidin-coated clear plate (Perkin Elmer, Item: AAAND-0005)] 17.5 μL of ATP/substrate mixture, 5 μL of aqueous solution of a test compound (μL of pure water only were added to the control and blank), and 7.5 μL of the kinase solution prepared above (4× buffer only was added to the control) were added. Each well was mixed sufficiently, then incubated at room temperature (27° C.) for 50 minutes, washed with wash buffer, and dried three times, then HRP conjugated antibody [Phospho-Tyrosine Mouse mAb (P-Tyr-100) (HRP Conjugate, Cell signaling Technology, Catalog Number: 5465)] was added, and incubated for 1 hour. The microplate was washed with wash buffer and dried three times, and then TMB (Sigma, Catalog Number: T4444) was added and incubated for 5 to 15 minutes to allow for color change. Stop solution (1 N sulfuric acid solution) was added to stop the reaction. Absorbance was measured on a Novostar microplate reader at a wavelength of 450 nm. IC50 values of test compounds were calculated from the data of the test compounds for inhibiting the activity of JAK3 kinase at different concentrations. CYP450 Enzyme Inhibition Test Experimental Method: 4-hydroxydiclofenac (the substrate for CYP450 and 2C9 enzymes) and different doses of compounds were added into human liver microsome (Xenotech, LLC), then NADPH (Reduce dcoenzyme II, Chem-impex international, Inc.) was added, mixed, and then incubated in 37° C. water bath, at the terminal time, the stop solution (200 ng/mL tolbutamide and 200 ng/mL labetalol dissolved in acetonitrile) was added to stop the reaction, methanol or ethanol was used to precipitate proteins, the concentration of the metabolites of substrates was determined by using LC-MS/MS to obtain the IC50 values of compounds on CYP450 and 2C9 enzyme. Enzyme Inhibition Assay The enzyme reaction was started by the addition of enzyme to the reaction mixture containing substrate and test compounds. The reaction was stopped by spotting an aliquot onto a silica gel plate that had been prespotted with thymine and thymidine. The plate was developed in ethyl acetate-water-formic acid (60:35:5), and the spots were visualized under UV light (254 nm) and cut out for radioactivity determination in toluene-based scintillation cocktail. Kinetic constants Km and Ki were determined from the Lineweaver-Burk and Dixon plots. Data based on results from at least four independent experiments were evaluated by the nonlinear regression method (GOSA, Bio-Log). Receptor Binding Assay A prepared WP was homogenated and a membrane fraction was collected with high-speed centrifugation. A compound of the present invention was added to the plate and [3H]-PGD2 was also added. A platelet membrane, a protein concentration is 2 mg/mL, was added and mixed in the plate, and placed on ice for 2 hours. The reaction solution was transferred to a low protein-adsorptive filter and washed with a wash solution eight times using a cell harvester. After the final washing, water was removed sufficiently, and scintillator was added. DP inhibitory activity was investigated by measuring [3H] by using Micro Beta. ITK Enzyme Assay ITK activity was determined by measuring the effect of a test compound in an ITK enzyme assay. 1.0 M HEPES Buffer pH 7.5 solution was prepared as follows: 238.3 g HEPES free acid (Sigma) and 800 mL of water were combined, and the mixture was stirred until complete dissolution. The pH was adjusted to 7.5 via titration with 5N NaOH and the volume adjusted to 1000 mL. The solution was filtered and sterilized. ITK assay buffer was prepared as follows: 50 mL of HPLC-grade water was treated with 2 mL of 1.0 M HEPES Buffer, 500 μL of 2% Gelatin (Sigma), 1.0 mL of aqueous MgCl2 solution (1.0 M), and 1.0 mL of aqueous glutathione solution (0.5 M), and the solution was mixed. The solution was brought to 99 mL in a graduated cylinder by addition of water and sterilized through a 0.2 μm filter. 0.1 mL of Brij-35™ Surfact-Amps Detergent Solution (10% w/v aqueous solution, ThermoFisher) and 1.0 mL of ATP (Teknova, 100 mM) were added and the solution was mixed. Preparation of 1.33×ITK enzyme solution was as follows: 49.99 mL of ITK assay buffer was treated with 4.1 μL of ITK enzyme (ITK FL (N-Flag and C-His tagged, 72 kDa) Lake Pharma, 0.25 mg/ml in a buffer containing 25 mM Tris pH 7.8, 150 mM NaCl, 10% glycerol and 2 mM TCEP) and the mixture was gently agitated. The resulting solution was stored on ice. 30 Minutes prior to use, the enzyme solution was removed from ice and equilibrated to RT by incubation in a RT water bath. Preparation of 4×ITK substrate solution was as follows: 50 mL of ITK assay buffer was treated with 100 μL of BTK peptide (China Peptide Company, 2 mM stock solution in DMSO). The tube was capped, mixed by gently inverting the tube, and then stored on ice. 30 Minutes prior to use, the substrate solution was removed from ice and equilibrated to RT by incubation in a RT water bath. At the time of assay, 7.5 μL of the 1.33×ITK enzyme solution was added to plate wells containing 0.1 μL of varying concentrations of test compound in DMSO. The plate was incubated 30 min at RT. The plate wells were each treated with 2.5 uL of the 4×ITK substrate solution and the plate was sealed (TopSeal, Perkin Elmer). The plate was spun at 1000 rpm for 30 sec and then incubated for 60 min at RT. The seal was removed, and each well was treated with 10 μL of Stop/Detect Buffer (20 mM HEPES pH 7.5, 0.01% gelatin, 1 nM LANCE PT66 (Perkin Elmer), 16.5 μg/ml Surelight APC (Perkin Elmer), 10 mM EDTA, 250 mM NaCl). The plate was again covered and was spun at 1000 rpm for 30 seconds. The plate was allowed to incubate overnight at RT and in a closed carrier to reduce dehydration. The seal was removed, and the fluorescence was read with a plate reader with an excitation wavelength of 665 nm and an emission wavelength of 615 nm. The concentrations and resulting effect values for the tested compound were plotted and the concentration of compound required for 50% effect (IC50) was determined with the four-parameter logistic dose response equation. Fluorescence Based Assay The fluorescence based assay to monitor the activity of human nSMase2 in the presence or absence of potential inhibitors has been described recently. Figuera-Losada, et al. Lysate of cells expressing recombinant nSMase2 is used to catalyze the hydrolysis of sphingomyelin (SM) to ceramide and phosphorylcholine. Phosphorylcholine undergoes dephosphorylation in a reaction catalyzed by alkaline phosphatase (4 U/mL) to produce choline which in turn is oxidized by choline oxidase (0.1 U/mL) to betaine and hydrogen peroxide (H2O2). Hydrogen peroxide is made to react with Amplex red (50 μM) in the presence of horseradish peroxidase (HRP, 1 U/mL) to generate the fluorescent molecule resorufin. Generation of fluorescence is monitored by measuring relative florescence units (RFU) with excitation at 530 nm and emission at 590 nm. Extent of fluorescence is directly proportional to the extent of SM hydrolysis. Substrate stock solution is prepared in 2% Triton X-100 and sonicated for 1 min. Reactions are carried out for 1 h at 37° C. in 100 mM Tris-HCl pH 7.4, 10 mM MgCl2, 0.2% Triton X-100. This assay has been optimized in 384-well format (50 μL total volume per well) under conditions where nSMase2-catalyzed hydrolysis of SM is linear with respect to nSMase2 concentration, time of incubation and SM concentration (FIGS. 3A-3C). The assay has high reliability (Z′=0.8-0.9). It is used for compound screening, IC50 determinations and mode of inhibition studies AChE and BChE Inhibition Assay In vitro anti-AChE activity was performed based on the modified Ellman's method [Ellman et al., Biochem. Pharmacol., 71:88-95]. For this purpose, synthesized compounds 6 were dissolve in a mixture of DMSO (5 mL) and methanol (5 mL) and diluted in 0.1 M KH2PO4/K2HPO4 buffer (pH 8.0). Each well contained 50 μL potassium phosphate buffer (KH2PO4/K2HPO4, 0.1 M, pH 8), 25 μL prepared sample as described above, 25 μL enzyme with final concentration of 0.22 U/mL in buffer. They were preincubated for 15 min at room temperature, and then 125 L DTNB (3 mM in buffer) was added. Characterization of the hydrolysis ofATCI catalyzed by AChE was performed spectrometrically at 405 nm followed by the addition of substrate (ATCI 3 mM in water). The change in absorbance was measured at 405 nm after 15 min. The IC50 values were determined graphically from inhibition curves (log inhibitor concentration vs. percent of inhibition). A control experiment was performed under the same conditions without inhibitor and the blank contained buffer, water, DTNB, and substrate. The described method was also used for BChE inhibition assay. For all compounds, four different concentrations were tested for each compound in triplicate to obtain the range of 20%−80% inhibition for AChE. Inhibition Assay Cytochrome P450 3A4 and 2D6:Recombinant enzymes, 3A4 and 2D6, generated using the ABL yeast expression system were used. For CYP3A4, the enzyme amount was 2 pmol per well, and the substrate (7-benzyloxy-4-(trifluoromethyl) coumarin; BFC) was used at 15 μM. The assay mixture also included K2HPO4 (pH 7.4; conc. 0.1 M) and NADPH (conc. 1 mM). The incubation time was 30 minutes, and the reference inhibitor was A-naphthoflavone. For CYP2D6, the enzyme amount was 2 pmol per well, and the substrate (7-methoxy-4-(aminomethyl)-coumarin; MAMC) was used at 20 μM. The assay mixture also included K2HPO4 (pH 7.4; conc. 0.1 M) and NADPH (conc. 0.06 mM). The incubation time was 35 minutes, and the reference inhibitor was quinidine.Briefly, 0.6 μL of serially diluted compound was added to 75 μL water. 10 μL of diluted compound in water was then added to each assay plate. 20 μL of a K2HPO4, enzyme, and substrate mixture was added. After 10 minutes of room temperature pre-incubation, 10 μL of NADPH was added, and the plates were placed at 37° C. for 30 or 35 minutes depending on the enzyme being tested. After the incubation was complete, 20 μL of 0.1% Tris/ACN was added to the plate. The plate was read on a FluorStar fluorescence plate reader. Inhibitory Activity Assay Assay 1:The assay was performed in the presence of OptiMEM (supernatant collected over 24 h and cleared from cellular debris by centrifugation) containing the ectodomain of BACE1, 25 μl water containing the desired 2-fold concentration of test compound and 2% DMSO, 1 μM substrate peptide, 20 mM NaOAc, pH 4.4, and 0.04% Triton-X100 in a total assay volume of 50 μl in a 384 well plate. In general, 25 μl of compound dilution were given to the plate followed by the addition of 10 μl of BACE1 containing OptiMEM diluted 1:10 in water with 0.2% Triton X-100. The reaction was started with the addition of 15 μl substrate in NaOAc buffer. The reaction was incubated at rt (dark) in an Envision multilabel reader (Perkin Elmer) and the cleavage of the substrate was recorded as kinetic for 60 min at ex: 485 nm, em: 538 nm. Blank wells containing no enzyme were included on each plate.The intensity of fluorescence was regressed against time in order to derive velocities of reaction in all 384 wells. These velocities were used for calculating percent control using an uninhibited control containing 1% DMSO as 100% and blank control performed in the absence of enzyme as 0%. IC50 values were calculated by fitting percent control vs. test compound concentration using Assay Explorer. 5-HT2A Receptor Binding Assay The binding affinities of disclosed compounds at the ketanserin binding site of the 5-HT2A receptor were determined in radioligand binding experiments, with the results summarized in Table 1. Disclosed compounds exhibited substantial binding affinity for the 5-HT2A receptor. The affinity of Compound 1 was much higher than the other pyridine isomer Compound 11 and the pyrazine Compound 12, indicating the preferred positioning of the pyridine nitrogen in the compounds of the invention. Generally, compounds with an ethyl substituent alpha to the basic amine were less potent than compounds bearing a hydrogen or methyl substituent at this position. Longer alkyl or fluoroalkyl substituents at position 5 of the pyridine also tended to increase potency compared to smaller substituents at this position. Inhibition Assay Assay buffers consist of 20 mM citric acid, 60 mM disodium hydrogen orthophosphate, 1 mM EDTA, 0.1% CHAPS, 4 mM DTT, pH 5.8 for legumain, 50 mM dihydrogen sodium orthophosphate, 1 mM EDTA, 5 mM DTT, pH 6.25 for cathepsin B and cathepsin Land 100 mM Tris, 0.1% CHAPS, 10% sucrose, 10 mM DTT, pH 7.4 for caspase-3. Concentrations of substrates during the measurement were 10 nM (legumain, cathepsin Land caspase-3) and 50 nM (cathepsin B) and concentration of enzymes were 100 nM for cathepsin Land caspase-3, 270 nM for legumain and 360 nM for cathepsin B. Each enzyme was incubated with inhibitor concentrations ranging from 1 nM to 1 mM in the presence of the substrates. Inhibition Assay The degree of inhibition activity for PTP1B was investigated by using 2 mM p-nitrophenyl phosphate (p-NPP) as a substrate to measure the dephosphorylation degree. First, PTP1B diluted with distilled water was reacted with 2 mM p-nitrophenyl phosphate {p-NPP, 0.1 M NaCl, 1 mM EDTA, 50 mM citrate pH 6.0, and 1 mM dithiothreitol (DTT)} and Compound 1 at various concentrations at 30° C. for 30 minutes, and then the reaction was terminated with a 1 N-sodium hydroxide (NaOH) solution. The absorbance of the samples thus prepared was measured to confirm the inhibition degree (IC50: The half maximal inhibitory concentration) for PTP1B activity according to the concentration of Compound 1. Mitogenesis inhibition method To measure beta2-AR mediated inhibition of mitogenesis, HEK-beta2-AR, 1321N1 or U87MG cells were seeded in a 96-well plate at approximately 5,000 cells/well. After 48 hours, the wells were rinsed twice and the medium was replaced with fresh medium containing 10 uL of drug in sterile water. After another 24 hours of incubation at 37° C., 0.25 uCi of [3H]-thymidine was added to each well. The cells were incubated for an additional 2 hours at 37° C., at which point 10 uL of 10x trypsin was added, and the resuspended cells were harvested using a Tomtec 96 harvester through glass fiber filters. Biochemical Assay Buffer pH7.4: Prepare 1 (M) KH2PO4 and 1 (M) K2HPO4. Titrate 1(M) K2HPO4 with 1 (M) KH2PO4 to obtain pH 7.40. Dilute this buffer 10 fold in Water (30 ml buffer+270 ml of water) to obtain 100 mM phosphate buffer. Adjust pH to 7.40±0.02 using 5(N) HCl or 5(N) NaOH. NADPH Regeneration System (NRS): Prepare a solution containing 13 mM NADP, 33 mM Glucose-6-phosphate, 33 mM MgCl2and 4 U/ml Glucose-6-phosphate dehydrogenase in buffer. Liver Microsome (LM) suspension: Thaw LM vial on ice, then mix 1.0 ml LM (20 mg/ml) with 19 ml buffer [final LM Conc: 1 mg/ml] LM+NRS suspension: Mix 5.0 ml NRS with 20 ml LM suspension [final LM Conc: 0.8 mg/ml] System suitability standard: a synthesized compound having Mol wt 686.2 used as System suitability standard. Dissolve this compound in ice-cold acetonitrile to obtain concentration of 0.1 μg/ml and store at 4° C. Compound Dilution: Compound Stock: 10 mM in DMSO Sub stock (100 μM): 4 μl of 10 mM Compound Stock+398 μl Acetonitrile. Working plate (2 μM): 10 μl of 100 μM Sub stock+490 μl buffer Assay Procedure Incubate all plastic materials including tips at 37° C. overnight. Incubate LM suspension and NRS at 37° C. for 15 min before use. Add 40 μl buffer to the wells of blank plate. Add 40 μl compound (from working plate) to 0, 5, 10, 20, 30 and 60 min plates. Initiate reaction by adding 40 μl of LM+NRS suspension in each plate. Terminate reaction by adding 240 μl ice-cold acetonitrile containing system suitability standard at designated time points. For T=0 add 240 μl ice-cold acetonitrile containing system suitability standard before LM+NRS addition. Centrifuge (3500 rpm, 20 min and 15° C.) the plates. Mix 110 μl supernatant with 110 μl water and quantitate amount of Compound in the solution using LC-MS/MS. CA Inhibition Assay An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561-2573.Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10-20 mM Hepes (pH 7.5, for α-CAs) or TRIS (pH 8.3 for β-CAs) as buffers, and 20 mM Na2SO4 (for α-CAs) or 20 mM NaBF4 for β-CAs (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10-100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5%-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. CA Inhibition Assay An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity [Khalifah et al., J. Biol. Chem., 246:2561-2573]. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10-20 mM Hepes (pH 7.5, for α-CAs) or TRIS (pH 8.3 for β-CAs) as buffers, and 20 mM Na2SO4 (for α-CAs) or 20 mM NaCl− for β-CAs (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10-100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by nonlinear least-square methods using PRISM 3, whereas the kinetic parameters for the uninhibited enzymes from Lineweaver-Burk plots, as reported earlier. CA Inhibition Assay An Applied Photophysics stopped-flow instrument has been used for assaying the CA-catalyzed CO2 hydration activity [Khalifah et al., J. Biol. Chem., 246:2561-2573]. Phenol red (at a concentration of 0.2 mM)has been used as indicator, working at the absorbance maximum of 557 nm with 10-20 mM Hepes (pH 7.5, for α-CAs) or TRIS (pH 8.3 for β-CAs) as buffers, and20 mM Na2SO4 (for α-CAs) or 20 mM NaCl for β-CAs (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10-100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled deionized water and dilutions up to 0.01 nM were done thereafter with distilled-deionized water. Inhibitor andenzyme solutions were preincubated together for 15 min at room temperature prior to assay in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, whereas the kinetic parameters for the uninhibited enzymes from Lineweaver-Burk plots,as reported earlier. Enzymatic Assay The test compounds are dissolved in 100% DMSO at a concentration of 10 mM and in a first step diluted in DMSO to a concentration of 5 mM, followed by serial dilution steps in 100% DMSO. Dilution factor and number of dilution steps may vary according to needs. Typically 8 different concentrations by 1:5 dilutions are prepared, a further intermediate dilutions of the substances is carried out with assay buffer resulting in 1% final DMSO concentration in the assay.0.1 nM of FLAG-tagged Vanin-1 (AA 22-493, T26I, produced internally) and test compounds are incubated at room temperature for 20 minutes in assay buffer (1 mM DTT, 0.0025% Brij-35, 50 mM HEPES, pH7.5). D-Pantethine (Sigma, Cat #P2125-5G) in assay buffer is added (final concentration 3 μM) and incubated for additional 30 minutes at room temperature. Total assay volume typically is 40 μl but might be adjusted according to needs. Reaction is stopped by adding equal volume of stop solution as the reaction mixture to reach 100 nM HD-pantothenic acid (as an internal standard) and 1% TFA. Assay plates are centrifuged for 2 minutes and the formation of pantothenic acid is detected by RapidFire Mass Spectrometry (mobile phase A: 0.1% formic acid and 0.01% trifluoroacetic acid in water; mobile phase B: 47.5% acetonitrile, 47.5% methanol, 0.1% formic acid and 0.01% trifluoroacetic acid in water) using a C18, 12 μL cartridge (Agilent Cat #G9205A). Kinase Inhibition Assay (Inhibition of Enzymatic Axl Kinase Activity) Compounds of Formula (I) as obtained above were initially diluted to 10 nM in 100% DMSO (CALBIOCHEM™) for storage and made into kinase buffer solution to create a compound concentration ranging from 1 uM and 10 uM. Serial dilutions of the compounds were dispensed into a 96-well plate (GREINER BIOSCIENCES™) at 6 μL each. Truncated human Axl (CARNA BIOSCIENCES™) were diluted in kinase buffer and added to the compound solutions and pre-incubated for 30 minutes at room temperature. Next, ATP (TEKNOVA™) and substrate solution (suggested manufacture substrates of PERKINELMER™, for example, ULIGHT™-TK peptide) for Axl (PERKINELMER™) was added (12 μL each) to the wells containing the compound solution and enzyme. The reaction mixture was incubated for 1 hour. Following the incubation, the stop solution made with EDTA, water, and Lance detection buffer (PERKINELMER™) were added (12 μL each) to stop phosphorylation. Following the addition of the stop solution and 5 minutes of shaking, the detection solution containing the Europium-labeled antibody (suggested manufacture substrates of PERKINELMER™, for example, PT66 for Axl), water, and Lance detection buffer were added (12 μL) to the reaction mixture and incubated again for 50 minutes. Substrate phosphorylation was a function of the 665 nm emission measured following the addition of the detection solution and 50 minutes of incubation. ROS Inhibiting Drug A high-throughput screen was performed to find molecules that inhibit ROS production by neutrophils. Extracted human neutrophils were purified and kept in culture. The cells were then exposed to various drugs and ROS production was monitored over time. Compounds that also scavenged hydrogen peroxide (H2O2) and/or lowered neutrophil ATP levels (reflecting toxicity) were removed. The top hits from the screen were selected for further analysis.160 basal hits were tested for their ability to inhibit neutrophil ROS production. 64 molecules were able to inhibit ROS production in the presence of PMA activation. 67 molecules were able to inhibit ROS production in the presence of N-Formylmethionine-leucyl-phenylalanine (fMLP). Of those 47 molecules were able to inhibit ROS production by both stimulation methods. Binding Assay NK1: The following radioligand: [3H] substance P (PerkinElmer Cat#NET111520) was used in this assay. Binding assays were performed in a 50 mM Tris/5 mM MnCl2/150 mM NaCl/0.1% BSA at pH 7.4. Binding assays consisted of 25 μl of membrane suspension (approximately 5 μg of protein/well in a 96 well plate), 50 μl of compound or reference ligand (Substance P) at increasing concentrations (diluted in assay buffer) and 2 nM [3H] substance P. The plate was incubated 60 min at 25° C. in a water bath and then filtered over GF/C filters (Perkin Elmer, 6005174, presoaked in 0.5% PEI for 2 h at room temperature) with a Filtration unit (Perkin Elmer). DPP III Enzyme Activity Assay The inhibitory activity of flavonoids toward human DPP III was assayed in a 50 mM Tris-HCl buffer, pH 7.4. In brief, recombinant human DPP III (0.29 nM) was preincubated with increasing concentrations of flavonoid first for 1 min at 25°C and then for 3 min at 37°C. The enzymatic reaction was started with Arg-Arg-NA (40 μM) as a substrate, and after the 15 min incubation at 37°C in a water bath, the reaction was stopped and the absorbance was measured using the spectrophotometric method described above[Abramić et al., Biol. Chem. Hoppe-Seyler., 369:29]. The stock solutions of flavonoids were prepared daily in dimethyl sulfoxide (DMSO, Sigma-Aldrich). Determination of the value of binding affinity constant (KO between the compound and BRD4 BD2 protein The purity of BRD4 BD2 protein used in the experiment was greater than 95%, and the protein concentration was 46.33 uM. The 96-well plate was purchased from Corning (black, #3694). The multifunctional microplate reader was a product of TECAN, model: SPARK 10M. Buffer: 100 mM potassium phosphate (pH 6.5), 2% ethylene glycol (Sigma) and 0.01% Trition X-100 (Sigma). The experimental water was Millipore-Q pure water.The Ki value of the compound and BRD4 BD2 protein was measured according to the FP test procedures for detecting the Ki value of the compound and BRD4 BD1 protein except that the BRD4 BD1 protein was replaced with the BRD4 BD2 protein. In Vitro HPLC Assay This assay was used to determine the inhibiting effect of the inhibitors in a concentration of 1 mM. A standard incubation mixture consisted of 75 μl of 50 mM potassium phosphate buffer pH 7.4 and contained NMMA in a concentration of 200 μM. The inhibitors were used in final concentrations of 1 mM. By adding 2 μg of hDDAH-1, the reaction was started and the samples were incubated for 30 minutes at 37° C. in, a shaking water bath. The reaction was then stopped by adding 75 μl of acetonitrile, the samples were shaken for 5′ minutes and centrifuged (12,000 g, 5 min). The supernatant was added to the HPLC. Inhibition Assay The prepared substances were tested for TAFIa inhibition using the Actichrome plasma TAFI activity kit from American Diagnostica (Pr. No. 874). This entailed adding 29 μl of assay buffer (20 mM Hepes, 150 mM NaCl, pH 7.4) and 10 μl of TAFIa (American Diagnostica Pr. No. 874TAFIA; 2.5/ml) to 1 μl of 5 mM DMSO solution of the substance and incubating in a 96 half-well microtiter plate at room temperature for 15 minutes. The enzymic reaction was started by adding 10 μl of TAFIa developer (prediluted 1:2 with water). The time course of the reaction was followed at 420 nm in a microtiter plate reader (SpectraMax plus 384; Molecular Devices) for 15 minutes. Kinase-Glo Assay These assays are set up in duplicate 50 ul volumes in white, flat bottom 96 well plates. Inhibitors are added to the solution of 1× kinase buffer, 10 uM ATP, 100 uM Pim-1-specific substrate, 50 ng of active Pim-1 enzyme, and water in serial dilutions ranging from micromolar to nanomolar concentrations. This solution is incubated at 30 degrees Celsius at 360 rpm for two hours. Following the incubation, 50 ul of Kinase-Glo reagent is added to each well, including all positive and negative control wells, and incubated at room temperature for 15 minutes. The plate is then read by the Luminoskan Ascent instrument and the results displayed with the Ascent Software version 2.6. Inhibitory Activity Assay Two-fold dilution of each test compound solution (10 μM, 100% dimethyl sulfoxide) is carried out on 96-well V bottom plate (Costar 3363). After ten-fold dilution of each the test compound solution (100% dimethyl sulfoxide) with deionized distilled water, 10 μL of the diluted each mpound solution (10% dimethyl sulfoxide) was aliquoted to a black flat bottom 96-well plate (Costar 3915). 50 μL of 1.6× Assay solution (224 mM NaCl, 80 mM Tris-HCl (pH 8.0), 8 mM KCl, 1.6 mM CaCl2, 1.6 mM MgCl2, and 1.6 mg/mL fatty acid free BSA) was added thereto, and subsequently 20 μL of 20 nM human ENPP2 solution (buffer solution: 140 mM NaCl, 50 mM Tris-HCl (pH 8.0), 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 1 mg/mL fatty acid free BSA) and 20 μL of 5 μM FS-3 solution (buffer solution:deionized distilled water) were added thereto, respectively, followed by mixing. Under reaction for 30 minutes at 37° C., fluorescence intensity measurement (Ex: 485 mm, Em: 528 mm) was carried out at every 5 minutes by Envision Xcite Multilabel Reader. ΔCFU30 min value (CFU value measured at 30 minutes−CFU value measured at 0 minute) is obtained for each test solution and based on the equation of 100−(ΔCFU30 min of test solution/Mean value of ΔCFU30 min of control group)×100, the inhibitory activity percentage ratio (i.e., % inhibition) is obtained Inhibitory Activity on Human ENPP2 Two-fold dilution of each test compound solution (10 μM, 100% dimethyl sulfoxide) is carried out on 96-well V bottom plate (Costar 3363). After ten-fold dilution of each the test compound solution (100% dimethyl sulfoxide) with deionized distilled water, 10 μL of the diluted each compound solution (10% dimethyl sulfoxide) was aliquoted to a black flat bottom 96-well plate (Costar 3915). 50 μL of 1.6× Assay solution (224 mM NaCl, 80 mM Tris-HCl (pH 8.0), 8 mM KCl, 1.6 mM CaCl2, 1.6 mM MgCl2, and 1.6 mg/mL fatty acid free BSA) was added thereto, and subsequently 20 μL of 20 nM human ENPP2 solution (buffer solution: 140 mM NaCl, 50 mM Tris-HCl (pH 8.0), 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 1 mg/mL fatty acid free BSA) and 20 μL of 5 μM FS-3 solution (buffer solution: deionized distilled water) were added thereto, respectively, followed by mixing. Under reaction for 30 minutes at 37° C., fluorescence intensity measurement (Ex: 485 mm, Em: 528 mm) was carried out at every 5 minutes by Envision Xcite Multilabel Reader. ΔCFU30 min value (CFU value measured at 30 minutes−CFU value measured at 0 minute) is obtained for each test solution and based on the equation of 100−(ΔCFU30 min of test solution/Mean value of ΔCFU30 min of control group)×100, the inhibitory activity percentage ratio (i.e., % inhibition) is obtained. Biochemical Assays for Wild-Type IDH1 and IDH2 Enzymes IDH1 and IDH2 enzymes catalyze the conversion of isocitrate to αKG. Wild-type IDH1 (National Center for Biotechnology Information, Accession: NP_001269316.1) and IDH2 (National Center for Biotechnology Information, Accession: EAX02082.1) proteins containing N-terminal His-tag are expressed in E. coli and purified using nickel affinity chromatography. The enzyme assays are carried out in V-bottom 96 well polypropylene plates containing 100 mM Tris-HCl buffer at pH 7.5, 1 mM DTT, 0.005% TRITON X-100, 120 mM NaCl. For the IDH1 wild-type assay isocitrate, NADP+ and MnCl2 are included at the concentrations of 85 μM, 50 μuM and 20 μM respectively. For the IDH2 wild-type assay isocitrate, NADP+ and MnCl2 are included at the concentrations of 30 μM, 50 μM and 10 μM respectively. Inhibitors dissolved in a DMSO stock solution are diluted in the reaction mixture at a final DMSO concentration of 4%. The enzyme assay is terminated (quenched) by adding ACN (50:50) containing d6-2-ketopentanedioic acid (d6-αKG) as an internal standard for mass spectrometry analysis. Ten microliters of reaction mixture is combined with 100 μL of water, 50 μL of 1 M O-benzylhydroxylamine in pyridine buffer (8.6% pyridine, pH 5), and 50 μL of 1 M N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC) in pyridine buffer. Following derivatization at room temperature for one hour, samples are extracted with 600 μL of EtOAc. Four hundred μL of the upper layer is removed, dried under heated nitrogen and reconstituted with 100 μL of MeOH/water (1:1). Ten μL of derivatized sample is injected onto an LC-MS system consisting of a Shimadzu Prominence 20A HPLC system and a The Thermo Quantum Ultra triple quadrupole mass spectrometer. Analytes are separated on a Waters XBridge C18 column (2.1×50 mm, 3.5 μm) with a flow rate of 0.6 mL/minute. Mobile phase A is 0.1% formic acid in water and mobile phase B is MeOH. The αKG signal detected is transformed into analyte concentration using a calibration curve generated using known αKG concentrations. For each compound tested, the % inhibition is calculated using a DMSO control sample as 0% inhibition and a no enzyme control as 100% inhibition. Inhibition Assay A study on inhibition of protein kinases activity at in vitro biochemical levelMaterial and method: kinases such as c-Met, VEGFR-2, Axl and RET, obtained from Invitrogen; HTRF KinEASE; TK kit (Cisbio Company); 384-well plate (Greiner Company); ATP (Sigma Company), MgCl2 (Sigma Company); PHERAstar FS multifunctional plate reader (BMG Company); conventional centrifuge (StaiteXiangyi Company); incubator (Binder Company). Dilution and storage of compounds: depending on solubility, the test compounds are prepared as stock solutions of 0.5-10 mmol/L with dimethyl sulphoxide (DMSO), and aliquots of the stock are stored at −20° C.;Preparation of working solution of the compounds: Before test, aliquots of the compounds are removed from the refrigerator, and diluted with pure DMSO to 50× of the level needed; and then the compounds are diluted with deionized water to 4× of the level needed;Preparation of 1.33× enzymatic buffer: 5× enzymatic buffer (from HTRF kit) is diluted to 1.33× with deionized water, and appropriate components of the final level 1.33× are added: 1.33 mmol/L dithiothreitol (DTT) and 1.33 mmol/L MgCl2;Preparation of kinase working solution: Met is diluted to 2× final level required, 0.2 ng/μL, with 1.33× enzymatic buffer;Preparation of substrate working solution: the substrate labeled with biotin (from HTRF kit) and ATP (10 mM) are diluted with 1.33× enzymatic buffer to prepare a mixture at 4× of the final level required;Preparation of the testing working solution: Streptavidin-XL665, 16.67 μmol/L is diluted with HTRF test buffer to 4× of the final level required, and then mixed with antibody-Cryptate of the same volume (both from the HTRF kit).Enzymatic reaction step: 4 μl of kinase working solution is added to each well of the low volume 384-well plate. Meanwhile, 4 μL 1.33× enzymatic buffer is added as the negative control. 2 μL compound working solution is added into each well, and 2 μL 8% DMSO in water is added as zero compound level control (i.e., positive control). At 25° C. (or 30 C), incubate for 5-10 min; add 2 μL substrate working solution into each well to initiate the enzymatic reaction. Allow to react while shaking at 25° C. (or 30° C.) for 15-60 min. ABTS Free-Radical Cation Scavenging Assay The ABTS free radical cation scavenging ability of the synthe- sized compounds was determined according to the procedure described earlier [Free Radical Biol. Med. 26(9-10):1231-1237]. ABTS was dissolved in distilled water (7x10^3 M) and potassium persulphate (2.45x10^3 M) was added. This reaction mixture was left overnight (12-16 h) in the dark, at room temperature. Various concentrations of test substances (1000 µg/mL 500 µg/mL, 250 µg/mL, 100 µg/mL, 50 µg/mL, 25 µg/mL, 10 µg/mL concentrations) were incubated with the ABTS+P solution for 30min. The absorbance was measured at 734 nm, and the % inhibition was calculated using the formula described in Eq. (1) and the IC50 was calculated. Ascorbic acid was used as the positive control. Binding Assay NK2: Binding assays were performed in a 25 mM HEPES/1 mM CaCl2/5 mM MgCl2/0.5% BSA/10 μg/ml saponin, at pH 7.4. Binding assays consisted of 25 μl of membrane suspension (approximately 3.75 μg of protein/well in a 96 well plate), 50 μl of compound or reference ligand (Neurokinin A) at increasing concentrations (diluted in assay buffer) and 0.1 nM [125I]-Neurokinin A. The plate was incubated 60 min at 25° C. in a water bath and then filtered over GF/C filters (Perkin Elmer, 6005174, presoaked in assay buffer without saponine for 2 h at room temperature) with a Filtration unit (Perkin Elmer). The radioactivity retained on the filters was measured by using the TopCount-NXT reader (Packard). Biological Assay Human liver Cathepsin L (Sigma) was preincubated with test compounds at various concentrations for 5 minutes at 25C. The assay was initiated by addition of substrate Z-Phe-Arg-aminomethylcoumarin (Z-F-R-AMC Bacchem) and the final assay conditions were 1 nM cathepsin L, 50 uM Z-F-R-AMC, 100 mM sodium acetate pH 5.5, 1 mM EDTA (Omnipure), 3 mM DTT (EMD), 0.01% BRIJ 35 (Sigma), and 2.0% DMSO (Acros). Test compounds were serially diluted with DMSO and water to include a final concentration range of 10 uM to 10 pM. The reaction was monitored fluorometrically for 5 minutes at 25C. using black 96-well Corning 3686 assay microplates with a Thermo Fluoroskan Ascent FL microplate reader at excitation and emission filter wavelengths of 355 nm and 460 nm, respectively. DXR Inhibition Assay H-DXR was pre-incubated during 2 min in the presence of the inhibitors at different concentrations and DXP (480 µM). NADPH (160 µM final concentration) was then added to measure the residual activity. The flavonoids are poorly soluble in water. Flavonoids solutions at different concentrations were prepared in DMSO by serial dilutions from 100 mM stock solutions. Aliquots of the solutions were added in the assays so that the concentration of DMSO did not exceed 0.4% (v/v). The inhibition mechanism of myricetin was studied by preincubating H-DXR with DXP at varied concentrations (96 480 µM) and fixed concentrations of myricetin (0.5, 1 and 2 µM). The enzymatic reaction was initiated by NADPH (final concentration160 µM). Fosmidomycin was used as a positive reference. Enzyme Assay Primary stock solutions of test compounds at a concentration of 6 mM were prepared from the 2 to 5 mg powder. The primary stock solutions of each test compound were prepared freshly in distilled water on the day of study to obtain a final concentration of 6 mM. For determination of IC50 values, 12 test compound concent rations were prepared as 3-fold serial dilutions. Concentration range of test compound utilized for nNOS were 0.001 to 300 μM and for eNOS were 0.003 to 10 00 μM. The vehicle of the test compound or inhibitor was used as blank control. For non-specific activity, 100 μM L-NMMA was used. Runs using the IC50 concentration of L-NAME were done in parallel as controls. All incubations are performed in duplicate. Fluorescence-Based CA II Binding Assay The assay was performed according to a previously reported protocol.[Banerjee et al., Biochemistry, 44:3673-3682] DNSA stock solution was prepared at 1 mm in DMSO/water (1:1). CA II (1 mm) was added to the test probe (over a 0.1-100 mm concentration range) in HEPES buffer [HEPES (25 mm) with acetonitrile (10 %), pH 7.0] and mixed well. DNSA (0.1 mm) was added, and the resulting solution was incubated at RT for 10 min and then transferred into a black-wall 96-well plate (final volume 100 mL/well). The fluorescence was measured with a Spectra Max 250 plate reader (Molecular Devices, Sunnyvale, CA, USA) at an excitation of 285 nm and emission of 465 nm at 25 °C. In Vitro Assay Chinese hamster ovary (CHO) cells expressing the human orexin-1 receptor and the human orexin-2 receptor, respectively, are grown in culture medium (Ham F-12 with L-Glutamine) containing 300 μg/ml G418, 100 U/ml penicillin, 100 μg/ml streptomycin and 10% heat inactivated fetal calf serum (FCS). The cells are seeded at 20′000 cells/well into 384-well black clear bottom sterile plates (Greiner). The seeded plates are incubated overnight at 37° C. in 5% CO2.Human orexin-A as an agonist is prepared as 1 mM stock solution in MeOH: water (1:1), diluted in HBSS containing 0.1% bovine serum albumin (BSA), NaHCO3: 0.375 g/l and 20 mM HEPES for use in the assay at a final concentration of 3 nM. In Vitro Protein Kinase Assay NSC 109555 was diluted in water. All other drugs were dissolved in DMSO, in which case the final DMSO concentration in reactions was 10%, and the controls were performed under comparable conditions. Recombinant Chk2 was incubated with either histone H1 (or GST-Cdc25C) in reaction buffer, and incubated at 30 deg C for indicated times. For the drug inhibition experiments, samples were coincubated with drug during the reactions. Reactions were stopped by adding sample buffer, and samples were boiled for 5 min. Reaction products were separated by 4-20% SDS-PAGE. Chk2 protein kinase activity, measured as 32P incorporation into Chk2, histone H1 (or GST-Cdc25C), was determined using a PhosphorImager (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). Densitometry was performed using Image-Quant. Ligand Binding Assay As Alt et al., 2002. Membranes (20 μg) are incubated in 50 mM Tris-HCl, pH 7.4 with [3H]diprenorphine or [3H]nociceptin in the absence or presence of varying concentrations of test compounds for 60 min in a shaking water bath at 25° C. Nonspecific binding is measured using 10 μM naloxone (MOR, DOR, KOR) or N/OFQ (NOP). Samples are filtered through GF/C glass-fiber filtermats mounted on a Brandel cell harvester and rinsed four times with 4° C. 50 mM Tris-HCl, pH 7.4 buffer. Filtermats are dried and 0.1 ml EcoLume scintillation cocktail added to each sample area to soak the filter. Each filtermat in a heat-sealed bag, is counted in a Wallac 1450 MicroBeta Liquid Scintillation and Luminescence Counter. Ligand Binding Assays As Alt et al., 2002. Membranes (20 ug) are incubated in 50 mM Tris-HCl, pH 7.4 with [3H]diprenorphine or [3H]nociceptin in the absence or presence of varying concentrations of test compounds for 60 min in a shaking water bath at 25° C. Nonspecific binding is measured using 10 uM naloxone (MOR, DOR, KOR) or N/OFQ (NOP). Samples are filtered through GF/C glass-fiber filtermats mounted on a Brandel cell harvester and rinsed four times with 4° C. 50 mM Tris-HCl, pH 7.4 buffer. Filtermats are dried and 0.1 ml EcoLume scintillation cocktail added to each sample area to soak the filter. Each filtermat in a heat-sealed bag, is counted in a Wallac 1450 MicroBeta Liquid Scintillation and Luminescence Counter. MAO Inhibition Assays The enzymatic reactions were carried out at pH 7.4 (K2HPO4/KH2PO4 100 mM, made isotonic with KCl) to a final volume of 500 µL. The reactions contained the different inhibitor concentrations spanning at least 3 orders of magnitude and the MAO-A/B mixed substrate kynuramine (45 µM for MAO-A and 30 µM for MAO-B). DMSO, as co-solvent (4%) was added to each reaction. The enzyme reactions were initiated with the addition of MAO-A or MAO-B (0.0075 mg protein/mL) and incubated for 20 min at 37 °C in a waterbath. After termination with the additionof 400 µL NaOH (2 N) and 1000 µL water, the concentrations of the MAO generated 4-hydroxyquinoline were measured by fluorescence spectrophotometry (excitation = 310 nM; emission = 400 nM). [35S]GTPγS Binding Assay Human CB2 expressing CHO-K1 membranes (5 μg per incubation mixture) were diluted in 50 mM Tris-HCl (pH 7.5) and 0.5 mM EDTA and added to HU compounds in a pre-mixed incubation cocktail. Final incubation concentrations were 55 mM Tris-HCl (pH 7.4), 1 mM EDTA, 100 mM NaCl, 5 mM MgCl2, 0.5% BSA, 50 μM GDP, 0.2 nM [35S]GTPγS (PerkinElmer) with varied HU compound concentration and 5 μg membrane. Incubations were continued for 60 minutes at 30° C. in a shaking water bath. Assays were terminated by addition of 2 ml ice cold wash buffer (50 mM Tris-HCl, pH 7.5 and 5 mM MgCl2) and filtration through pre-soaked GF/C filters (Whatman), followed by two further washes. [3H] cAMP SPA Assay The analysis of the activity of the enzyme Phosphodiesterase [3H] cAMP SPA (Perkin Elmer #7090 TRKQ) kit was used for the study of Phosphodiesterase 10. The kit contains:Yttrium scintillation proximity assay (SPA) in beads: yttrium silicate in microspheres resuspended pellet in MiliQ water containing 18 mM zinc sulfate. 10×PDE assay buffer: 500 mM Tris/HCl; 83 mM MgCl2; 17 mM EGTA; pH 7.5. Tracer [3H] cAMP: substrate for enzymatic reaction.The test is based on the preferential binding of linear nucleotides to the SPA beads in the presence of zinc sulfate, with respect to the binding of cyclic nucleotides, which is almost imperceptible. Therefore, in optimum conditions, the product of the enzymatic reaction binds directly to SPA, and the substrate of the enzyme does not. Biochemical Assays iFLiK and HTRF studies were carried out as described in Z. Fang, J. R. Simard, D. Plenker, H. D. Nguyen, T. Phan, P. Wolle, S. Baumeister, D. Rauh, ACS Chem. Biol. 2015, 10, 279-288.All reagents for HTRF experiments were purchased from Cisbio Bioassays, France. OriginPro 9.1G software (OriginLab Corporation, Northhampton, Mass.) was used for data analysis and data was fit to a sigmoidal dose-response model using the following four-parameter logistic equation:y = A 2 + ( A 2 - A 1 ) ( 1 + ( x IC 50 ) p ) ( 1 ) (A1: bottom asymptote; A2: top asymptote; IC50: half-maximal inhibitory concentration; p: Hill coefficient) Kinetic Characterization of Covalent Probe Compounds:Time-dependent IC50 measurements were performed with activated full-length Akt1 as described under Biochemical assays. Briefly, IC50 values were determined for twelve different incubation times and afterwards plotted versus accordingly. Data was analyzed according to literature procedure as described in B. F. Krippendorff, R. Neuhaus, P. Lienau, A. Reichel, W. Huisinga, J. Biomol. Screen. 2009, 14, 913-923. Ki and kinact were calculated with XLfit (Version 5.4.0.8, IDBS, Munich, Germany) defining the substrate concentration as 250 nM and the corresponding substrate KM as 150 nM.Mass Spectrometry:Purified full-length wtAkt1 was thawed under cold water and diluted to a final concentration of 1 mg/mL in storage buffer (50 mM HEPES, 200 mM NaCl, 10% Glycerol, pH 7.4). 20 μL of the respective mixture were mixed with 2 molar equivalents of the compounds of formulas (1a) and (2a), respectively (10 mM in DMSO); samples containing equal volumes of DMSO were individually prepared for control measurements. Following incubation for thirty minutes on ice, the samples were analyzed by ESI-MS using an Agilent 1100 Series HPLC System connected to a ThermoFinnigan LTQ Linear Ion Trap mass spectrometer. Therefore, 6 μL of sample were injected and separated using a Vydac 214TP C4 5 u column (150 mm×2.1 mm) starting at 20% of solvent B for five minutes followed by a gradient up to 90% of solvent B over 14 min (flow rate 210 μL/min) with 0.1% TFA in water as solvent A and 0.1% TFA in acetonitrile as solvent B. After washing the column for two minutes with 90% of solvent B, the concentration of solvent A was increased to 80% in 1 min and the column was washed for five additional minutes. During the complete experiment, a mass range of 700 to 2000 m/z was scanned and raw data was deconvoluted and analyzed with MagTran and mMass (Version 5.5.0) software.For ESI-MS/MS measurements, samples were denatured, separated via SDS-PAGE followed by staining with Coomassie Brilliant Blue and prepared according to standard tryptic in-gel digest protocols as described in A. Shevchenko, H. Tomas, J. Havlis, J. V. Olsen, M. Mann, Nat. Protoc. 2006, 1, 2856-2860. Subsequently, samples were thawed, dissolved in 20 μL of 0.1% TFA in water, sonicated at room temperature for 15 min, and centrifuged at 15000×g for 1 min shortly before analysis. 3 μL of sample were loaded onto a pre-column cartridge and desalted for 5 min using 0.1% TFA in water as eluent at a flow rate of 30 μL/min. The samples were back-flushed from the pre-column to the nano-HPLC column during the whole analysis. Elution was performed using a gradient starting at 5% B with a final composition of 30% B after 35 min (flow rate 300 nL/min) using 0.1% formic acid in water as eluent A and 0.1% formic acid in acetonitrile as eluent B and a column temperature of 40° C. The nano-HPLC column was washed by increasing the percentage of solvent B to 60% in 5 min and to 95% in additional 5 min, washing the columns for further 5 min, flushing back to starting conditions and equilibration of the system for 14 min. During the complete gradient cycle, a typical TOP10 shot-gun proteomics method for the MS and MS/MS analysis was used. Cytochrome P450 Inhibition Protocol 1: Studies were carried out in human liver microsomes. Human liver microsomes were purchased from BD Gentest. DMSO stocks were prepared for the test compounds. Aliquots of the DMSO solutions were diluted 1:3 by acetonitrile:ACN mixture (v/v: 40:60) to "400×" intermediate solutions, then further diluted by liver microsomes/buffer to "2×" intermediate solutions. "2×" intermediate solutions were mixed with "2×" NADPH/substrate solutions, which had been pre-warmed to 37° C. (final test compound concentrations were 10 μM, 3.3 μM, 1.1 μM, 0.37 μM, 0.122 μM, 0.041 μM, 0.0136 μM, and 0 μM). The plates were kept in a 37° C. water bath for the duration of the experiment. At the end of incubation (5 minutes for 3A4; 45 minutes for 2C19; 10 minutes for 1A2, 2C9, 2D6), 120 μL of acetonitrile was added into corresponding wells. After the final time point was sampled, the plates were shaken at a vibrator (IKA, MTS 2/4) for 10 mM (600 rpm/min) and then centrifuged at 5594 g for 15 mM (Thermo Multifuge×3R). Aliquots of the supernatant were removed, diluted 1:1 into distilled water, and analyzed by LC-MS/MS. The peak area response ratio to internal standard (PARR) of the compounds at 10 μM, 3.3 μM, 1.1 μM, 0.37 μM, 0.122 μM, 0.041 μM, and 0.0136 μM was compared to the PARR at 0 μM to determine the percent of metabolite generation from substrate at each test compound concentration. Fluorescence Polarization (FP) Assay Fluorescence polarization assay was performed on a Wallac Victor 3V multi-label counter/plate reader (PerkinElmer, Shelton, CT) using 484 nm excitation and 535 nm emission filters for the fluorophore used in the binding experiment. The plate used for the FP measurement was Corning 3575 384-well plate, loaded with 40 μL of assay solution per well. The buffer used in FP assays is 10 mM HEPES buffer, pH 7.4, containing 150 mM NaCl, 50 mM EDTA, and 0.005% Tween-20. Deionized water from a Millipore water purification used to prepare all aqueous solutions in FP assay. The fluorescent probe used in this assay is 9-mer Nrf2 ETGE motif derived peptide, FITC-LDEETGEFL-NH2. In each well, the final volume is 40 μL that consisted of 10 μL of 400 nM Keap1 Kelch domain protein, and 20 μL of 20 nM FITC-9mer Nrf2 peptide amide, and 10 μL of an inhibitor compound of different concentrations. The experiments were done in triplicates, with initial concentration of the inhibitor typically 5 μM and 50 μM. Then, the plate was centrifuged for 2 mM to ensure thorough mixing and get rid of any air bubbles in the solution. The plate was covered and shacked for 30 mM at room temperature and then centrifuged for 2 mM prior to FP measurements. The determination of FP is by measuring the parallel and perpendicular fluorescence intensity (F∥ and F⊥) with respect to the linearly polarized excitation light. The measurement of IC50 of the inhibitors was determined from the plot of % inhibition against concentration of the inhibitor analyzed by Sigma Plot 12.3 software. Inhibition of EGFR and HER2 Kinase Activity Assay The assay for the inhibition of EGFR and HER2 kinase activity by small molecular compounds was carried out using the method as follows:1) Dilution of the compoundsIn a 96-well plate a, the compounds were diluted with DMSO using a 3-fold gradient dilution to form 11 concentrations, the 12th concentration is pure DMSO (as a positive control); and in a new 96-well plate b the above solutions were diluted 25 times with ultrapure water (DMSO concentration is 4%).2) Transferring the compounds to 384-well plateThe compound solutions diluted with ultrapure water in the 96-well plate b above was transferred to the corresponding wells of a 384-well plate in duplicate.3) Addition of 4×kinase solution: 2.5 μl of the above 4× kinase solution was taken using multichannel pipette and added to the corresponding reaction wells of the 384-well plate, mixed well and pre-reacted at room temperature for 5 minutes.4) Addition of 2×substrate/ATP mixed solution: 5 μl of the above 2×substrate/ATP mixed solution was taken using multichannel pipette and added to the corresponding reaction wells of the 384-well plate.5) Negative control: negative control wells were set in the 384-well plate, and 2.5 μl 4×substrate, 2.5 μl 4×enzyme solution, 2.5 μl 1×Kinase Assay Buffer and 2.5 μl ultrapure water containing 4% DMSO were added to each well.6) Mixed by centrifugation and kept at room temperature for 2 hours in the dark.7) Termination of the enzymatic reaction:5 μl of the above 4× stop solution was pipetted to the corresponding wells of the 384-well plate, centrifuged and mixed, and reacted at room temperature for 5 minutes.8) Development reaction:5 μl of the above 4× detection solution was pipetted into the corresponding wells of the 384-well plate, centrifuged and mixed, and reacted at room temperature for 1 hour.9) The 384-well plate was placed into a microplate reader and the signal was detected using the corresponding program.10) IC50 analysis:Well reading value=10000*EU665 value/EU615 valueInhibition rate=(reading value of positive control well−reading value of experimental well)/(reading value of positive control well−reading value of negative control well)*100%Corresponding IC50s can be calculated by entering the drug concentrations and the corresponding inhibition rates into GraphPad Prism 5. Activation Assay The activity of sGC was evaluated by measuring the amount of a cyclic guanosine monophosphate (cGMP) which is produced by human purified sGC. A test compound was dissolved in DMSO and diluted 20-fold with ultrapure water. 2 μL of the diluted test compound solution (maximum concentration 100 μM), 2 μL of a substrate solution [0.5 μM TEBA, 0.03 μM dithiothreitol, 0.01 μM GTP, 0.04 μM MgCl2, and 0.03 μM sodium nitroprusside (SNP)], and 6 μL of a human enzyme suspension were added to 384-well plates (manufactured by Greiner Bio-One), and incubated at room temperature for one hour. The quantitative determination of cGMP is using HTRF which based on the competition between sample cGMP and fluorescent dye labeled cGMP for binding to a cGMP-specific antibody. Aldh1a2 Enzyme Inhibition Assay Recombinant protein extraction: pET-Aldh1a2 transformed BL21-DE3 cultures induced at 20° C. for 19 h with 0.3 mM IPTG rocking. Cultures were spun at 3500 g for 10 min, supernatants were poured off and allowed to drain fully. Cells were resuspended in 10 mM HEPES pH 7.4, 10 mM KCl. Cells were freeze-thawed in liquid nitrogen and then a 37° C. water bath for 10 cycles followed by ultrasonication at 50% amplitude, 3 sec on, 9 sec off for 10 cycles at 4° C. Cell extracts were spun at 16000×g for 5 minutes.Reaction performed at 20° C. in reaction buffer (10 mM HEPES pH 7.4, 10 mM KCl, 0.1 M Resazurin, 1 mg/mL BSA, 200 uM NAD+, diaphorase and aldehyde substrate). Recombinant enzyme and inhibitor added immediately before assay. Reaction rate measured by resorufin fluorescence. BChE Inhibition Assay BChE inhibitions by the carbamate inhibitors were assayed by the Ellman method [Ellman et al., Biochem. Pharm., 7:88-95]. BChE-catalyzed hydrolysis of BTCh in the presence of the carbamate inhibitors and DTNB was followed continuously at 410 nm on a UV-visible spectrometer. The temperature was maintained at 25.0°C by a refrigerated circulating water bath. All inhibition reactions were performed in sodium phosphate buffer (1 mL, 0.1 M, pH 7.0) containing NaCl (0.1 M), acetonitrile (2% by volume), triton X-100 (0.5% by weight), substrate (ATCh for AChE or BTCh for BChE) (50 μM), DTNB, and varying concentrations of inhibitor. Requisite volumes of stock solution of substrateand inhibitors in acetonitrile were injected into the reaction buffer via a pipet. BChE was dissolved in sodium phosphate buffer (0.1 M, pH 7.0). Biochemical ATX Assay 5 nM recombinant ATX (Cayman Chemicals) was supplemented to 50 mM Tris buffer (pH 8.0) containing 3 mM KCl, 1 mM CaCl2), 1 mM MgCl2 0.14 mM NaCl, and 0.1% bovine serum albumin. Test compounds were dissolved in DMSO and tested in the range of 0.1 nM to 10 μM. The enzymatic reaction (22.5 μL) was started by addition of 2.5 μL 10 μM 18:1 LPC (Avanti Lipids, Alabaster, AL, USA). After 2-h incubation at room temperature, the reaction was stopped by addition of 20 μL water containing 500 nM 20:4 LPA as internal standard and 100 μL 1-butanol for extracting LPA. Subsequently, the plates were centrifuged at 4000 rpm, 4° C., for 2 min. The resultant upper butanol phase was directly used for injection at a RapidFire system (Agilent). Biochemical ATX Assay 5 nM recombinant ATX (Cayman Chemicals) was supplemented to 50 mM Tris buffer (pH 8.0) containing 3 mM KCl, 1 mM CaCl2, 1 mM MgCl2 0.14 mM NaCl, and 0.1% bovine serum albumin. Test compounds were dissolved in DMSO and tested in the range of 0.1 nM to 10 μM. The enzymatic reaction (22.5 μL) was started by addition of 2.5 μL 10 μM 18:1 LPC (Avanti Lipids, Alabaster, Ala., USA). After 2-h incubation at room temperature, the reaction was stopped by addition of 20 μL water containing 500 nM 20:4 LPA as internal standard and 100 μL 1-butanol for extracting LPA. Subsequently, the plates were centrifuged at 4000 rpm, 4° C., for 2 min. The resultant upper butanol phase was directly used for injection at a RapidFire system (Agilent). Biochemical Assay The autoparsylation activity of the TNKS 1/2 or PARP1/2 enzymes was measured by the liquid chromatography-mass spectrometry (LC/MS) detection of nicotinamide as readout. Compound activity in inhibiting the TNKS and PARP autoparsylation was evaluated by IC50 measurements. In the compound screening assays, the reaction is composed of 5 uL of compound in beta-point serial dilutions with concentrations ranging from 0.0086 to 18.75 uM, 20 nM of purified enzyme, and 250 nM of beta-NAD+ in the 1x Assay Buffer. After 60 min incubation at room temperature, the reactions were quenched by the addition of 10 uL of 5x quenching solution (20% formic acid and 500 nM [d4]-nicotinamide in water). For the background control wells, 10 uL of the 5x quenching solution per well was added prior to the addition of beta-NAD+. Biological Assay CHO-K1 cells stably transfected with ROR t are maintained in MEM-EBS with 5% FBS, 1% penicillin streptomycin solution and 1 mg/mL G418. The cells are seeded at a density of 200000 cells/mL in white 96 well flat bottom plate. Post 16-18 hours incubation, the cells are transiently transfected with pFR Luc (50 ng/well DNA) for four hours. The cells are then treated with different doses of the test compounds in MEM EBS media with 10% FBS and 1% penicillin streptomycin. DMSO is used as vehicle control. After 18-20 hours treatment, the cells are lysed with lysis buffer (40 mM HEPES, 20 mM EGTA, 50 mM -glycerophosphate, 10% glycerol and 1% Triton X-100 in distilled water) for 0.5 hour and luminescence is read using Tecan Safire reader at 1000 milli second integration time. In Vitro AKT1 Kinase Assay Following addition of compound or control to the assay plate, 6p1 peptide mix containing 3 μM substrate (5-FAM-GRPRTSSFAEG-CONH2; CRB) and 40 μM ATP in Kinase base buffer (100 mM Hepes pH 7.5, 0.015% Brij-35) and 6p1 enzyme mix containing 8 nM AKT1/PKBα active enzyme (Upstate Biotechnology, Cat No. 14-276), 8 mM DTT and 20 mM MgCl2 in kinase base buffer was added. All buffers were made up with 18MΩ water. The plates were sealed and incubated at room temperature for 50 minutes. The reaction was stopped by the addition of 10 μl stop buffer (100 mM Hepes pH 7.5, 0.015% Brij-35 solution, 0.1% coating reagent #3, 40 mM EDTA, 5% DMSO) to each well (N.B. plates can be frozen after stopping and read later). In-Vitro Fluorescence Polarization Assay The fluorescence polarization assay tests the ability of compounds to inhibit the self-aggregation of α-synuclein peptide fragments. Peptides were incubated for 60 min at room temperature in the presence or absence of test compounds (compound concentrations were 33.3 to 0.3 μM). Samples were read on a BMG Pherastar plate reader in fluorescence polarization mode using excitation at 485 nm and emission at 520 nm. Data was analyzed using a four-parameter logistic fit (XLFit, IDBS Software). Peptide 4F (CTGFVKKDQLGK (SEQ ID NO: 1)) was prepared by American Peptide. Fresh peptide samples were reconstituted in purified water at 5 mM and diluted into 50 mM HEPES pH 7.4 with 50 mM NaCl to 100 nM final concentration. Solid compounds were dissolved in DMSO (10 mM), and then diluted in buffer. [3H]NMS/Carbachol Binding Assay In brief, binding reactions containing ~10 μg of membrane protein per tube were carried out for 2 h (22 °C) in 1 ml of binding buffer containing 25 mM sodium phosphate and 5 mM MgCl2 (pH 7.4). In saturation binding assays, we employed six different [3H]NMS concentrations (200 to 7,000 pM). In competition binding assays, we used a fixed concentration of [3H]NMS (500 pM) in the presence of 10 different concentrations of carbachol, the cold competitor. Nonspecific binding was defined as binding observed in the presence of 10 μM atropine. Binding reactions were terminated by rapid filtration over GF/C Brandel filters, followed by three washes (~4 ml/wash) with ice-cold distilled water. The amount of radioactivity that remained bound to the filters was determined by liquid scintillation spectrometry. BACE Assay For each compound being tested, the BACE activity was monitored in a fluorescence quenching assay (FRET) using the ectodomain of BACE (aa 1-454) fused to a myc-his tag and secreted from HEK293/BACEect. cells into OptiMEM (Invitrogen) as enzyme source and a substrate peptide derived from the APP-Swedish mutation which possesses a Cy3-fluorophore at the N-terminus and a Cy5Q-quencher at the C-terminus (Cy3-SEVNLDAEFK-Cy5Q-NH2; Amersham). The substrate was dissolved at 1 mg/mL in DMSO.The assay was performed in the presence of 5 μl OptiMEM (supernatant collected over 24 hours and cleared from cellular debris by centrifugation) containing the ectodomain of BACE, 25 μl water containing the desired concentration of test compound and 1% DMSO, 1 μM substrate peptide, 20 mM NaOAc, pH 4.4 and 0.04% Triton-X100 in a total assay volume of 50 μl in a 384 well plate. In general, 25 μl of compound dilution were given to the plate followed by the addition of 10 μl of BACE containing OptiMEM diluted 1:2 in water with 0.2% Triton X-100. The reaction was started with the addition of 15 μl substrate in NaOAc buffer. The reaction was incubated at 30° C. in a fluorimeter and the cleavage of the substrate was recorded as kinetic for 60 min. at ex: 530 nm, em: 590 nm. Blank wells containing either no inhibitor or no enzyme were included on each plate.The intensity of fluorescence was regressed against time in order to derive velocities of reaction in all 384 wells. These velocities were used for calculating percent inhibition using an uninhibited control containing 1% DMSO and a fully inhibited control incubations performed in the absence of enzyme. IC50 values were calculated by fitting percent inhibition vs. inhibitor concentration using standard software like GraphPadPrism. Biochemical Assay A reagent buffer was prepared in filtered and autoclaved water according to the following:50 mM Tris-buffer pH 7.5 (1 M Tris-buffer pH 7.5, Invitrogen, Cat. No. 15567-027);50 mM NaCl (5 M NaCl, Sodium Chloride Solution, Sigma, 59222C-);5 mM MgCl2 (1 M MgCl2, Sigma, M1028);0.1 mM ZnCl2 (Zinc Chloride [7646-85-7], powder, Cell Culture Tested, Sigma, Z-0152); and0.001% Tween 20 (TWEEN 20, Sigma Aldrich, P1379-).A buffer for the cGAS enzyme was prepared in filtered and autoclaved water according to the following:50 mM Tris-buffer pH 7.5;5 mM MgCl2; and0.001% Tween 20.Compounds were dispensed to a 386 well plate. The human truncated cGAS enzyme (4.2 mg/mL 147-522 human cGAS, MW 43,909 g/mol) was stored in 50 mM Tris, 500 mM NaCl, 5% (v/v) glycerol at pH 8 and diluted in the cGAS buffer enzyme shortly before use. The enzyme solution was transferred into the reagent buffer to give a final concentration of 30 nM. The reaction was started by mixing the enzyme with ISD (a 45 bp double stranded DNA, MW 27,670 g/mol, 5 mM), GTP and ATP to a final concentration of 5 μM, 0.5 mM and 0.5 mM respectively in a final volume of 10 μl. The reaction plates were then centrifuged at 1000 rpm for 1 minute and incubated at room temperature for 1 h. After 1 h of incubation, [5Ns]-2′3′-cGAMP to a final concentration of 200 nM and 30 μL of 100% acetonitrile/0.175% of TFA were added to the reaction mixture. The plates were centrifuged at 1000 rpm for 1 minute before being sealed for 3 seconds at 170° C. using a ThermoScientific sealer (ALPS™ 50V) and an aluminum sealing cover (Pierce Seal, 4titude, Product Code: 4TI-0531). Biochemical Assay Wild-Type IDH1: Enzymes catalyze the conversion of isocitrate to αKG. Wild-type IDH1 (National Center for Biotechnology Information, Accession: NP_001269316.1) and IDH2 (National Center for Biotechnology Information, Accession: EAX02082.1) proteins containing N-terminal His-tag are expressed in E. coli and purified using nickel affinity chromatography by methods commonly used and well known to those skilled in the art. The enzyme assays are carried out in V-bottom 96 well polypropylene plates containing 100 mM Tris-HCl buffer at pH 7.5, 1 mM DTT, 0.005% TRITON X-100, 120 mM NaCl. For the IDH1 wild-type assay isocitrate, NADP+ and MnCl2 are included at the concentrations of 85 μM, 50 μM and 20 μM respectively. For the IDH2 wild-type assay isocitrate, NADP+ and MnCl2 are included at the concentrations of 30 μM, 50 μM and 10 μM respectively. Inhibitors dissolved in a DMSO stock solution are diluted in the reaction mixture at a final DMSO concentration of 4%. The enzyme assay is terminated (quenched) by adding ACN (50:50) containing d (d6-αKG) as an internal standard for mass spectrometry analysis. Ten microliters of reaction mixture is combined with 100 μL of water, 50 μL of 1 M O-benzylhydroxylamine in pyridine buffer (8.6% pyridine, pH 5), and 50 μL of 1 M EDC in pyridine buffer. Following derivatization at room temperature for one hour, samples are extracted with EtOAc (600 μL). Four hundred L of the upper layer is removed, dried under heated nitrogen, and reconstituted with MeOH/water (1:1) (100 μL). Ten μL of derivatized sample is injected onto an LC-MS system consisting of a Shimadzu Prominence 20A HPLC system and a Thermo Quantum Ultra triple quadrupole mass spectrometer. Fluorescence Quench Assay The inhibitory activity of compounds was assessed by a fluorescence quench assay of BACE1 activity using commercially available substrate HiLyte Fluor 488-Glu-Val-Asn-Leu-Asp-Ala-Glu-Phe-Lys-(QXL 520)-OH (SEQ ID NO:1) AnaSpec, San Jose, Calif.) and truncated human beta-secretase, BACE1 (amino acids 1-454) fused to a myc-his tag and secreted from HEK293/BACEect. cells into OptiMEM (Invitrogen). The substrate was dissolved at 1 mg/ml in DMSO.The assay was performed in the presence of OptiMEM (supernatant collected over 24 h and cleared from cellular debris by centrifugation) containing the ectodomain of BACE1, 25 ul water containing the desired 2-fold concentration of test compound and 2% DMSO, 1 uM substrate peptide, 20 mM NaOAc, pH 4.4, and 0.04% Triton-X100 in a total assay volume of 50 ul in a 384 well plate. In general, 25 ul of compound dilution were given to the plate followed by the addition of 10 ul of BACE1 containing OptiMEM diluted 1:10 in water with 0.2% Triton X-100. The reaction was started with the addition of 15 ul substrate in NaOAc buffer. The reaction was incubated at rt (dark) in an Envision multilabel reader (Perkin Elmer) and the cleavage of the substrate was recorded as kinetic for 60 min at ex: 485 nm, em: 538 nm. Blank wells containing no enzyme were included on each plate. The intensity of fluorescence was regressed against time in order to derive velocities of reaction in all 384 wells. These velocities were used for calculating percent control using an uninhibited control containing 1% DMSO as 100% and blank control performed in the absence of enzyme as 0%. IC50 values were calculated by fitting percent control vs. test compound concentration using Assay Explorer. MAO-A/MAO-B Inhibition Assay Recombinant human MAO-A and MAO-B (5 mg/mL) were purchased from Sigma-Aldrich, pre-aliquoted and stored at -80 °C. Solutions of test compounds were prepared in DMSO in 2.5 mM for storage and diluted with potassium phosphate buffer (100 mM, pH 7.40, containing KCl 20.2 mM) before use. All the enzymatic reactions were conducted in potassium phosphate buffer (100 mM, pH 7.40, containing KCl 20.2 mM) to a final volume of 500 μL containing kynuramine (45 μM for MAO-A and 30 μM for MAO-B) and various concentrations of test compounds (0-100 μM) with the concentration of DMSO lower than 4%. The reactions were initiated by the addition of MAO-A or MAO-B (7.5 μg/mL) and the solutions were incubated at 37 °C for 30 min. The enzymatic reactions were terminated by the addition of 400 μL NaOH (2 N) and then 1000 μL water, centrifuged for 10 min at 16,000 g. The concentrations of the MAO generated 4-hydroxyquinoline in the reactions were determined by measuring the fluorescence of the supernatant on a Varioskan Flash Multimode Reader (Thermo Scientific) with excitation and emission wavelengths at 310 nm and 400 nm, respectively. A linear calibration curve was constructed by preparing samples containing 4-hydroxyquinoline (0.047-1.56 μM) dissolved in 500 μL potassium phosphate buffer. To each calibration standard, 400 μL NaOH (2 N) and 1000 μL water were added. The appropriate control samples were included to confirm that the test compounds do not fluoresce or quench the fluorescence of 4-hydroxyquinoline under the assay conditions. Time-Dependent Inhibition of Enzymatic Activity of Site of Metabolism of Midazolam in Human Liver Microsome CYP3A4 Table 7: A 100 mM PBS buffer was prepared. A 0.25 mg/mL microsome solution, a 7.5 mM MgCl2 and a 5 mM NADPH solution were prepared using the buffer. A 30 mM stock solution was diluted with DMSO to obtain 30 mM, 10 mM, 3 mM, 1 mM, 0.3 mM, 0.1 mM, 0.03 mM and 0 mM serial solutions I, which were further diluted 200-fold with phosphate buffer (PBS) to obtain serial test solutions II (150, 50, 15, 5, 1.5, 0.5, 0.15 and 0 μM). A midazolam working solution was diluted with PBS to 15 μM.The serial test solutions prepared above were well mixed by shaking and aliquoted in 20 μL portions into corresponding reaction plates (+NADPH, T0 and -NADPH groups were set). Three parallel tests were set. 40 μL of the liver microsome working solution was added to each 96-well plate. 20 μL of a corresponding substrate solution was added to the TO plate. 20 μL of NADPH was added to the TO plate and +NADPH group. A timer was started, and the plates were incubated in a water bath at 37° C. After 30 min of incubation, the TO plate was taken out, and the reaction was terminated with 250 μL of an internal standard-containing ACN solution. The +NADPH group was supplemented with 20 μL of the corresponding substrate solution, and the −NADPH group was supplemented with 20 μL of the corresponding substrate solution and 20 μL of NADPH. A timer was started, and the plates were incubated in a water bath at 37° C. After 30 min of incubation, the plates were taken out, and the reactions were terminated with 250 μL of an internal standard-containing ACN solution. cytomteric bead array (CBA) assay The Fix buffer I was warmed up to 37° C. in an incubator or water bath prior to use. The Perm Buffer III was chilled in a 20° C. freezer prior to use. The cells were collected at the end of treatment with testing compounds. One volume of the pre-warmed Fix Buffer I was mixed with one volume of cell suspension. If the volume of the cell suspension is greater than 100 uL, the cells were spun and resuspended in 100 uL medium or PBS. The buffer and the cell suspension were mixed well and incubated in a 37° C. water bath for 10 min. The cells were spun down at 250 g for 10 min and the supernatant was aspirated. The cells were washed once with BD Stain Buffer. The pellet was spun and the supernatant was removed. The cells were vortexed to be loosened, and permeabilized by slowly adding cold Perm Buffer III while vortexing or mixing. Subsequently, the cells were incubated on ice for 30 min. The cells were then spun down and washed twice with Stain Buffer. The supernatant was spun and aspirated. The cells were resuspended in a small volume of Stain buffer (50 or 100 uL containing from 200,000 to 1 million cells). Anti-IKFZ3 antibody was added to the cell suspension at 1:1000 dilution and incubated for 45 min at 4° C. The cells were then spun down and washed once with stain buffer. Secondary antibody was added to the cells at 1:5000 dilution and incubated at room temperature for 20 min in the dark. The cells were washed once with stain buffer prior to analysis by FACS. Biochemical Assay Affinity evaluation of the tested compounds and their selectivity with respect to the different PARP isoforms of interest was assessed in a displacement assay. The identification of compounds capable of binding several PARP proteins is carried out through a screening method including the steps of a) providing a reaction mixture containing: the PARP protein isoform under investigation, a compound of formula (IP): wherein R11 is hydrogen atom or a methyl group, B is (CH2)n NH group wherein n is 2 to 6; m is 0 or 1 and X- is a counterion, andserial dilutions of the test compound; b) comparing the polarization signal generated in the absence of the test compound with the one generated in the presence of different concentrations of the test compound, andc) evaluating the ability of the test compound to displace the compound of formula (IP) as defined above indicated from a decreased fluorescence polarization level. Biochemical Assay Affinity evaluation of the tested compounds and their selectivity with respect to the different PARP isoforms of interest was assessed in a displacement assay.The identification of compounds capable of binding several PARP proteins is carried out through a screening method including the steps ofa) providing a reaction mixture containing:the PARP protein isoform under investigation,a compound of formula (IP): wherein R10 is hydrogen or methyl, B is (CH2)nNH group, n is 2 to 6; m is 0 or 1 and X− is a counterion, and serial dilutions of the test compound;b) comparing the polarization signal generated in the absence of the test compound with the one generated in the presence of different concentrations of the test compound, andc) evaluating the ability of the test compound to displace the compound of formula (IP) as defined above indicated from a decreased fluorescence polarization level. The polarization signal can be measured, e.g., by a plate reader such as the Saphire2 (Tecan). Biological Assay All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≤2%. Biological Assay All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≦2%. Biological Assay SSAO: All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≤2%. Biological Assay SSAO: All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration≤2%. Biological Assays All primary assays were performed at RT. with purified recombinantly expressed human SSAO. Enzyme was prepared essentially as described in hman et al. (Protein Expression and Purification 46 (2006) 321-331). In addition, secondary- and selectivity assays were performed using SSAO prepared from various tissues or purified rat recombinant SSAO. The enzyme activity was assayed with benzylamine as substrate by measuring either benzaldehyde production, using 14C-labeled substrate, or by utilizing the production of hydrogen peroxide in a horseradish peroxidase (HRP) coupled reaction. Briefly, test compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM. Dose-response measurements were assayed by either creating 1:10 serial dilutions in DMSO to produce a 7 point curve or by making 1:3 serial dilutions in DMSO to produce 11 point curves. The top concentrations were adjusted depending on the potency of the compounds and subsequent dilution in reaction buffer yielded a final DMSO concentration ≤2%. Dundee MALDI-TOF Mass Spectrometry Assay (ICs) USP30 (25 ng/μl) tested against K48-linked diubiquitin (5.6 μM). USP30 was diluted in a buffer containing 40 mM Tris, 0.01% BSA, 1 mM DTT and K48 in 40 mM Tris, 0.01% BSA. The compounds were pre-incubated with the USP30 for 5 mins at room temp before the K48 dimer addition. The assay mixture was then incubated for 45 mins at room temp. The assay was stopped by the addition of TFA to a final concentration of 2% (v/v). Acidified samples of the DUB assays were mixed with 0.5 mM 15N-ubiquitin and then with one part of 2% (v/v) TFA and one part of 2.5 DHAP matrix solution (7.6 mg of 2.5 DHAP in 375 ml ethanol and 125 ml of an aqueous 12 mg ml 1 diammonium hydrogen citrate). Then 250 nl of these solutions were spotted onto an MTP AnchorChip 1,536 TF and this is analysed on the Bruker rapifleX MALDI-TOF. Inhibition Assay The DAAO inhibitory activity was measured by assaying the amount of hydrogen peroxide (H2O2) produced by reacting DAAO protein with flavin adenine dinucleotide (FAD) and D-alanine. The amount of H2O2 was determined by measuring the fluorescence generated on conversion of Amplex red (manufactured by Invitrogen Co.) into resorufin by the reaction of H2O2 with horseradish peroxidase (HRP). 4 uL of 4% dimethyl sulfoxide (DMSO) buffer (50 mM sodium phosphate (pH 7.5), 0.02% CHAPS) solution of the test compound was added to 384-well black, low volume plate, a mixed solution (4 uL) of recombinant human DAAO protein (15 nM), which had been expressed in Escherichia coli and purified, and 18 uM FAD was added, and the mixture was incubated at room temperature for 15 min. After incubation, a mixed buffer (4 uL) of 2.25 mM D-alanine, 1.5 U/mL HRP and 150 uM Amplex red was added, the mixture was incubated at room temperature for 30 min. Biochemical Assay Recombinant DHX9-Human recombinant DHX9 protein was custom ordered from Viva.RNA substrateOligos for double-stranded RNA substrate were custom ordered from IDT Technologies. (Oligol-GCCUGGUCCCUGUCCUUGUUAUUUUCCUUGGUUAAUU (SEQ ID NO:1). Oligo2-GAAUUAACCAAGGAAAAUAACAAGGACAGGGACCAGG (SEQ ID NO:2)). Each oligo was reconstituted in RNAse/DNAse-free water to achieve a 100 uM solution. The two oligo solutions were mixed in 1:1 ratio and annealed by heating at 70° C. for 5 minutes and cooling gradually to room temperature on the benchtop.Chemicals and Assay ComponentsAurintricarboxylic Acid and dimethylsulfoxide were purchased from Sigma-Aldrich (St. Louis, MO). White 384-well assay plates (Catalog #781075) were obtained from Greiner Bio-One (Frickenhausen, Germany). The ADP-Glo kinase assay kit from Promega Corporation is composed of ADP-Glo reagent, kinase detection reagent (made by mixing kinase detection buffer with a lyophilized kinase detection substrate), Ultra Pure ATP and ADP. Biochemical Assay The autoparsylation activity of the TNKS 1/2 or PARP1/2 enzymes was measured by the liquid chromatography-mass spectrometry (LC/MS) detection of nicotinamide as readout. Compound activity in inhibiting the TNKS and PARP autoparsylation was evaluated by IC50 measurements. In the compound screening assays, the reaction is composed of 5 μL of compound in 8-point serial dilutions with concentrations ranging from 0.0086 to 18.75 μM, 20 nM of purified enzyme, and 250 μM of β-NAD+ in the 1× Assay Buffer. After 60 min incubation at room temperature, the reactions were quenched by the addition of 10 μL of 5× quenching solution (20% formic acid and 500 nM [d]-nicotinamide in water). For the background control wells, 10 μL of the 5× quenching solution per well was added prior to the addition of β-NAD+. Chemiluminescent PARP Assay PARP assays are conducted using a chemiluminescent PARP assay kit (Trevigen). Briefly, reactions are performed in Histone-coated strip wells, by adding 10 microliters test compound dissolved in 1x PARP Buffer (prepared by mixing 20x PARP buffer diluted with high-purity water) and 15 microliters diluted PARP-HSA enzyme (diluted in 1x PARP buffer, 0.1 unit per well) to 25 microliters PARP cocktail (prepared from 10x PARP cocktail and 10x activated DNA, both 2.5 microliters per well and 20 microliters per well of 1x PARP buffer). The reactions are incubated at ambient temperature for 60 minutes, then the liquid was removed. After washing the wells four times with PBS (200 ul), 50 microliters of STREP-HRP (Horseradish Peroxidase) solution (diluted 500-fold in 1x Strep-Diluent) was added and the reactions were allowed to incubate for 30 minutes at ambient temperature. Enzyme Assay The aim of this assay is to determine the affinity of a test compound for the mPGES-1 enzyme. 47 ul of recombinant human mPGES-1 (0.5 ug protein/well) containing microsomal suspension in a buffer containing GSH, (2.5 mmol/L L-Glutathione reduced, dissolved in 0.1 mol/L Phosphat Buffer pH 7.4) is dispensed in a 384-well plate and thereafter 1 ul of the test compound(s) is/are added and incubated for 25 minutes at room temperature. The enzyme reaction is started by the addition of 2 ul PGH2 (final conc. 2 uM) dissolved in water-free Diglyme. After 60 seconds the reaction is terminated by addition of a stop solution containing FeCl2 (10 uL 0.074 mol/l FeCl2). The samples are diluted between 1:25 in PBS (Phosphate Buffered Saline). 10 ul of the diluted samples are transferred to 384-well low volume plate. Enzyme Assay The assay buffer is prepared to give a final concentration of 50 mM H2NaPO3 HNa2PO3, 0.01% bovine serum albumin and 0.01% Triton X-100 in water, at pH 7. Compounds to be tested are diluted in pure dimethyl sulfoxide (DMSO) using ten point concentration response curves. Maximal compound concentration in the reaction mixture is 30 or 1 μM. Compounds at the appropriate concentration are pre-incubated with OGA enzyme for 30 minutes before the reaction is started by the addition of substrate. The final enzyme concentration is 3.24 nM or 0.5 nM, for the 30 or 1 μM maximal compound concentration, respectively. Reactions are allowed to proceed for 60 minutes at room temperature. Then, without stopping the reaction, fluorescence is read. IC50 values are calculated by plotting the normalized data vs. log of the compound and fitting the data using a four parameter logistic equation. Human NR2B calcium influx assay The medium was removed and the cells were loaded with 200 μl loading buffer (Molecular Devices) in Mg2+-free HBPS containing 100 μM 7-CKA at 37° C. for one (1) hour. The test compounds were then solubilized in 100% DMSO and diluted to yield eight (8) different concentrations in 100% DMSO. A 96 well drug plate was prepared by diluting with water and glycine/glutamate to a 5-fold of final test concentration. Fluorescence intensity of the cells in the plate was measured in a FlexStation using an excitation wavelength of 485 nm and an emission wavelength of 525 nm. Twenty (20) seconds after starting the recordings the compounds together with the agonists glycine (100 μM) and glutamate (100 μM) were added into the wells and the fluorescence measured for ninety (90) seconds in summary. Inhibition Assay Various concentrations of the substances to be tested are dissolved in dimethyl sulphoxide and diluted with water. In white 96-well plates having a flat bottom, 20 μl of substance dilution are mixed with 20 μl of ecarin solution (ecarin reagent, from Sigma E-0504, final concentration 20 mU per batch) in Ca buffer (200 mM Hepes+560 mM sodium chloride+10 mM calcium chloride+0.4% PEG) or with 20 μl of Ca buffer (as unstimulated control). Furthermore, 20 μl of fluorogenic thrombin substrate (from Bachem I-1120, final concentration 50 μmol/l) and 20 μl of citrate plasma (from Octapharma) are added and homogenized thoroughly. The plate is measured in a SpectraFluorplus Reader using an excitation filter of 360 nm and an emission filter of 465 nm each minute over a period of 20 minutes. beta-Galactosidase Assay Enzymatic activity of beta-galactosidase from Escherichia coli (1.2 x 10^-10 M) was measured using the colorimetric substrate ortho-nitrophenyl-b-galactoside ([ONPG]: 2500, 2000, 1500, 1000, 500, 250, 125 uM) in Z-Buffer (100 mM sodium phosphate at pH = 7, 10 mM KCl, 1 mM MgSO4) to which was added fresh 50 mM beta-mercaptoethanol at 30 °C. In a 96-well plate were combined a 5x stock solution of Z-buffer (containing 250 mM beta-mercaptoethanol) and 5x stock solutions of ONPG. Samples were adjusted to a final volume of 180 uL with water. Samples were equilibrated at 30 °C for 5 minutes on a Synergy H1 microplate reader. A 10x stock solution of enzyme (20 uL) was then injected into each well and the production of 2-nitrophenol was quantified at 410 nm over a period of 10 minutes. Biological Enzymatic Assay Flag tagged Human recombinant MetAP2 expressed and isolated for use as the enzyme source. 10 mM stock solutions of compounds were prepared in 100% DMSO and further diluted in 100% DMSO required concentration to 1 mM stocks. The stock compound solutions and DMSO vehicle controls were diluted to target final compound concentrations using assay buffer to a final concentration of 50 mM HEPES containing 100 mM NaCl, pH adjusted to 7.5. The MAS peptide was formulated to a 7.5 mM stock in distilled water and prior to use further diluted 1:4. Amino acid oxidase was prepared as a stock solution (6.2 mg/ml) and prior to use further diluted 1:49.6 in distilled water. A 250 μM solution of MnCl2 was prepared in advance of thawing an aliquot of MetAP2 enzyme. 40 μl of enzyme was mixed with 100 μl of MnCl2 then further diluted in assay buffer to a final concentration of 16 μg/ml. To test for compound effect on MetAP2 enzyme activity, 5 μl of test compound, 10 μl of MAS substrate/amino acid oxidase mixture, 10 μl of MetAP2 was added to test wells in a 384 well black plate with blank wells containing no enzyme, replaced with 10 μl of assay buffer. All compounds were tested in duplicate on two occasions on the same day. The final in well concentrations of the assay were: 1% DMSO, 0.272 μg/ml MetAP2, 10 μM MnCl2, 50.0 μg/ml (0.225 U/ml) amino acid oxidase, and 0.75 mM MAS. The plate was sealed with a TopSeal A cover and mixed briefly on an orbital mixer at 900 rpm. The plate was incubated for a further 25 minutes at 25° C. A 5× stock of Amplex buffer was prepared (0.25M sodium phosphate, pH 7.4) and stored at 4° C. When preparing for use the stock was diluted with distilled water Amplex Ultraread stock solution was prepared at 2.57 mg/ml in 100% DMSO and stored in 50 μl aliquots at −20° C. 20 μl of 505 U/ml. Horse radish peroxidase was diluted in 990 ml of Amplex buffer, 100 μl of this was combined with 50 μl of Amplex Ultrared in 4850 ml of 1× Amplex buffer to generate sufficient detection reagent for a 384 well plate. 25 μl detection reagent was added to each well of the test plate, which was re-sealed and mixed briefly on an orbital shaker. The plate was transferred to an Envision Multi-label reader and RFU measured corresponding to excitation 531 nm and emission 595 nm At the end of the MetAP2 incubation 25 μl Amplex/HRP mixture per well was added and the plate read plate on a plate reader. Homogeneous Time Resolved Fluorescence (HTRF) Assay (1) Each compound to be tested was prepared using gradient dilution method with DMSO and water to obtain a solution with the concentration of 50 nM, 10 nM, 2 nM, 0.4 nM, and 0.08 nM. The concentration of DMSO in the solution of each compound to be tested was 2%.(2) PARP7 enzyme (Cell Chemical Biology 27, 877-887, Jul. 16, 2020; the fusion tags was N-His6-TEV-AviMHHHHHHSSGVDLGTENLYFQSNAGLNDIFEAQKIEWHE) was dissolved in the buffer solution (the pH of the buffer solution was 7.4, and the buffer solution contained 25 mM HEPES (N-(2-hydroxyethyl) piperazine-N′-2-sulfonic acid), 120 mM NaCl, 5 mM MgCl2, 2 mM DTT (Dithiothreitol), 0.002% (ml/ml) Tween-20, 0.1% (ml/ml) BSA (bovine serum albumin) and water) to obtain a PARP7 enzyme solution with the concentration of 6 nM.(3) The RBN011147 (Cell Chemical Biology 27, 877-887, Jul. 16, 2020), MAb Anti His-T b cryptate Gold (Cisbio, Cat. No 61GSTTLF, Lot. No 09A), and Streptavidin-d2 (Cisbio, Cat. No 610SADLF, Lot. No 19G) were diluted with buffer solution (the pH of the buffer solution was 7.4, and the buffer solution contained 25 mM HEPES (N-(2-hydroxyethyl) piperazine-N′-2-sulfonic acid), 120 mM NaCl, 5 mM MgCl2, 2 mM DTT (Dithiothreitol), 0.002% (ml/ml) Tween-20, 0.1% (ml/ml) BSA (bovine serum albumin) and water) to obtain the solution containing fluorophore with the concentration of 10 nM, 0.7 nM, and 2.5 nM respectively. The MAb Anti His-Tb cryptate Gold was the donor fluorophore, and the Streptavidin-d2 was the acceptor fluorophore.(4) 2.5 μl of the solution of the compound to be tested was transferred into 384-well plate, 2.5 μl of the PARP7 enzyme solution was added. The resulting solution was incubated for 15 mins, and then 5 μl of the solution containing fluorophore was added. The resulting mixture was incubated at 25° C. for 3 hrs to obtain the final solution to be tested.(5) The fluorescence signal was read on SPARK plate reader (Tecan), the wavelength of the excitation spectrum of the SPARK plate reader was 320 nm, and the wavelength of the emission spectrum of the SPARK plate reader was 620 nm and 665 nm. The ratio of absorbance at 620 nm to absorbance at 665 nm was calculated for the solution in each well. The ratio was calculated according to the following formula: Ratio=absorbance at 665 nm/absorbance at 620 nm×104.(6) The activation of the compounds to be tested was calculated according to the following formula: Activation (%)=100×(ratiocompound−rationegative)/(ratiopositive−rationegative). Inhibition (%)=100−Activation (%). β-1,4-Galactosyltransferase I Assay β4GalT activity was assayed using UDP-Gal as glycosyl donor and (6-esculetinyl) β-D-glucopyranoside (esculine) as glycosyl acceptor. Assays were performed in a total volume of 200 μL. The reaction mixtures contained reagents in the following final concentrations: 50 mM Hepes buffer (pH 5.4), 10 mM MnCl2, 2.0 mg/mL BSA, 200 μM esculine, 40 μM UDP-Gal, 10 μL MeOH and potential inhibitors 1-6 at 0.8 mM concentration. The enzymatic reactions were initiated by the addition of 1 mU β4GalT and incubated at 30 °C for 60 min. Inactivation was quickly done by placing of the reaction solutions for 3 min in a thermo block set to 90 °C. Thesolutions were diluted with water (300 μL) and centrifuged for 20 min, filtered through M.E. Cellulose filter (0.2 μm × 13 mm) and the filtrate was injected into RP-HPLC system. AChE Inhibition Bioassay AChE enzymatic activity was measured using an adaptation of the method previously described [Ingkaninan et al., J. Ethnopharmacol., 89:261-264]; 98 μl of 50 mM Tris-HCl buffer (pH 8), 30 μl of a solution sample of the inhibitor, at different concentrations in methanol, and 7.5 μl of AChE solution containing 0.26 U/ml were mixed in a microplate and left to incubate for 15 min. Subsequently, 22.5 μl of 0.023 mg/ml AChI and 142 μl of 3 mM DTNB were added. The initial rate of the enzymatic reaction was followed by reading the absorbance at 405 nm during the first 5 min of reaction. Samples were prepared in a range of concentrations of the compounds in water (choline caffeate, choline trolox, choline cinnamate) or in an aqueous solution of 50% methanol (choline 3,4-dimethoxicinnamate, choline rosmarinate). A control reaction was carried out using the sample solvent instead of sample and it was considered 100% activity. AChE and BChE Inhibition Assay In vitro AChEI activity was performed based on the modified Ellman's methodas previously reported by our group using a 96-well plate reader (BioTek ELx808). For this purpose, compounds 8 were dissolve in a mixture of DMSO (5 mL) and methanol (5 mL) and diluted in 0.1 M KH2PO4/K2HPO4 buffer (pH 8.0). Each well contained 50 uL potassium phosphate buffer (KH2PO4/K2HPO4, 0.1 M, pH 8), 25 uL prepared sample as described above, 25 uL enzyme with final concentration of 0.22 U/mL in buffer. They were preincubated for 15 min at rt, and then 125 uL DTNB (3 mM in buffer) was added. Characterization of the hydrolysis of ATCI catalyzed byAChE was performed spectrometrically at 405 nm followed by the addition of substrate (ATCI 3 mM in water). The change in absorbance was measured at 405 nm after 15 min. The IC50 values were determined graphically from inhibition curves (log inhibitor concentration vs. percent of inhibition). Binding Assay The affinity of compounds of the invention for the NK-1 receptor was evaluated in CHO recombinant cells which express the human NK-1 receptor. Membrane suspensions were prepared from these cells. The following radioligand: [3H] substance P (PerkinElmer Cat#NET111520) was used in this assay. Binding assays were performed in a 50 mM Tris/5 mM MnCl2/150 mM NaCl/0.1% BSA at pH 7.4. Binding assays consisted of 25 μl of membrane suspension (approximately 5 μg of protein/well in a 96 well plate), 50 μl of compound or reference ligand (Substance P) at increasing concentrations (diluted in assay buffer) and 2 nM [3H] substance P. The plate was incubated 60 min at 25° C. in a water bath and then filtered over GF/C filters (Perkin Elmer, 6005174, presoaked in 0.5% PEI for 2 h at room temperature) with a Filtration unit (Perkin Elmer). The radioactivity retained on the filters was measured by using the TopCount-NXT reader (Packard). Binding Assay The affinity of compounds of the invention for the NK1 receptor was evaluated in CHO recombinant cells which express the human NK1 receptor. Membrane suspensions were prepared from these cells. The following radioligand: [3H] substance P (PerkinElmer Cat#NET111520) was used in this assay. Binding assays were performed in a 50 mM Tris/5 mM MnCl2/150 mM NaCl/0.1% BSA at pH 7.4. Binding assays consisted of 25 μl of membrane suspension (approximately 5 μg of protein/well in a 96 well plate), 50 μl of compound or reference ligand (Substance P) at increasing concentrations (diluted in assay buffer) and 2 nM [3H] substance P. The plate was incubated 60 min at 25° C. in a water bath and then filtered over GF/C filters (Perkin Elmer, 6005174, presoaked in 0.5% PEI for 2 h at room temperature) with a Filtration unit (Perkin Elmer). The radioactivity retained on the filters was measured by using the TopCount-NXT reader (Packard). Biological Assay Recombinant human procathepsin K was obtained from Enzo Life Sciences. Activation of the proenzyme was performed in 32.5 mM sodium acetate pH 3.5, EDTA 1 mM, NaCl 500 mM, human procathepsin K 5.5 uM, at room temperature. Activation times were optimized and varied between 35 and 150 minutes. Cathepsin K was preincubated with test compounds at various concentrations for 5 minutes at 25C. The assay was initiated by addition of substrate Z-Phe-Arg-aminomethylcoumarin ("Z-F-R-AMC" Bacchem) and the final assay conditions were 1.5 nM cathepsin K, 50 uM Z-F-R-AMC, 150 mM sodium acetate pH 5.5, 2.5 mM EDTA (Omnipure), 2.5 mM DTT (EMD), 0.01% BRIJ 35 (Sigma), and 4.0% DMSO (Acros). Test compounds were serially diluted with DMSO and water to include a final concentration range of 10 uM to 10 pM. The reaction was monitored fluorometrically for 5 minutes at 25C. using black 96-well Corning 3686 assay microplates with a Thermo Fluoroskan Ascent FL microplate reader. Competitive Binding Assay Competitive binding assays were performed by incubating rhER alpha (α) and beta 1 (β1) receptors with 10 nM [3H]estradiol (the radio ligand) in the presence or absence of increasing concentrations, 0.25 to 250,000 nM, of the phenolic test compounds of Tables 1 to 3 (nM is nano molar). Each data point is the average of at least two assays. Stock solutions of the compounds of Tables 1 to 3 were prepared at 10×E-2 M in 100% ethanol, water or DMSO (dimethyl sulfoxide). Compounds were diluted 10 fold in binding buffer and then 1:4 in the final assay mix. The final concentration of ethanol or DMSO in the assay well was 5%. The highest concentration of the hydrolysis test compound was 2.5×E-4 M (250,000 nM). The residual monomers of Tables 1 to 3 were tested at seven concentrations over log increments. The lowest concentration was 2.5×E-10 M (0.25 nM). Cyclooxygenase Assay 100 mM Tris HCl buffer, pH 8.0 containing 1 µM heme and COX-1 (ovine) or COX-2(human recombinant), which was preincubated for 10 min in a water bath at 37 °C was used for reaction mixture preparation. Addition of 10 µL arachidonic acid to the reaction mixture (final concentration 100 µM) was used to initiate reaction. After 2 min reaction was stopped by addition of 1 M HCl. PGE2 in the reaction mixture was determined using ELISA method. 100 mM potassium phosphate buffer (pH 7.4) was used to dilute the DMSO stock solutions of the test compounds to the desired concentration. This reaction mixture was transferred to a 96-well platethat was precoated with a mouse anti-rabbit IgG. Tracer prostaglandin acetylcholine esterase and primary antibody (mouse anti PGE2) were also added to the plates followed by overnight incubation at room temperature. After overnightincubation, reaction mixtures were pipetted out and the wells were washed with 10 mM potassium phosphate buffer containing 0.05% Twe Enzymatic Assay r-AC protein samples were pre-incubated with various concentrations of test compounds or vehicle control in 100 mM NaH2PO4/citrate buffer pH 4.5, 0.1% Nonidet P-40, 3 mM DTT for 30 min at 37° C. Samples were incubated with 100 μM N-lauroyl ceramide (Nu-Chek Prep, Elysian, Minn.) at 37° for 30 min. The reaction was stopped by addition of a mixture of chloroform/methanol (2:1 vol/vol) containing 1 nmol of heptadecanoic acid (HDA; NuChek Prep). The organic phases were collected, dried under nitrogen, and analyzed by LC/MS in the negative-ion mode using heptadecanoic acid (HDA) as internal standard (m/z=199 for lauric acid, m/z=269 for HDA). HDA was eluted on an XDB Eclipse C18 column isocratically at 2.2 mL/min for 1 min with a solvent mixture of 95% methanol and 5% water, both containing 0.25% acetic acid, and 5 mM ammonium acetate. The column temperature was 50° C. Enzyme Assay The aim of this assay is to determine the affinity of a test compound for the mPGES-1 enzyme. 47 μl of recombinant human mPGES-1 (0.5 μg protein/well) containing microsomal suspension in a buffer containing GSH, (2.5 mmol/L L-Glutathione reduced, dissolved in 0.1 mol/L Phosphat Buffer pH 7.4) is dispensed in a 384-well plate and thereafter 1 μl of the test compound(s) is/are added and incubated for 25 minutes at room temperature. The enzyme reaction is started by the addition of 2 ul PGH2 (final conc 2 μM) disolved in water-free Diglyme. After 60 seconds the reaction is terminated by addition of a stop solution containing FeCl2 (10 μL 0.074 mol/l FeCl2). The samples are diluted between 1:25 in PBS (Phosphate Buffered Saline). 10 μl of the diluted samples are transferred to 384-well low volume plate. Enzyme Assay The aim of this study was to evaluate TK-112690 in vivo as an inhibitor of uridine phosphorylase (UPase) enzyme activity. The range of TK-112690 doses studied for ability to prevent metabolic breakdown of uridine, through the in vitro inhibition of mouse and human small intestinal UPase enzyme, was 0, 0.1, 0.5, 1, 5, 10, 50, 100, 500, 1000, 5000 and 10000 uM). Detection of UPase activity was determined by HPLC analysis using UV detection of uracil concentration (UPase catabolizes uridine into uracil and ribose-1-phosphate).The UPase enzyme material was prepared from homogenized mouse and human being small intestinal tissue. TK-112690 was dissolved in water (50 mg/ml) and analyzed for UPase inhibition in aqueous solution containing 5 mM uridine, 0.01 M Tris, 0.01 M phosphate, 1 mM EDTA, and 1 mM DTT. Reactions were performed at 37 C. at pH of 7.3.TK-11260 inhibition of mouse and human UPase was analyzed by reverse phase HPLC using UV detection. FLIPR Assay HEK 293 Cells were grown in media containing DMEM, 10% FBS pen/strep/L-Glutamine and non-essential amino acids. The cells were plated in 384-well PDL coated plates at 12000 cells/well and incubated overnight at 37° C./5% CO2. Media was then removed from the cells, which were then incubated with buffer (Hank's containing HEPES and Chaps) containing FLIPR calcium-5 dye, made with buffer containing probenecid, for 60 minutes at 37° C. Varying concentrations of compound in a final concentration of 5% DMSO were then added to the cells and incubated at 25° C. for 30 minutes. The plates were then added to the FLIPR Tetra and the device added a concentration of a PAR1 selective receptor-activating peptide with the sequence Ala-parafluoroPhe-Arg-Cha-Cit-Try-Nh2 (prepared in water) at a concentration equal to the effective concentration that achieves 80% activation of signaling on the day of the experiment. Fluorescence Polarization Assay The fluorescence polarization assay tests the ability of compounds to inhibit the self-aggregation of α-synuclein peptide fragments. Peptides were incubated for 120 min at room temperature in the presence or absence of test compounds (compound concentrations were 33.3 to 0.015 □M). Samples were read on a Beckman Coulter DTX 880 plate reader in fluorescence polarization mode using excitation at 485 nm and emission at 520 nm. Data was analyzed using a four-parameter logistic fit (XLFit, IDBS Software). Peptide 4F (CTGFVKKDQLGK (SEQ ID NO: 1)) was prepared by American Peptide. Fresh peptide samples were reconstituted in purified water at 5 mM and diluted into 50 mM HEPES pH 7.4 with 50 mM NaCl to 100 nM final concentration. Solid compounds were dissolved in DMSO (10 mM), and then diluted serially in DMSO (300×) followed by dilution in buffer (1×) to provide solutions with a consistent final DMSO concentration of 0.33%. GLO1 Activity Assay Assays were carried out in 100 mM sodium phosphate, pH 7.0 buffer using 96-well Clear UV Plate (Corning UV Transparent Microplates; catalog #3635). A fresh solution of glutathione (pre-substrate 1, 100 mM) as well as methylglyoxal (pre-substrate 2, 100 mM) was prepared in deionized water. The substrate was prepared by adding 14.5 ml of buffer and 0.99 ml of each pre-substrate components. The substrate mixture was vortexed vigorously for 15 s, then allowed to sit at room temperature for 20 min. Initial well volume was 50 μl containing GLO1 (40 ng) and inhibitor. This protein and inhibitor mixture was incubated for 15–20 min before the addition of substrate. To this was then added substrate (150 μl), yielding a maximum amount of 5% DMSO per well. The enzyme activity was measured using a BioTek Synergy H4 plate reader by measuring absorbance at 240 nmevery 1 min for 8 min. High throughput discovery of novel modulators of ROMK K+ channel activity: Analog Library Testing Assay Provider: Jerod Denton Assay Provider Affiliation: Vanderbilt University Grant Title: High throughput discovery of novel modulators of ROMK K+ channel activity Grant Number: R21 NS057041-01 The Renal Outer Medullary Potassium channel (ROMK, Kir1.1) is expressed in the renal tubule where it critically regulates fluid and electrolyte homeostasis (1). An emerging body of evidence suggests that ROMK could be a target for a novel class loop diuretic that lowers blood pressure while preserving plasma potassium levels (2). Furthermore, homozygous loss-of-function mutations in the gene encoding ROMK (KCNJ1) cause antenatal Bartter syndrome, a severe salt and water wasting disease in infants (3). ROMK is thus an important pharmacological target for the management of disease. Its actual therapeutic value and drugability, however, are unknown due to the lack of small-molecule probes targeting the channel. The discovery of ROMK modulators will provide important new tools for studying In Vitro Inhibition Assay The reagents used have the following composition: Enzyme buffer (EB): 50 mM HEPES (pH: 7.0) (Sigma H7523), 100 mM NaCl (Sigma S7653), NaN.sub.3 at 0.01% (Sigma S8032), BSA at 0.005% (Sigma A2153), 0.05 mM sodium orthovanadate (Calbiochem 567540). Detection buffer (DB): 50 mM HEPES (pH: 7.0), BSA at 0.1%, 0.8 M KF (Fluka 60239), 20 mM EDTA (Sigma E5134).The peptide used is the one described in Biochemistry, 2005, 44, 8533-8542; A-21-K(biotin)NH.sub.2, obtained from NeoMPS (reference SP081233). All the HTRF reagents Mab PT66-K (61T66KLB) and streptavidin-XL665 (610SAXLB), and the SEB reagent, are purchased from Cisbio.The test is carried out in a 384-well plate (Greiner 784076). The serial dilutions are carried out in pure DMSO, and then an intermediate one-in-three dilution in water is carried out, with 1 microliter of each concentration being distributed, all these operations being performed using the Zephyr apparatus (Caliper Life Sciences). In-Vitro Fluorescence Polarization Assay The fluorescence polarization assay tests the ability of compounds to inhibit the self-aggregation of α-synuclein peptide fragments. Peptides were incubated for 120 min at room temperature in the presence or absence of test compounds (compound concentrations were 33.3 to 0.015 μM). Samples were read on a Beckman Coulter DTX 880 plate reader in fluorescence polarization mode using excitation at 485 nm and emission at 520 nm. Data was analyzed using a four-parameter logistic fit (XLFit, IDBS Software). Peptide 4F (CTGFVKKDQLGK (SEQ ID NO: 1)) was prepared by American Peptide. Fresh peptide samples were reconstituted in purified water at 5 mM and diluted into 50 mM HEPES pH 7.4 with 50 mM NaCl to 100 nM final concentration. Solid compounds were dissolved in DMSO (10 mM), and then diluted serially in DMSO (300×) followed by dilution in buffer (1×) to provide solutions with a consistent final DMSO concentration of 0.33%. In-Vitro Fluorescence Polarization Assay The fluorescence polarization assay tests the ability of compounds to inhibit the self-aggregation of α-synuclein peptide fragments. Peptides were incubated for 120 min at room temperature in the presence or absence of test compounds (compound concentrations were 33.3 to 0.015 DM). Samples were read on a Beckman Coulter DTX 880 plate reader in fluorescence polarization mode using excitation at 485 nm and emission at 520 nm. Data was analyzed using a four-parameter logistic fit (XLFit, IDBS Software). Peptide 4F (CTGFVKKDQLGK (SEQ ID NO: 1)) was prepared by American Peptide. Fresh peptide samples were reconstituted in purified water at 5 mM and diluted into 50 mM HEPES pH 7.4 with 50 mM NaCl to 100 nM final concentration. Solid compounds were dissolved in DMSO (10 mM), and then diluted serially in DMSO (300×) followed by dilution in buffer (1×) to provide solutions with a consistent final DMSO concentration of 0.33%. Inhibition Assay A mixture containing 100 mM MES-sodium hydroxide (pH 6.5), 1 mM magnesium acetate, 0.5 mM EGTA, 5 mM beta-mercaptoethanol, 0.02% Tween 20, 10% glycerol, 12 ug/ml P-GS1, 41.7 uM [gamma-32P] ATP (68 kBq/ml), bovine cerebral TPK1 and a compound shown in Table (a final mixture contained 1.7% DMSO deriving from a solution of a test compound prepared in the presence of 10% DMSO) was used as a reaction system. The phosphorylation was started by adding ATP, and the reaction was conducted at 25 C. for 2 hours, and then stopped by adding 21% perchloric acid on ice cooling. The reaction mixture was centrifuged at 12,000 rpm for 5 minutes and adsorbed on P81 paper (Whatmann), and then the paper was washed four times with 75 mM phosphoric acid, three times with water and once with acetone. The paper was dried, and the residual radioactivity was measured using a liquid scintillation counter. The results are shown in the table below. Inhibition Assay CYP17 activity was assayed according to the following procedure. Solutions of each test compound and isozyme inhibitor (ketoconazole) were separately prepared at concentrations of 2700, 540, 90, 18, 3, 0.6 and 0.1 uM by serial dilution with DMSO:ACN (50:50 v/v). The individual test compound and isozyme inhibitor solutions were then diluted 20-fold with deionized water (50:950 v/v) to concentrations of 135, 27, 4.5, 0.9, 0.15, 0.03 and 0.005 uM. The percent of organic solvent attributable to the test compound or inhibitor mixture in the final reaction mixture is 1%. Pooled rat testicular microsome suspension (20 mg/mL) was diluted with phosphate buffer to obtain a 1.25 mg/mL suspension. A solution of NADPH was prepared in phosphate buffer at a concentration of 2.5x. A stock solution of the substrate was prepared in DMSO:MeCN (50:50 v/v), mixed, and diluted in phosphate buffer to obtain a single solution containing the substrate at 5 uM. Inhibition Assay Compounds of the invention were initially diluted to 10 mM in 100% DMSO (CALBIOCHEM) for storage and made into kinase buffer solution to create a compound concentration ranging from 1 uM and 10 uM. Serial dilutions of compounds of the invention were dispensed into a 96-well plate (GREINER BIOSCIENCES) at 6 uL each. Purified full-length human SYK (CARNA BIOSCIENCES) were diluted in kinase buffer and added to the compound solutions and pre-incubated for 30 minutes at room temperature. Next, ATP (TEKNOVA) of Km (15 uM) and substrate solution (suggested manufacture substrates of PerkinElmer, Ulight-TK peptide for SYK) was added (12 uL each) to the wells containing the compound solution and enzyme. The reaction mixture was incubated for 1 hour. Following the incubation, the stop solution made with EDTA, water, and Lance detection buffer (PERKINELMER) was added (12 uL each) to stop phosphorylation. Following the addition of the stop solution and 5 minutes of shaking. Inhibition Assay Mixtures of isozyme inhibitors (sulfaphenazole, tranylcypromine, and ketoconazole as specific inhibitors of isozymes 2C9, 2C19, and 3A4, respectively) were prepared containing each inhibitor at concentrations of 6000, 2000, 600, 200, 60, 20, 6, and 2 uM by serial dilution with DMSO:ACN (50:50 v/v). The mixed inhibitor solutions were then diluted 20-fold with DMSO:MeCN:deionized water (5:5:180 v/v/v) to concentrations of 300, 100, 30, 10, 3, 1, 0.3, and 0.1 uM. The percent of organic solvent attributable to the test compound or inhibitor mixture in the final reaction mixture was 2% v/v. Pooled human liver microsome suspension (20 mg/mL) was diluted with phosphate buffer to obtain a 5 mg/mL suspension. A solution of NADPH was prepared in phosphate buffer at a concentration of 5 mM. Separate stock solutions of each substrate were prepared in DMSO:MeCN (50:50 v/v), mixed, and diluted in phosphate buffer to obtain a single solution. Inhibition Assay The potential inhibition of enzyme activities of human cytochromes P450 (CYP) of Compound 1, 2, or 105 was evaluated using pooled human liver microsomes.Methods: The competitive inhibition potential of Compounds 1, 2, and 105 was determined by assessing at multiple concentrations on probe CYP reactions near their respective Km values to create IC50 curves in human liver microsomes (HLM). The time-dependent inactivation (TDI) potential was also assessed for CYP3A4/5 by evaluating KI and kinact values when appropriate.A suspension containing PB, HLM, CYP-selective probe substrate, and the inhibitor being tested was added to a 96-well plate. The plates were preincubated in a 37 ° C. water bath for approximately 2 minutes. The reaction was initiated by the addition of NADPH to each well of the 96-well plate. The final concentrations for PB, HLM, and NADPH were 100 mmol/L (pH 7.4), 0.1 mg/mL, and 2.3 mmol/L, respectively. The CYP probe substrates and CYP inhibitors used as positive controls. Inhibition Assay Various concentrations of the substances to be tested are dissolved in dimethyl sulphoxide and mixed with an aqueous refludan solution (10 μg/ml). In clear 96-well plates having a flat bottom, 30 μl of citrate plasma (Octapharma) are mixed with 10 μl of the substance dilution. Then, either 20 μl of a solution of a rattlesnake toxin (Russel viper venom (RVV); RVV reagent: Pentapharm 121-06, final concentration 0.6 mU) in an aqueous calcium chloride solution buffer (final concentration of calcium chloride 0.05 M) or 20 μl of the aqueous calcium chloride solution (final concentration of calcium chloride 0.05 M) without RVV reagent (as reference for an unstimulated sample) are added. After addition of 20 μl of ChromozymX substrate (final concentration 1.6 mmol/l, Bachem L-1565, diluted in water) the samples are measured in a SpectraFluor Reader using a measurement filter of 405 nm each minute over a period of 20 minutes. Inhibition Kinetics Assay Full length porcine calpain (156 nM), or papain (236 pM) was added to a solution of 100 mM NaCl, 50 mM HEPES, pH 7.6, 1 mM TCEP, 30 μM Suc-LLVY-AMC (SEQ ID NO: 8) substrate, and inhibitor (0.5 to 50 μM). Calpain reactions also contained CaCl2 (1 mM and 100 mM for porcine and rat respectively). Both substrate and inhibitors were dissolved in acetonitrile/DMSO (1:1) with the exception of E-64, dissolved in water. Organic solvent remained <2% in all reactions, and most often <1%. Reactions were carried out in microtiter 96-well plates, with 150 μL per well, 30° C., and product formation was monitored over time by fluorescence (Ex/Em 346/444 nm, with 420 nm cutoff filter). Kinetic values of kobs were determined via non-linear regression using one-phase association analysis and linear plots of 1/kobs vs. 1/[I] provided kinetic constants ki and KI. Inhibition of SIK; Abl and Src Kinases Briefly, a radiometric protein kinase assay (33PanQinase® Activity Assay) was used for measuring the kinase activity of the five protein kinases. All kinase assays were performed in 96-well FlashPlates™ from PerkinElmer (Boston, MA, USA) in a 50 uL reaction volume. The reaction cocktail was pipetted in four steps in the following order:25 uL of assay buffer (standard buffer/[gamma-33P]-ATP)10 uL of ATP solution (in water)5 uL of test compound (in 10% DMSO)20 uL enzyme/substrate mixThe assay for all protein kinases contained 70 mM HEPES-NaOH pH7.5, 3 mM MgCl2, 3 mM MnCl2, 3 μM Na-orthovanadate, 1.2 mM DTT, ATP (variable concentrations, corresponding to the apparent ATP-Km of the respective kinase, see Table 2A), [gamma-33P]-ATP (approx. 8×105 cpm per well), protein kinase (variable amount, see Table 2A), and substrate (variable amounts, see Table 2A). Inhibiton Assay For studying inhibition of Plasmodium or human DHODH enzyme, two assays that are in routine use are described, for example, in Baldwin, et al. (2002) JBiol Chem., 277, 41827-41834, and Baldwin, et al. (2005) J. Biol. Chem., 280. 21847-21853.Briefly, this colorimetric assay monitors the reduction of 2,6-dichloroindophenol (DCIP) at 600 nm (e = 18.8 mM-1cm-1) for measuring DHOD inhibition. The assay was carried out using a solution containing 100 mM HEPES, pH 8.0, 150 mM NaCl, 10% glycerol, 0.1% Triton X-100, 20 micro molar CoQD (coenzyme QD), 200 micro molar L-dihydroorotate, and 120 micro molar DCIP. Reactions are initiated by addition of enzyme to a final concentration in the range of about 5 nM to about 50 nM while maintaining the temperature of a circulating water bath at 25 ° C. Alternatively, for potent compounds, activity was determined by directly measuring the production of orotic acid at 296 nm ( µ = 4.3 mM-1 cm-1). Inhibitory Activity Assay Streptavidin (manufactured by Vector Laboratories) was dissolved in a 0.1M carbonate buffer solution (composition: 90 nM Na2CO3, 10 mM NaHCO3) to a concentration of 40 μg/ml, Each 50 μl of this solution was added to a well of an immunoplate (manufactured by NUNC), this is allowed to stand at 4° C. overnight to adsorb. Then, each well was washed with a phosphate buffer (composition: 13.7 mM NaCl, 0.27 mM KCl, 0.43 mM Na2HPO4, 0.14 mM KH2PO4) two times, and 300 μl of a phosphate buffer containing 1% skim milk to block it for 30 minutes. Further, each well was washed with a phosphate buffer two times, 50 μl of a substrate DNA solution (2 pmol/μl) was added to adsorb at room temperature for 30 minutes while shaking, and this was washed with a phosphate buffer two times and, then, distilled water once. Inhibitory Assay For the measurement of CH24H inhibitory activity, using the human CH24H lysate prepared in Experimental Example 2, the amount of 24-HC produced from cholesterol by catalytic activity of CH24H was measured in the presence of a test compound, and compared with that measured in the absence of the test compound. That is, a test compound solution at various concentrations was mixed with a reaction buffer (50 mM potassium phosphate containing 0.1% BSA and Complete, EDTA-free, pH 7.4) and human CH24H lysate. Then, [14C] cholesterol (53 mCi/mmol specific activity, 15 μM) was added, and CH24H reaction was performed at 37° C. for 5 hr. After completion of the reaction, a quenching solution consisting of chloroform/methanol/distilled water (2:2:1 v/v) was added, and the resulting 24-HC was extracted by shaking. The extract was applied to silica gel thin layer chromatography (ethyl acetate:toluene=4:6), and the obtained 14C-24HC fraction was measured with BAS2500 (Fujifilm Corporation). KHK Inhibition Assay Determination of Human and Rat recombinant KHK-A and KHK-C isozyme IC50 ValuesAn assay reagent cocktail was prepared by combining NADH, water, TEA, KCl, MgCl2, PEP, ATP, DTT, coupling enzymes (pyruvate kinase and lactate dehydrogenase, LDH) to final concentrations as shown in Table B.TABLE BReagent Final ConcentrationNADH 300 μMWater —TEA 33 mMKCl 100 mMMgCl2 6 mMPEP 1.33 mMATP 0.1 mMDTT 12 mMPyruvate kinase 1.0 U/mLLDH 1.0 U/mL[0367]To this was added the relevant KHK isozyme to a final concentration of 6 nM. Aliquots of each inhibitor compound were diluted via 5-fold serial dilutions to produce final concentrations ranging from 1000 nM to 0.064 nM. The inhibitor aliquots were added to the assay reagent cocktail containing KHK with fructose (at a concentration of 2 mM) in a 96-well plate. The absorbance at 340 nm was measured via spectrophotometry and inhibition was analyzed using non-linear regression. Kinase Inhibition Assay Compounds of the invention were initially diluted to 10 mM in 100% DMSO (CALBIOCHEM) for storage and made into kinase buffer solution to create a compound concentration ranging from 1 uM and 10 uM. Serial dilutions of compounds of the invention were dispensed into a 96-well plate (GREINER BIOSCIENCES) at 6 uL each. Purified full-length human SYK, and KDR (CARNA BIOSCIENCES) were diluted in kinase buffer and added to the compound solutions and pre-incubated for 30 minutes at room temperature. Next, ATP (TEKNOVA) of Km (15 uM) and substrate solution (suggested manufacture substrates of PerkinElmer, for example, Ulight-TK peptide for SYK and Ulight-JAK1 for KDR (PERKINELMER)) was added (12 uL each) to the wells containing the compound solution and enzyme. The reaction mixture was incubated for 1 hour. Following the incubation, the stop solution made with EDTA, water, and Lance detection buffer (PERKINELMER) was added (12 uL each) to stop phosphorylation. LOX Activity Assay Enzyme activity was monitored by UV analysis. 0.05 cm^3 of enzyme solution was added to a cuvette containing 2 cm^3 linoleic acid solution and the appropriate amounts of buffer and inhibitor solutions in thermostatic water bath at 25°C. There was no pre-incubation time of the enzyme with inhibitor solution. The activity of the enzyme was determined by monitoring the increase in the absorption caused by the oxidation of linoleic acid at 234 nm and 25°C (ε = 25 000 M^−1 cm^−1). One standard solution of complexes in DMSO (10^-2 M) was used for theinhibition activity experiments (three sets). In this case, the substrate concentration was kept constant (0.3 mM), while the amounts of buffer and inhibitor solutions varied according to the inhibitor final concentration needed(0.5-30 μM or 0.15-9 μL from standard solutions). The total experiment volume was 3 cm^3. Molecular Docking The atomic co-ordinates of CK2α were retrieved from the Protein Data Bank [PDB ID: 3PE1] by deleting the heteroatoms except for water molecules within 6.5 of the ligand. Compounds 12, 23, 10, and 27 were constructed with the SYBYL 8.1 programa using compound CX-4945 as the template and were energy minimized with the Tripos force field. The chemical structures and IC50 values of the tricyclic quinoline analogs are shown in Figure 1. Four compounds were docked into the active site of CK2α using the molecular docking program Genetic Optimization for Ligand Docking (GOLD) version 3.0. The active site of CK2α was defined as a 6.5- -radius sphere centered on CX-4945, which comprise all complete amino acids there in. The crystal structure of CK2a in complex with compound CX-4945 was used to optimize docking parameters and evaluate the reliability of the docking methods. The standard default genetic algorithm (GA) parameters were used for all calculations and applied to predi NMR Assay All NMR spectra were acquired at 298 K on a Bruker DRX 600 MHz spectrometer equipped with a cryoprobe. Typically, NMR samples contained 0.1-0.2 mM protein in 50 mM KH2PO4 and 50 mM Na2HPO4, pH 7.4, containing 150 mM NaCl and 5 mM β-Mercaptoethanol. Water suppression was carried out using the WATERGATE sequence. NMR data were processed using the Bruker program Xwin-NMR version 3.5. NMR ligand binding experiments were carried out in an analogous way to those previously described. See D'Silva L., et. al., J. Am. Chem. Soc. 2005, 127, 13220-13226 and Popowicz G. M., et. al., Cell Cycle. 2007, 6, 2386-2392. The maximum concentration of DMSO at the end of titration experiments was less than 1%. The pH was maintained constant during the entire titration. The 1H-15N-HSQC spectra were recorded using fast HSQC pulse sequence as described by Mori et. al., J. Magn. Reson. B 1995, 108, 94-98. PDE5 assay Experimental protocol: The test compound, i.e the compound of the present invention, reference compound or water (control) are added to a buffer containing 40 mM Tris/HCl (pH 7.8), 3 mM MgCl2, 1.4 mM DTT, 0.21% BSA, 200 mM NH4Cl, 1 μM cGMP and 0.1 μCi [3H]cGMP. Thereafter, the reaction is initiated by addition of the enzyme and the mixture is incubated for 60 min at 22° C. For basal control measurements, the enzyme is omitted from the reaction mixture. Following incubation SPA beads are added. After 20 min at 22° C. under shaking, the amount of [3H]5′GMP is quantified with a scintillation counter (Topcount, Packard).The results shown in Table 1 are expressed as a percent inhibition of the control enzyme activity. The standard inhibitory reference compound is dipyridamole, which is tested in each experiment at several concentrations to obtain an inhibition curve from which its IC50 value is calculated. Radioligand Binding The binding of [3H]MLA was measured using a modification of the methods of Davies et al., Neuropharmacol. 38: 679 (1999). [3H]MLA (Specific Activity=25-35 Ci/mmol) was obtained from Tocris. The binding of [3H]MLA was determined using a 2 h incubation at 21 C. Incubations were conducted in 48-well micro-titre plates and contained about 200 g of protein per well in a final incubation volume of 300 uL. The incubation buffer was PBS and the final concentration of [3H]MLA was 5 nM. The binding reaction was terminated by filtration of the protein containing bound ligand onto glass fiber filters (GF/B, Brandel) using a Brandel Tissue Harvester at room temperature. Filters were soaked in de-ionized water containing 0.33% polyethyleneimine to reduce non-specific binding. Each filter was washed with PBS (3x1 mL) at room temperature. Non-specific binding was determined by inclusion of 50 uM non-radioactive MLA in selected wells. Radiometric Assay ALKl (Invitrogen, Carlsbad, CA, USA) kinase activity was measured at ProQinase GmbH ( Freiburg. Germany) in a radiometric assay using gamma-33P-ATP and casein (Sigma, St. Louis, MO, USA) as substrate in 96-weli PerkinElmer FlashPlates (Boston, MA, USA). The compounds were tested at 10 concentrations in the range of 1 x 10"4 M to 3 x 10"9 M in a total volume of 50 ul with a finalDMSO concentration of 1 % each. The assay components were mixed in the order: 20 ul assay buffer (70 mM HEPES-NaOH, pi I 7.5. 3 mM MgCl2, 3 mM MnCl2, 2uM sodium orthovanadate, 1.2 mM DTT); - 5ul gamma-33P-ATP (1.0ug/50ug in water, approx. 6 x10E+5 cpm per well); 5 ul test compound solution (in 10% DMSO);10 ul substrate (200 ug/ml, 1.0ug/50ml final concentration) / enzyme (4 ug/ml, 20 ng/50ul = 5.5 nM final concentration) solution (1 : 1 mixture). Receptor Assay Competitive binding test for receptors: 20 μl of each of the test compounds and 20 μl of the radioactive ligand together with 160 μl of the receptor proteins were added into the reaction tubes, and the final concentrations of the test compound and the positive drug were all 10 μmol/L. After 50 min of incubation in 30° C. water bath, the tubes were immediately moved to ice bath to terminate the reactions. GF/C glass fiber filter papers were used for rapid sucking filtration on a Millipore cell sample collector, elution buffer (50 mM Tris-HCl, PH 7.4) was applied for 3 ml×3 times, and microwave was applied for 4-5 min for drying. The filter papers were moved into 0.5 ml centrifuge tubes, and 500 μl of lipid-soluble scintillation solution was added. The tubes were allowed to stand still for over 30 min in dark, and the intensities of radioactivity were measured by a counter. Receptor Binding Assay Materials for the Receptor Binding AssayIsotope ligand [3H]-Ketanserin (67.0 Ci/mmol) was purchased from PerkinElmer Company; Methysergide was purchased from RBI Company; GF/C glass fiber filter paper was purchased from Whatman Company; Tris was imported and divided into aliquots; PPO, POPOP were purchased from Shanghai No. 1 Reagent Factory; lipid-soluble scintillation solution. Beckman LS-6500 Multi-function Liquid Scintillation Counter was used.Procedures(1) The prepared membrane was first applied with appropriate amount of buffer (0.05 M Tris-HCl buffer: 6.05 g of Tris was dissolved in 1000 ml of double-distilled water, and concentrated HCl was used to adjust to pH 7.5), and homogenizer was used for evenly dispersing. 15 tubes were mixed into a 100 ml container, and appropriate amount of homogenized liquid was added to give 50 ml of membrane suspension, which was reserved for future use.(2) 100 uL of membrane preparation and 100 uL of buffer. Stopped-flow Assay Osmotic water permeability was measured at 22-24 °C bymonitoring 90° scattered light intensity at 520 nm wavelength. Measurements were made using a PiStar 180 (Applied Photophysics, UK), with a dead time of 1-2 milliseconds. Thirty minutes prior to the assay, stripped red blood cell ghosts or proteoliposomes containing purified AQP1 and stored in ice were treated with mercuric chloride (50-100 µM; positive control for inhibition) or with the various compounds usually at a concentration of 150 µM. Stock solutions of compounds were in 100% ethanol (for stripped erythrocyte membrane experiments) and in 100% DMSO (for proteoliposome experiments). An inwardly directed osmotic gradient (hyperosmotic shock) was created by mixing the vesicles maintained in Buffer A (50 mM sodium phosphate buffer pH, 100 mM NaCl, 1 mM EDTA, 0.025% sodium azide) with an equal volume of 200 mM mannitol. On the other hand, vesicles preincubated with 200 mM mannitol were mixed with Buffer A to create an outwardly VPS34 Enzyme Assays 100 nL compounds in DMSO are added to wells of a 384 well microtitre plate (Greiner 780076). At room temperature: 5 ul VPS34 reaction buffer (Invitrogen Assay Buffer Q (diluted 1 in 5 with nanopure water) plus 2 mM DTT and 2 mM MnCl2) containing ATP (20 uM, Promega) and 200 uM PI-PS substrate (Invitrogen PV5122) is added followed immediately by 5 ul VPS34 reaction buffer (as above) containing VPS34 (5 nM, Millennium Protein Sciences Group) and the mixture is incubated with shaking at room temperature for 1 hour. Then 5 ul VPS34 stop-detect mix (as per Invitrogen Adapta Assay kit (PV5009) instructions (contains kinase quench buffer, TR-FRET buffer, Adapta Eu anti-ADP antibody and Alexa Fluor 647 ADP tracer)) is added to quench the reaction. The plates are then incubated for 30 minutes at room temperature with shaking and then read on a BMG PheraStar Plus reader. cAMP HRTF Assay The test compound or water (control) is mixed with the human recombinant PDE4B1 enzyme (4.8 U) in a buffer consisting of 44.4 mM tris-HCl, 5.28 mM MgCl2, 2.64 mM DTT and 0.044% Tween 20 (pH 7.8). After adding the cAMP enzyme substrate (final concentration 40 nM) the mixture is incubated for 30 minutes at room temperature. Then a fluorescence acceptor (Dye2 marked with cAMP), a fluorescence donor (anti-cAMP antibody marked with a europium cryptate) and the non-specific phosphodiesterase inhibitor IBMX (3-isobutyl-1-methylxanthine; final concentration 1 mM) are added. After 60 minutes the fluorescence transfer, which correlates with the amount of remaining cAMP, is measured with a microplate reader (Rubystar, BMG) at λex=337 nm, λem=620 nm and λem=665 nm. The enzyme activity is calculated from the quotient formed from the measured signal at 665 nm and that at 620 nm. Enzymatic Assay IDO1: Recombinant full-length human IDO1 with a N-terminal hexahistidine tag expressed in E. coli and purified to homogeneity is incubated at a final concentration of 2 nM in assay buffer consisting of 37.5 mM phosphate buffer at pH6.5 supplemented with 10 mM sodium L-ascorbate, 0.45 μM methylene blue, 50 U/ml catalase, 0.01% BSA, and 0.01% Tween 20 (protocol modified from Seegers et al, JBS 2014). Example compounds are serially diluted in DMSO, further diluted in phosphate buffer, and added to the enzyme at final concentrations ranging from 10 μM to 0.5 nM. The final DMSO concentration is 0.6%. Following a pre-incubation of 30 minutes at RT, the reaction is started by the addition of L-tryptophan at a final concentration of 5 μM in assay buffer. After 30 minutes of incubation at RT, 3 μL of the reaction mixture are transferred to a 384 deep well plate containing 25 μL of deionized water. 100 μl of 200 nM L-Tryptophan-(indole-d5) in cold 100% methanol are added followed by a 10 minutes centrifugation at 4,000 rpm at 4° C. An additional 75 μL of deionized water are then added and followed by a 10 minutes centrifugation at 4,000 rpm at 4° C. The product of the reaction N′-Formylkynurenine (NFK) is quantified by LCMS and normalized to the L-Tryptophan-(indole-d5) signal. Samples with 0.6% DMSO (0% effect) and a TDO/IDO inhibitor (100% effect) are used as control samples to set the parameters for the non-linear regression necessary for the determination of the half-maximal inhibitory concentration (IC50) for each compound. For each compound concentration the percentage of activity compared to 0% and 100% effect is calculated as average ±STDEV (each concentration measured in duplicate). IC50 values and curves are generated with XLfit software (IDBS) using Dose-Response One Site model 203 (four parameter logistic curve model). When compounds are measured multiple times, mean values are given. Enzymatic Assay TDO2: Recombinant human TDO comprising amino acids 19-407 with a N-terminal hexahistidine tag expressed in E. coli and purified to homogeneity is incubated at a final concentration of 15 nM in assay buffer consisting of 75 mM phosphate buffer at pH7 supplemented with 100 μM ascorbic acid, 50 U/ml Catalase, 0.01% BSA, and 0.01% Tween 20 (protocol modified from Seegers et al, JBS 2014). Example compounds are serially diluted in DMSO, further diluted in phosphate buffer, and added to the reaction mixture at final concentrations ranging from 10 μM to 0.5 nM. The final DMSO concentration is 0.6%. Following a pre-incubation of 30 minutes at RT, the reaction is started by the addition of L-tryptophan at a final concentration of 200 μM in assay buffer. After 30 minutes of incubation at RT, 3 μL of the reaction mixture are transferred to a 384 deep well plate containing 25 μL of deionized water. 100 μl of 200 nM L-Tryptophan-(indole-d5) in cold 100% methanol are added followed by a 10 minutes centrifugation at 4,000 rpm at 4° C. An additional 75 μL of deionized water are then added and followed by a 10 minutes centrifugation at 4,000 rpm at 4° C. The product of the reaction N′-Formylkynurenine (NFK) is quantified by LCMS and normalized to the L-Tryptophan-(indole-d5) signal. Samples with 0.6% DMSO (0% effect) and a TDO/IDO inhibitor (100% effect) are used as control samples to set the parameters for the non-linear regression necessary for the determination of the half-maximal inhibitory concentration (IC50) for each compound. For each compound concentration the percentage of activity compared to 0% and 100% effect is calculated as average±STDEV (each concentration measured in duplicate). IC50 values and curves are generated with XLfit software (IDBS) using Dose-Response One Site model 203 (four parameter logistic curve model). When compounds are measured multiple times, mean values are given. MOR cAMP Agonist and Antagonist Assays CHO Tag-lite human mOR stable cell line from Cisbio (NCBI accession number: NM_000914.3, Bedford, Mass.) were seeded and grown to approximately 80% confluence in Ham F-12 with 10% FBS, 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mM Hepes, and 1 mg/ml geneticin (Invitrogen, Carlsbad, Calif.). Cells were then harvested using accutase (Corning. Coming, N.Y.), centrifuged at 1300 RPM for 5 min, and plated at 5000 cells per 5 μL per well in a 5× dilution of stimulation buffer consisting of the HTRF cAMP Gi kit, water and IBMX at 0.5 mM (Cisbio, Bedford, Mass.) in white HTRF low volume 384 well plates (Cisbio, Bedford, Mass.). Plates were then incubated at 37° C. in 5% CO2 for 10 minutes. For the agonist assay, forskolin was added to a final concentration of 4 μM. For the antagonist assay, forskolin (4 μM) and DAMGO at EC90 final concentration were added. Test compounds were dissolved in DMSO and water, and then serially diluted to working concentrations such that the concentration of DMSO was less than 0.1%. Diluted test compounds were added at 2.5 μL per well, and plates were incubated at 37° C., and 5% CO2 for 15 minutes, and then at room temperature for 15 minutes. Next 5 μl/well of cAMP Eu-cryptate and 5 μl/well of anti-cAMP-d2 (both diluted 1:20 in lysis buffer) were added, and plates were incubated at room temperature for 1 hour. Following incubation, plates were read in a Synergy Neo2 multi-mode reader (Biotek, Winooski, Vt.). Plate reader settings were set to time resolved fluorescence with excitation at 330 nm and emissions of 620 nm and 665 nm. Emission fluorescence was normalized (665/620 nm signal×1000). For the agonist assay, data was normalized using the maximal DAMGO response. Measurements were performed in triplicate and the dose response was fit using nonlinear regression. Plaque Reduction Assay Hep-G2 cells (ECACC, 85011430) were passaged in flasks and seeded in 24-well plates in DMEM containing antibiotics and supplemented with 10% FBS. During inoculation and subsequent incubation, cells were cultured in DMEM containing 2% FBS. 100 plaque forming unit/well of RSV (RSV A2 ECACC, 0709161v) was mixed with eight serial dilutions of compound. Subsequently, 100 μL of the virus/compound mixtures was added to confluent Hep-G2 cell monolayers. The cells and virus/compound mixtures were incubated at 37° C. in a humidified 5% CO2 incubator for 2 h prior to removal of the inoculum and addition of 1 mL of overlay (DMEM containing 2% FBS and 0.8% CMC) containing compound dilutions. The cells and were incubated at 37° C. in a humidified 5% CO2 incubator for 2 days.Cells were washed with PBS before adding 75/25% v4v EtOH/MeOH, for 3 min. Fixative was removed and plates were washed with PBS. A pre-titrated amount of the primary antibody was added in 200 μL PBS/2% milk powder, and plates incubated for 90 min at 37° C. The plates were washed 3 times with PBS/0.05% Tween20 before addition of rabbit anti-goat horse radish peroxidase in 200 μL PBS/2% milk powder, and incubated for 1 h at 37° C. Following three wash steps with PBS/0.05% Tween20, 200 μL ready-to-use TrueBlue was added and plates were incubated at rt for 10-15 min before washing with water. After removal of water, plates were air-dried in the dark.Plates were scanned and analysed using the Immunospot S6 Macro analyser, which is equipped with BioSpot analysis software for counting immunostained plaques (virospots). Plaque counts were used to calculate % c infection relative to the mean of the plaque count in the virus control wells for RSV. The EC50 value was calculated as 50% reduction in signal, respectively, by interpolation of inhibition curves fitted with a 4-parameter nonlinear regression with a variable slope in Dotmatics. Screen Quest Fluo-8 No Wash Calcium Assay Kit Ca2+ influx was measured in HEK-293 cells stably transfected with the receptor using Screen Quest Fluo-8 No Wash Calcium Assay Kit (AAt Bioquest ). Briefly, once inside the cells, the lipophilic blocking groups of Fluo-8 are cleaved by non-specific cell esterases, resulting in a negatively-charged fluorescent-dye that stays inside cells. Its fluorescence increases upon binding to calcium. When HEK-293/P2X7 cells were stimulated with BzATP, Ca2+ entered the cells and the fluorescence of Fluo-8 NW increaseed. The dye absorption spectrum was compatible with excitation at 488 nm by argon laser sources and its emission wavelength was in the range of 515-575 nm.To routinely test the compounds, HEK-293 cells stably transfected with rat P2X7R were seeded overnight in growth medium at 10000, 15000 or 20000 cells/well in 384-well plate, according to the level of response after thawing. 24 hours later, the medium was removed and the cells were pre-loaded at RT for 1 hour with 20 μL/w of Fluo-8 NW prepared in Tyrode 0.3 mM Ca2+/Mg2+-free. Compounds of the invention were tested at 8 concentrations (4 replicates for each concentration): 10-3.16-1-0.316-0.1-0.0316-0.01 and 0.00316 μM, in the same plate.Compounds were tested at FLIPRTETRA according to the following method: first injection at FLIPRTETRA of 10 μL of 3× test compound (in Tyrode's buffer 0.3 mM Ca2+/Mg2+-free+DMSO 0.5% final concentration) 5′ incubation second injection at FLIPRTETRA of 15 □L of 3× BzATP at ECK) (in Tyrode's buffer 0.3 mM Ca2+/Mg2+-free+BSA 0.0003% final concentration) Fluorescence recording for 3′Between one plate and the following, tips were extensively washed with water, then with 100% DMSO and finally with water to avoid carry-over inside the tips.The effect of the test compounds was measured as percent inhibition vs a reference antagonist and IC50 values were calculated accordingly. In vitro MKP-1 Phosphatase Dose Response Active/Probe Assessment Assay - Effects of Catalase The MKP-1 dose response Active/Probe assessment-Catalase assay has been developed to evaluate the effects of adding 100 U/mL of Catalase on the MKP-1 inhibition of actives identified in the MH-76391 In vitro MKP-1 HTS assay AID #374, and subsequently confirmed in the HTS dose response confirmation assay AID #551. Protein tyrosine phosphatases have an active site cysteine that is very susceptible to inactivation by oxidation. In addition, a number of compounds such as quinone-like compounds are capable of generating reactive oxygen species via redox cycling in the presence of DTT. Adding Catalase to inactivate hydrogen peroxide (H2O2)does not affect the activity of MKP-1 in the assay but can reverse the inhibition of some inhibitors or significantly increase their IC50 values.The MKP-1 Phosphatase Dose Response Active/Probe Assessment Assay - Effects of Catalase has been Developed and Run at the University of Pittsburgh Molecular Screening Center (PMLSC) part of the Molecular Library S Choline Release Assasy The purpose of this assay is to detect autotaxin inhibition using a choline release assay.Test compound (10 mM stocks in 100% DMSO) is serially diluted in 100% DMSO resulting in 10 concentrations of 100× inhibitor in half area 96 well plates (Corning 3992). Each of these 10 wells in 100% DMSO is diluted 1:33.33 in assay buffer in round bottom 96 well plates (Fisher 12565502) resulting in 3× concentrations in well containing 3% DMSO. The assay buffer is 50 mM Tris pH8.0, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 0.01% TRITON X-100 (Sigma T9284) and 0.01% fatty acid free bovine serum albumin (Sigma A8806). A 20 μl aliquot of each 3× test compound is then added to black flat bottom 96 well plates (Corning 3991) in singlicate. A 20 μl aliquot per well of 3× recombinant human autotaxin, (Echelon, E-4000) (full length human autotaxin with a C-terminal His tag transfected into 293E cells and purified via nickel chelate and size exclusion chromatography) is then added to every well except for the no enzyme control wells. A 20 μl aliquot per well of assay buffer is added to the no enzyme control wells. A 20 μl aliquot of a 3× cocktail containing choline oxidase (Sigma C5896), horseradish peroxidase (Sigma P8125), amplex ultrared (Invitrogen A36006) and the autotaxin substrate lysophosphatidylcholine (LPC) 16:0 (Avanti Polar Lipids 855675P) is added to each well while avoiding exposure to light. The final concentrations in the well of choline oxidase, horseradish peroxidase, amplex ultrared and LPC 16:0 are 0.4 units/ml, 4 units/ml, 40 μM and 30 μM respectively. The plate is then sealed with aluminum foil seals and incubated at 37° C. for 1 hour in a Labline Imperial III incubator. During this incubation, LPC is cleaved by autotaxin resulting in Lysophosphatidic Acid (LPA) 16:0 and choline. The choline that is released is oxidized by choline oxidase resulting in betaine and hydrogen peroxide. The hydrogen peroxide reacts with the horseradish peroxide and amplex ultrared to form the fluorescent molecule resorufin. In Vitro DAAO Enzyme Assay The functional activity of compounds inhibiting the DAAO enzyme was determined by utilizing the co-product of the catalysis of D-Serine, H2O2 which can be quantitatively measured using the Amplex (trade mark) Red (Invitrogen) detection. Amplex Red reagent is a colorless substrate that reacts with hydrogen peroxide (H2O2) with a 1:1 stoichiometry in the presence of hydrogen peroxide to produce highly fluorescent resorufin (excitation/emission maxima=570/585 nm). The changes in fluorescence were monitored by a fluorescence plate reader, Envision (Perkin Elmer) and increases in DAAO activity were readily detected upon addition of D-Serine and suppression of this response observed with the application of test compounds.Human DAAO enzyme was supplied by the Takeda Pharmaceutical Company (Osaka) and each batch was tested and used at concentrations giving comparable levels of activity. The Km of D-Serine was measured for each enzyme batch to maintain consistency; this Km was used in subsequent assays.On the day of the assay compounds were serially diluted in DMSO before being diluted 1:20 with assay buffer (20 mM Tris ph 7.4). A 5 μl portion of assay buffer was added to the wells of a 384 clear base black-walled plate (Corning), 5 μl of diluted compound was then added via automated plate to plate transfer using the Bravo liquid handler (Agilent technologies) followed by 5 μl of human DAAO enzyme and then 5 μl D-Serine 50 mM was added to all but the negative control wells (final concentration of 10 mM). Finally 5 μl Amplex red reagent (Invitrogen) was added to all wells as per manufacturer's protocol. The plate was incubated for 60 minutes in the dark at 25° C. and the fluorescence in each well was measured in the Envision plate reader.The IC50 values for compounds were determined from ten point half log scale dose-response studies and represent the concentration of compound required to prevent 50% inhibition of DAAO activity in the presence of 10 mM D-Serine. Concentration response curves were generated using the average of duplicate wells for each data point and analyzed using non-linear regression and four parameter curve fit. In Vitro DAAO Enzyme Assay The functional activity of compounds inhibiting the DAAO enzyme was determined by utilizing the co-product of the catalysis of D-Serine, H2O2 which can be quantitatively measured using the Amplex Red (Invitrogen) detection. Amplex Red reagent is a colorless substrate that reacts with hydrogen peroxide (H2O2) with a 1:1 stoichiometry in the presence of hydrogen peroxide to produce highly fluorescent resorufin (excitationemission maxima=570/585 nm). The changes in fluorescence were monitored by a fluorescence plate reader, Envision (Perkin Elmer) and increases in DAAO activity were readily detected upon addition of D-Serine and suppression of this response observed with the application of test compounds.Human DAAO Enzyme was supplied by the Takeda Pharmaceutical Company (Osaka) and each batch was tested and used at concentrations giving comparable levels of activity. The Km of D-Serine was measured for each enzyme batch to maintain consistency; this Km was used in subsequent assays.On the day of the assay compounds were serially diluted in DMSO before being diluted 1:20 with assay buffer (20 mM Tris ph 7.4). A 5 μl portion of assay buffer was added to the wells of a 384 clear base black walled plate (Corning), 5 μl of diluted compound was then added via automated plate to plate transfer using the Bravo liquid handler (Agilent technologies) followed by 5 μl of human DAAO enzyme and then 5 μl D-Serine 50 mM was added to all but the negative control wells (final concentration of 10 mM). Finally 5 μl Amplex red reagent (Invitrogen) was added to all wells as per manufacturer's protocol. The plate was incubated for 60 minutes in the dark at 25° C. and the fluorescence in each well was measured in the Envision plate reader.The IC50 values for compounds were determined from ten point half log scale dose-response studies and represent the concentration of compound required to prevent 50% inhibition of DAAO activity in the presence of 10 mM D-Serine. Concentration response curves were generated using the average of duplicate wells for each data point and analyzed using nonlinear regression and four parameter curve fit. Malachite Green ATPase Assay (1) Greiner 384-well (Greiner 781101) or Costar 384-well flat-bottomed polystyrene multiwell plates (VWR). (2) Assay buffer of (a) 100 mM Tris-HCl, pH 7.4, (b) 150 mM KCl, (c) 6 mM MgCl2. Stored at room temperature. (3) 0.0812% (w/v) malachite green (M 9636, Sigma Aldrich Ltd., Poole, UK). Stored at room temperature. (4) 2.32% (w/v) polyvinyl alcohol USP (P 1097, Sigma Aldrich Ltd, Poole, UK) in boiling water (see Comment 1), allowed to cool, and stored at room temperature. (5) 5.72% (w/v) ammonium molybdate in 6 M hydrochloric acid. Stored at room temperature. (6) 34% (w/v) sodium citrate. Stored at room temperature. (7) 100 mM ATP, disodium salt, special quality (47699, Sigma Aldrich). Stored at −20° C. (8) E. coli expressed yeast HSP90 protein, purified >95% (see, e.g., Panaretou et al., 1998) and stored in 50 uL aliquots at −80° C. Method 1. Dilute test compounds to 500 μM in AR water (DMSO concentration will be 2.5%). Transfer 2.5 μl of these compounds directly from the daughter plate to the assay plate, giving a final assay concentration of 100 μM. To obtain 12 point IC50 values, perform serial dilutions 1:2 to produce a range of assay concentrations from 100 μM to 97.6 nM (2.5% DMSO), and transfer 2.5 μl of each concentration into the assay plate. Column 1 in the assay plate contains no compound, as a negative control. An additional row with no compound is also used as a background. 2. Prepare ATP by diluting 100 mM stock to 925 μM with assay buffer, and aliquot 5 μl of diluted ATP to each well including controls (final assay concentration 370 μM). 3. Add 5 μl of buffer to background row. 4. Dilute enzyme preparation to 1.05 μM with assay buffer, and aliquot 5 μl into each compound well and to the negative control column. 5. Collect the reagents to the bottom of the well, cover plate with plate seal and incubate overnight at 37 deg C. 6. First thing in the morning prepare the Malachite Green Reagent. Add 2 parts of Malachite Green Solution, 1 part of Polyvinyl Alcohol Solution, 1 part of Ammonium Molybdate Solution, and 2 parts of AR water. 7. Invert to mix, and leave for approximately 1 hour until the colour turns from brown to golden yellow. 8. Add 40 μl of Malachite Green Reagent to each well, allow 5 mins for colour to develop. 9. Add 5 μl of Sodium Citrate Reagent to each well (see comment 2) 10. Re-cover with plate seal and shake on plate shaker for at least 15 mins. 11. Measure Absorbance at 620 nM using a suitable plate reader (e.g. Victor, Perkin Elmer Life Sciences, Milton Keynes, UK). Under these conditions, the control absorbance is 0.9 to 1.4, and the background is 0.2-0.35 giving a signal to noise ratio of 12. The Z′ factor calculated from data obtained using these conditions is between 0.6 and 0.9. Comments Inhibition Assay The inhibition constants (K) the compounds for four CA isozymes, CA I, II, IX and XII were determined. An Applied Photophysics (Oxford, UK) stopped-flow instrument has been used for assaying the CA-catalyzed CO2 hydration activity (Khalifah, 1971). Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5) as buffer, 0.1 M Na2SO (for maintaining constant the ionic strength), following the CA-catalyzed CO2 hydration reaction for a period of 10-100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5-10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (1 mM) were prepared in distilled-deionized water with 10-20% (v/v) DMSO.
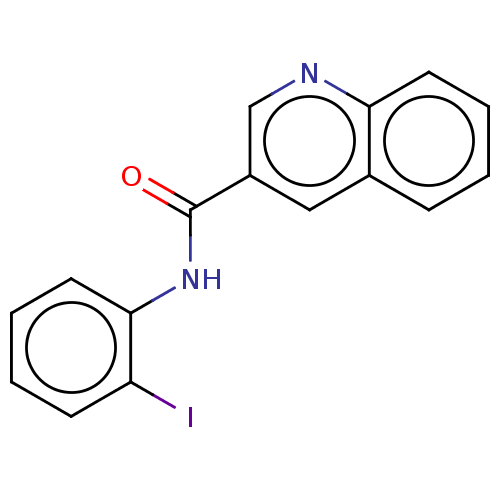 BDBM643937 US11866416, Compound HW-1-151
BDBM643937 US11866416, Compound HW-1-151 BDBM643938 US11866416, Compound HW-1-159
BDBM643938 US11866416, Compound HW-1-159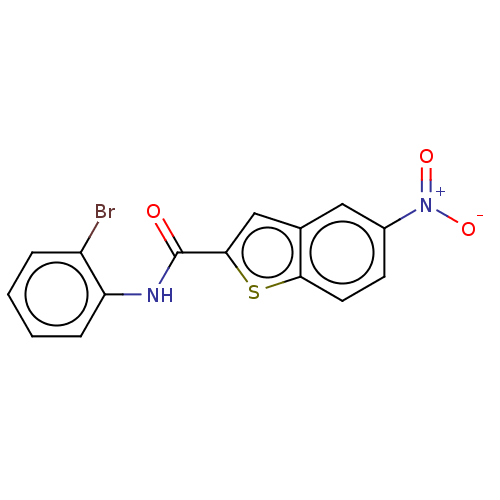 BDBM643939 US11866416, Compound HW-1-157
BDBM643939 US11866416, Compound HW-1-157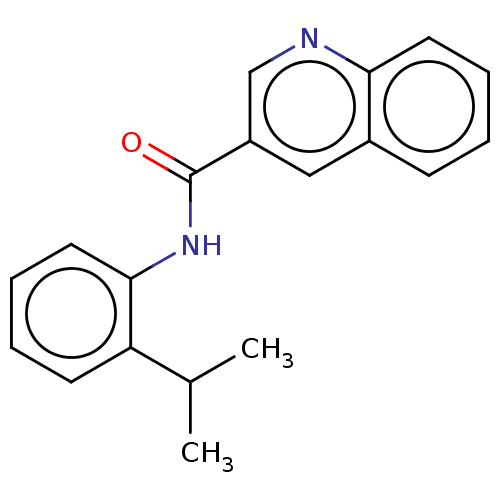 BDBM643941 US11866416, Compound HW-1-162
BDBM643941 US11866416, Compound HW-1-162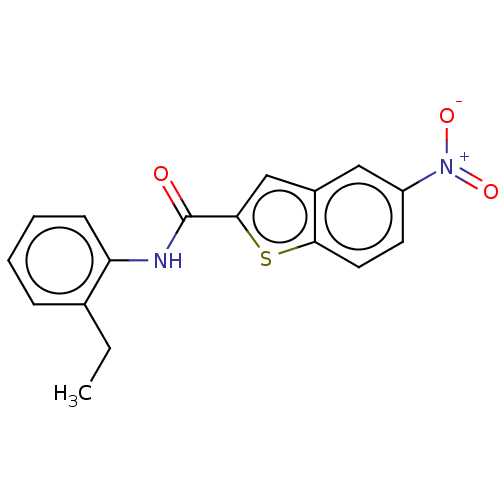 BDBM643942 US11866416, Compound HW-1-171
BDBM643942 US11866416, Compound HW-1-171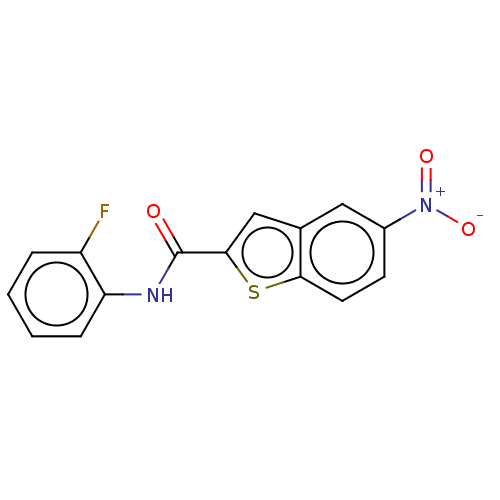 BDBM643943 US11866416, Compound HW-1-170
BDBM643943 US11866416, Compound HW-1-170 BDBM643998 US11866416, Compound HW-1-179
BDBM643998 US11866416, Compound HW-1-179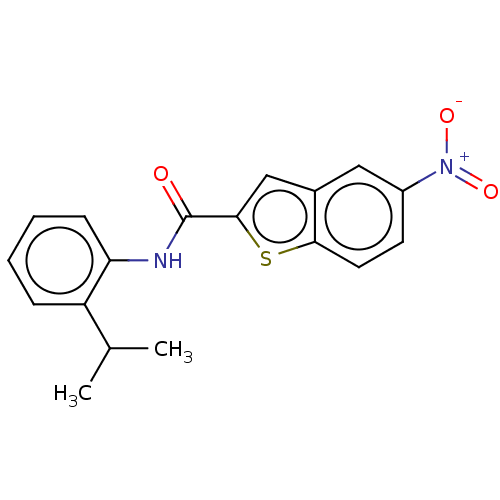 US11866416, Compound HW-1-169 BDBM643940
US11866416, Compound HW-1-169 BDBM643940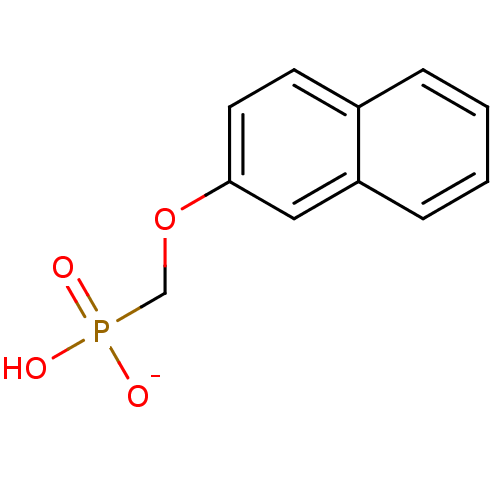 BDBM50086646 hydrogen (2-naphthyloxy)methylphosphonate
BDBM50086646 hydrogen (2-naphthyloxy)methylphosphonate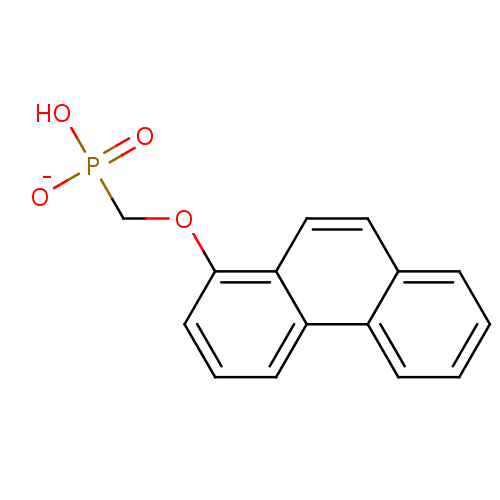 BDBM50086647 hydrogen (1-phenanthryloxy)methylphosphonate
BDBM50086647 hydrogen (1-phenanthryloxy)methylphosphonate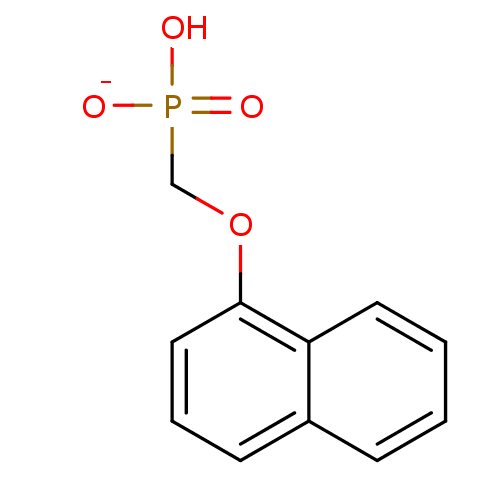 BDBM50086649 hydrogen (1-naphthyloxy)methylphosphonate
BDBM50086649 hydrogen (1-naphthyloxy)methylphosphonate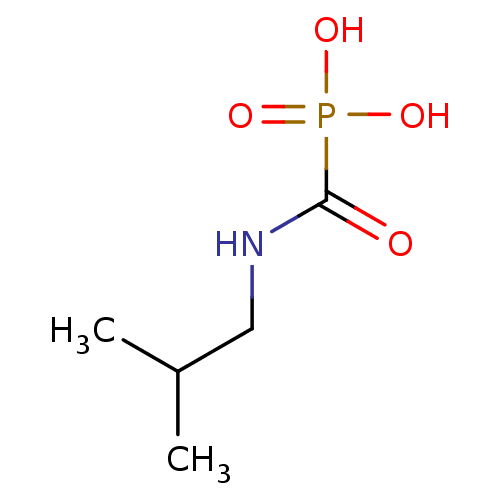 cyclohexanaminium hydrogen (isobutylamino)carbonylphosphonate BDBM50146806
cyclohexanaminium hydrogen (isobutylamino)carbonylphosphonate BDBM50146806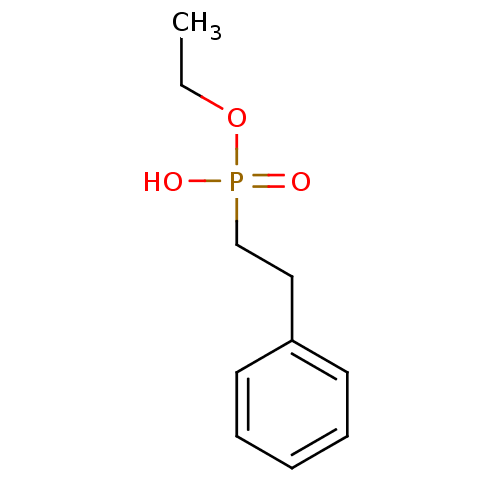 BDBM50195742 ethyl hydrogen 2-phenylethylphosphonate CHEMBL227517
BDBM50195742 ethyl hydrogen 2-phenylethylphosphonate CHEMBL227517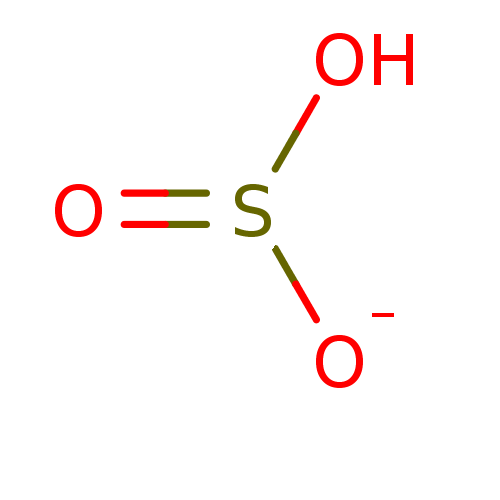 CHEMBL1689285 HSO3- bisulfite hydrogen sulfite BDBM26991
CHEMBL1689285 HSO3- bisulfite hydrogen sulfite BDBM26991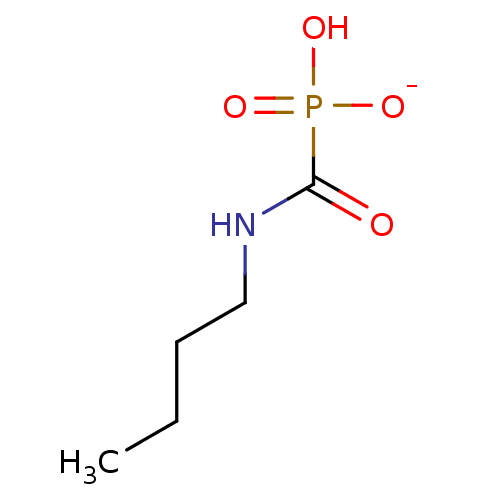 CHEMBL97389 sodium hydrogen (butylamino)carbonylphosphonate BDBM50146814
CHEMBL97389 sodium hydrogen (butylamino)carbonylphosphonate BDBM50146814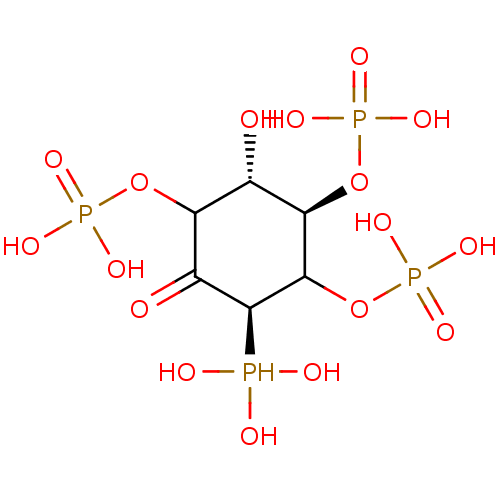 CHEMBL611046 BDBM50304359 (2R,3S,5S,6S)-6-(hydrogen phosphonato)-3,5-dihydroxycyclohexane-1,2,4-triyl tris(hydrogen phosphate)
CHEMBL611046 BDBM50304359 (2R,3S,5S,6S)-6-(hydrogen phosphonato)-3,5-dihydroxycyclohexane-1,2,4-triyl tris(hydrogen phosphate)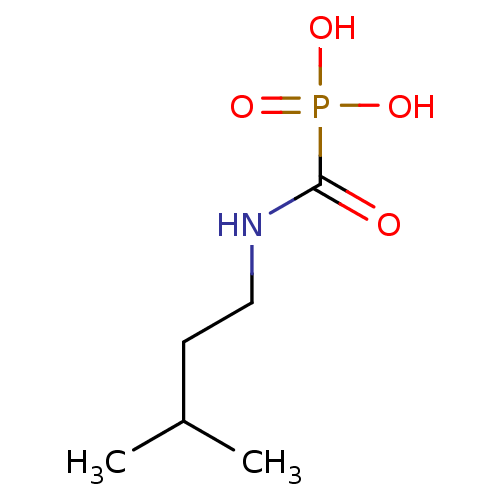 BDBM50146812 cyclohexanaminium hydrogen [(3-methylbutyl)amino]carbonylphosphonate
BDBM50146812 cyclohexanaminium hydrogen [(3-methylbutyl)amino]carbonylphosphonate [NNNH] triazoic acid N3H CHEMBL186537 hydrogen azide BDBM50153977 hydrido-1kappaH-trinitrogen(2N--N)hydrogen trinitride(1-) hydrazoic acid
[NNNH] triazoic acid N3H CHEMBL186537 hydrogen azide BDBM50153977 hydrido-1kappaH-trinitrogen(2N--N)hydrogen trinitride(1-) hydrazoic acid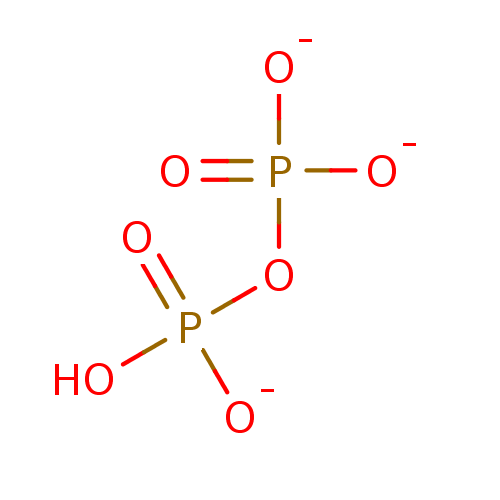 BDBM50079022 HP2O7(3-) hydrogen diphosphate diphosphate diphosphate(3-)
BDBM50079022 HP2O7(3-) hydrogen diphosphate diphosphate diphosphate(3-)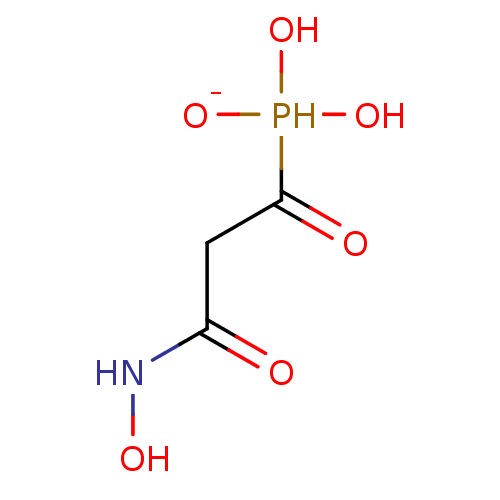 BDBM50198028 hydrogen 1-hydroxy-3-(hydroxyamino)-3-oxopropylphosphonate
BDBM50198028 hydrogen 1-hydroxy-3-(hydroxyamino)-3-oxopropylphosphonate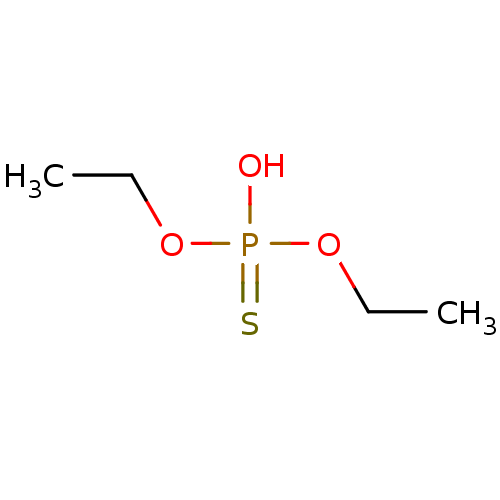 CHEBI:28006 BDBM50487243 O,O-DIETHYL S-HYDROGEN PHOSPHOROTHIOATE
CHEBI:28006 BDBM50487243 O,O-DIETHYL S-HYDROGEN PHOSPHOROTHIOATE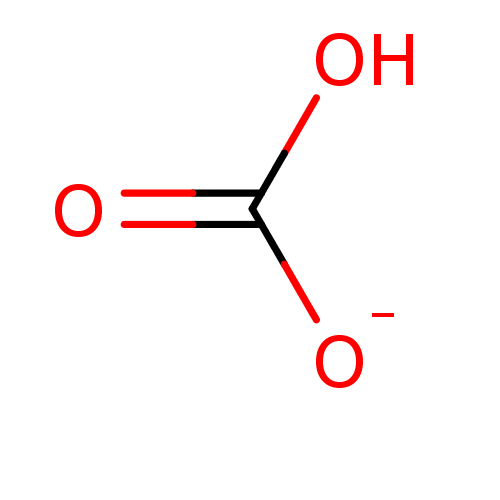 CHEMBL363707 SODIUM BICARBONATE bicarbonate ion hydrogen carbonate BDBM26986 HCO3-
CHEMBL363707 SODIUM BICARBONATE bicarbonate ion hydrogen carbonate BDBM26986 HCO3-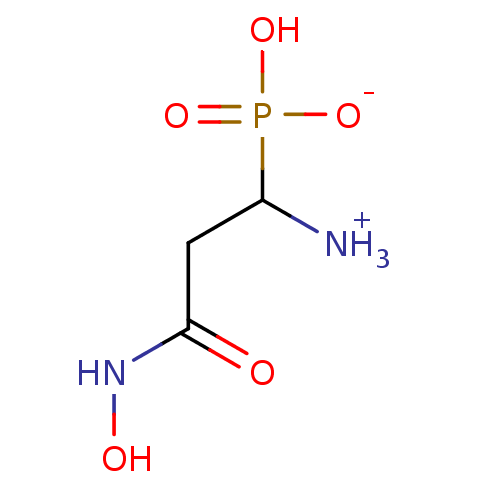 CHEMBL390655 BDBM50198029 hydrogen [1-azaniumyl-2-(hydroxycarbamoyl)ethyl]phosphonate
CHEMBL390655 BDBM50198029 hydrogen [1-azaniumyl-2-(hydroxycarbamoyl)ethyl]phosphonate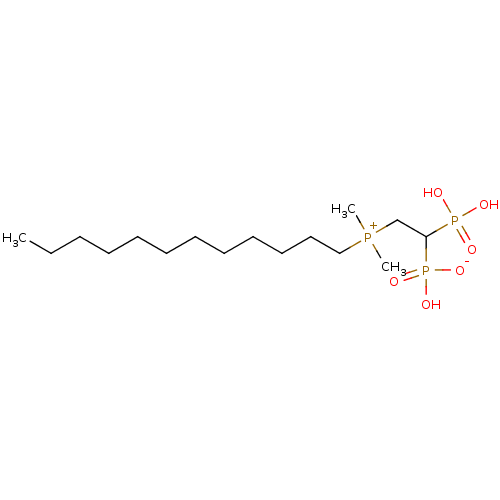 bisphosphonate, 9 hydrogen [2-(dodecyldimethylphosphanylium)-1-phosphonoethyl]phosphonate BDBM25256
bisphosphonate, 9 hydrogen [2-(dodecyldimethylphosphanylium)-1-phosphonoethyl]phosphonate BDBM25256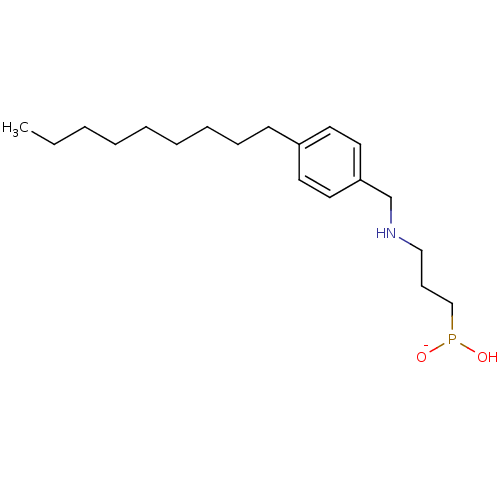 hydrogen (3-{[(4-nonylphenyl)methyl]amino}propyl)phosphonite BDBM50148402
hydrogen (3-{[(4-nonylphenyl)methyl]amino}propyl)phosphonite BDBM50148402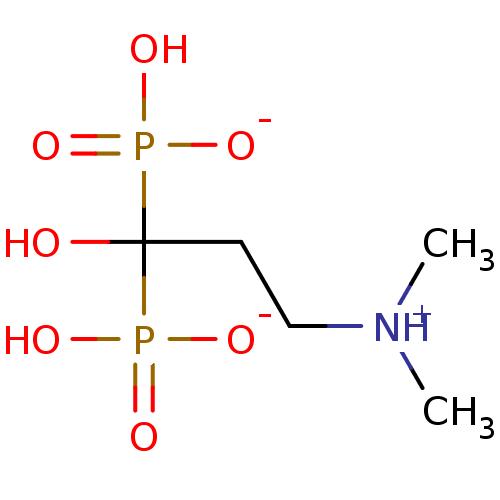 hydrogen 3-(dimethylammonio)-1-hydroxy-1-(hydroxyphosphinato)propylphosphonate BDBM50150877
hydrogen 3-(dimethylammonio)-1-hydroxy-1-(hydroxyphosphinato)propylphosphonate BDBM50150877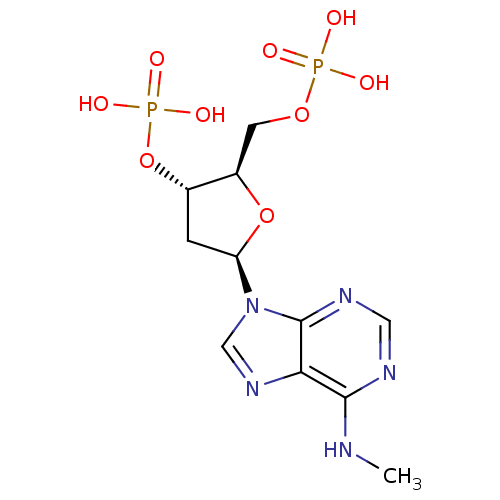 CHEMBL1096400 diammonium (2R,3S,5R)-2-[(hydrogen phosphonatooxy)methyl]-5-[6-(methylamino)-9H-purin-9-yl]oxolan-3-yl hydrogen phosphate BDBM50318029
CHEMBL1096400 diammonium (2R,3S,5R)-2-[(hydrogen phosphonatooxy)methyl]-5-[6-(methylamino)-9H-purin-9-yl]oxolan-3-yl hydrogen phosphate BDBM50318029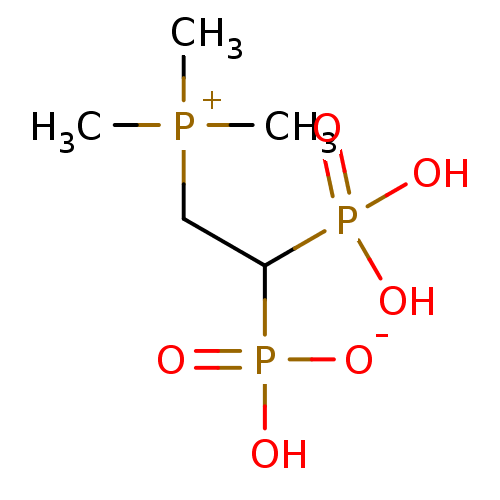 BDBM25309 hydrogen [1-phosphono-2-(trimethylphosphanylium)ethyl]phosphonate bisphosphonate, 58
BDBM25309 hydrogen [1-phosphono-2-(trimethylphosphanylium)ethyl]phosphonate bisphosphonate, 58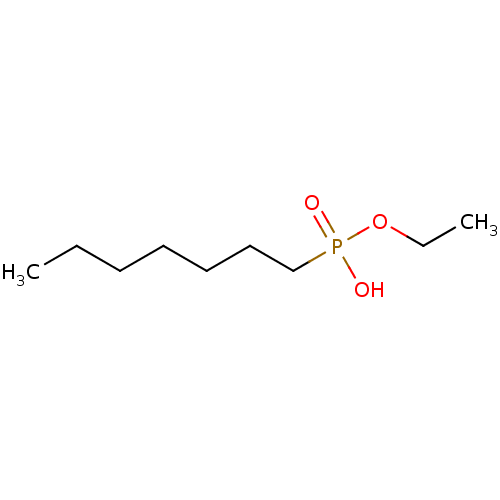 CHEMBL325304 Heptyl-phosphonic acid monoethyl ester BDBM50148588 ethyl hydrogen heptylphosphonate
CHEMBL325304 Heptyl-phosphonic acid monoethyl ester BDBM50148588 ethyl hydrogen heptylphosphonate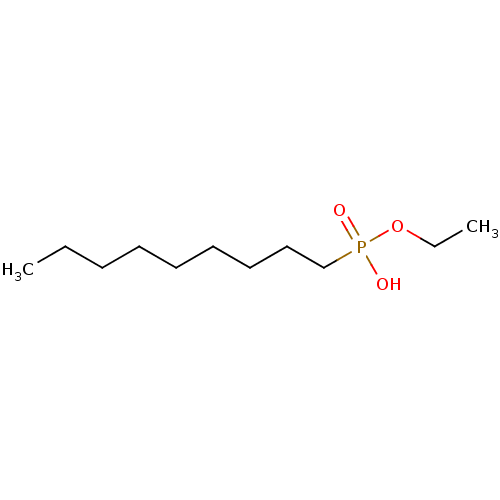 Nonyl-phosphonic acid monoethyl ester CHEMBL119480 BDBM50148595 ethyl hydrogen nonylphosphonate
Nonyl-phosphonic acid monoethyl ester CHEMBL119480 BDBM50148595 ethyl hydrogen nonylphosphonate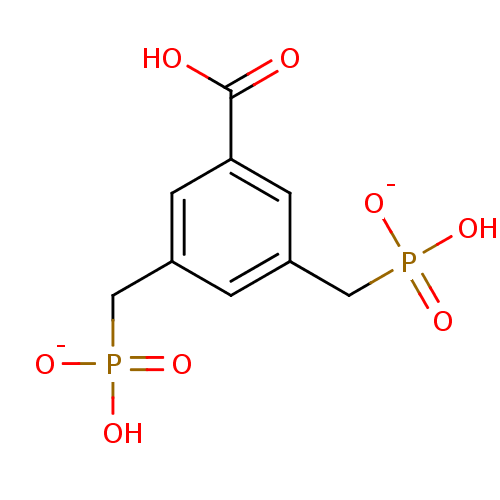 disodium hydrogen 3-carboxy-5-[(hydroxyphosphinato)methyl]benzylphosphonate BDBM50283584 CHEMBL88887
disodium hydrogen 3-carboxy-5-[(hydroxyphosphinato)methyl]benzylphosphonate BDBM50283584 CHEMBL88887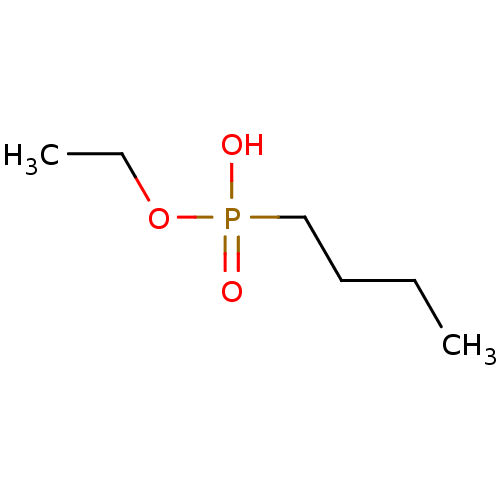 ethyl hydrogen butylphosphonate Butyl-phosphonic acid monoethyl ester CHEMBL118238 BDBM50148589
ethyl hydrogen butylphosphonate Butyl-phosphonic acid monoethyl ester CHEMBL118238 BDBM50148589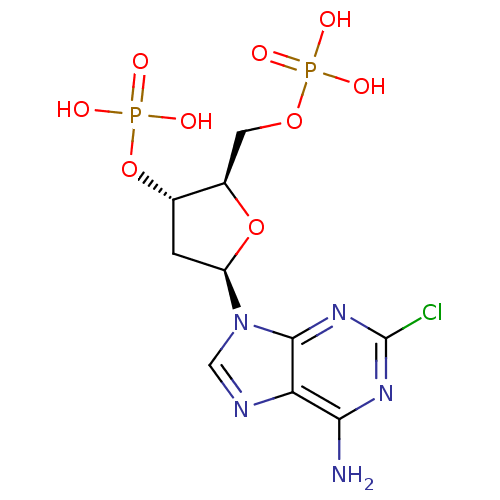 CHEMBL1094760 BDBM50318028 diammonium (2R,3S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-2-[(hydrogen phosphonatooxy)methyl]oxolan-3-yl hydrogen phosphate
CHEMBL1094760 BDBM50318028 diammonium (2R,3S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-2-[(hydrogen phosphonatooxy)methyl]oxolan-3-yl hydrogen phosphate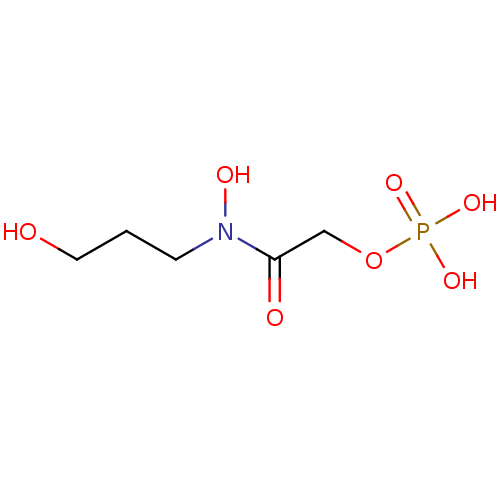 2-(hydroxy(3-hydroxypropyl)amino)-2-oxoethyl hydrogen phosphate BDBM50330436 CHEMBL1276292
2-(hydroxy(3-hydroxypropyl)amino)-2-oxoethyl hydrogen phosphate BDBM50330436 CHEMBL1276292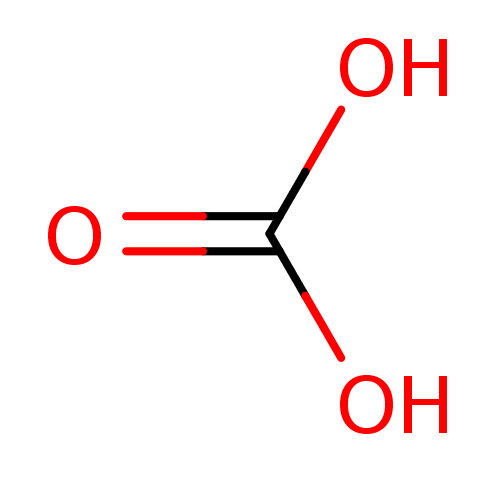 BDBM50147627 Bicarbonate Bicarbonate Ion CHEBI:28976 Carbonic Acid Anion Hydrogen Carbonate
BDBM50147627 Bicarbonate Bicarbonate Ion CHEBI:28976 Carbonic Acid Anion Hydrogen Carbonate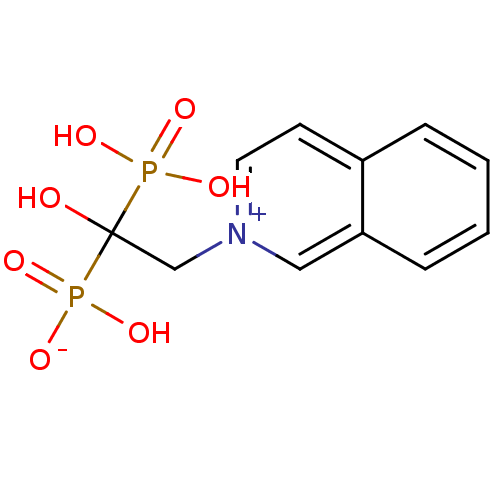 BDBM50165348 hydrogen 1-hydroxy-2-isoquinolinium-2-yl-1-phosphonoethylphosphonate CHEMBL425896
BDBM50165348 hydrogen 1-hydroxy-2-isoquinolinium-2-yl-1-phosphonoethylphosphonate CHEMBL425896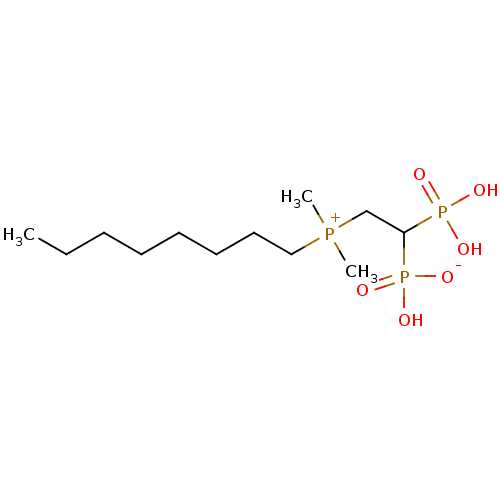 bisphosphonate, 22 hydrogen {2-[dimethyl(octyl)phosphanylium]-1-phosphonoethyl}phosphonate BDBM25268
bisphosphonate, 22 hydrogen {2-[dimethyl(octyl)phosphanylium]-1-phosphonoethyl}phosphonate BDBM25268 hydrogen(sulfide)(-1) sulfanide BDBM26990 HS anion HS(-1) hydrosulfide CHEMBL1644699
hydrogen(sulfide)(-1) sulfanide BDBM26990 HS anion HS(-1) hydrosulfide CHEMBL1644699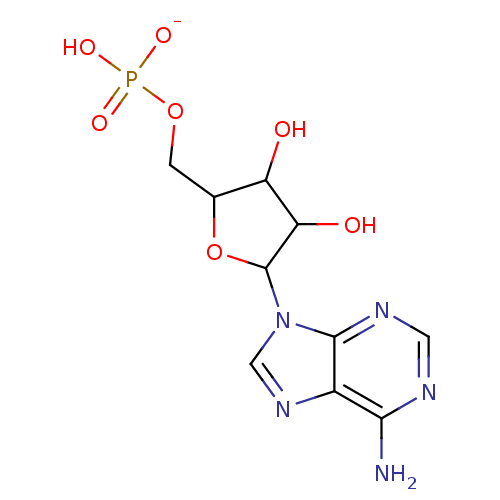 [5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl hydrogen phosphate;triethylazanium SMR000718807 [5-(6-aminopurin-9-yl)-3,4-dihydroxy-2-oxolanyl]methyl hydrogen phosphate;triethylammonium (5-adenin-9-yl-3,4-dihydroxy-tetrahydrofuran-2-yl)methyl hydrogen phosphate;triethylammonium BDBM61258 cid_16396156 MLS001306424 [5-(6-aminopurin-9-yl)-3,4-bis(oxidanyl)oxolan-2-yl]methyl hydrogen phosphate;triethylazanium
[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl hydrogen phosphate;triethylazanium SMR000718807 [5-(6-aminopurin-9-yl)-3,4-dihydroxy-2-oxolanyl]methyl hydrogen phosphate;triethylammonium (5-adenin-9-yl-3,4-dihydroxy-tetrahydrofuran-2-yl)methyl hydrogen phosphate;triethylammonium BDBM61258 cid_16396156 MLS001306424 [5-(6-aminopurin-9-yl)-3,4-bis(oxidanyl)oxolan-2-yl]methyl hydrogen phosphate;triethylazanium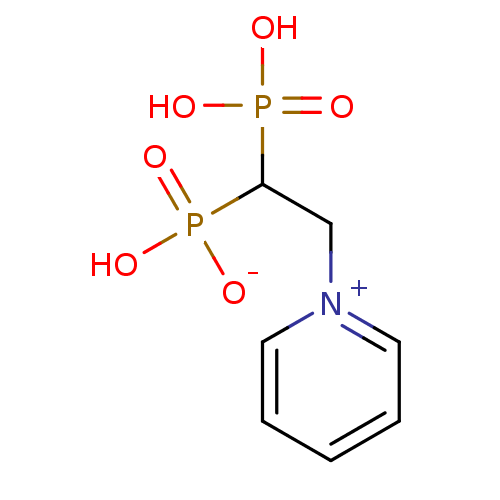 1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 60 BDBM25311
1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 60 BDBM25311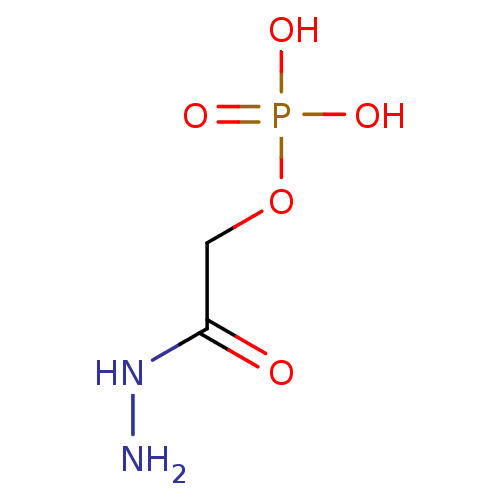 2-hydrazinyl-2-oxoethyl hydrogen phosphate BDBM50167772 Phosphoric acid monohydrazinocarbonylmethyl ester CHEMBL195520
2-hydrazinyl-2-oxoethyl hydrogen phosphate BDBM50167772 Phosphoric acid monohydrazinocarbonylmethyl ester CHEMBL195520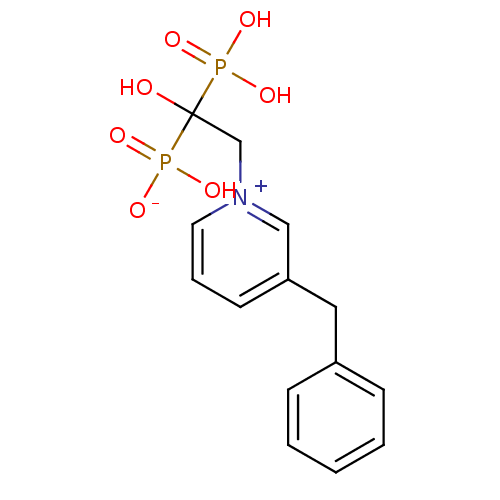 BDBM50165339 hydrogen 2-(3-benzylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL363434
BDBM50165339 hydrogen 2-(3-benzylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL363434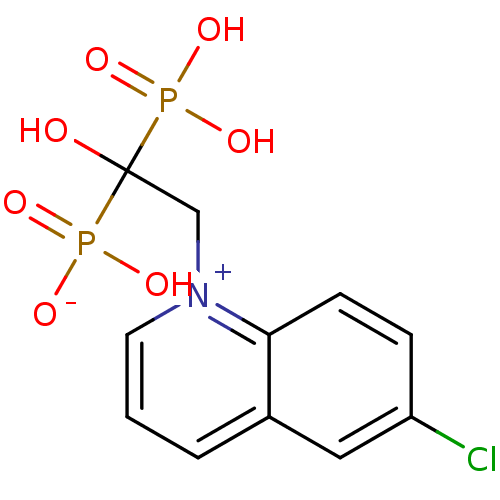 BDBM50165341 hydrogen 2-(6-chloroquinolinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL193131
BDBM50165341 hydrogen 2-(6-chloroquinolinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL193131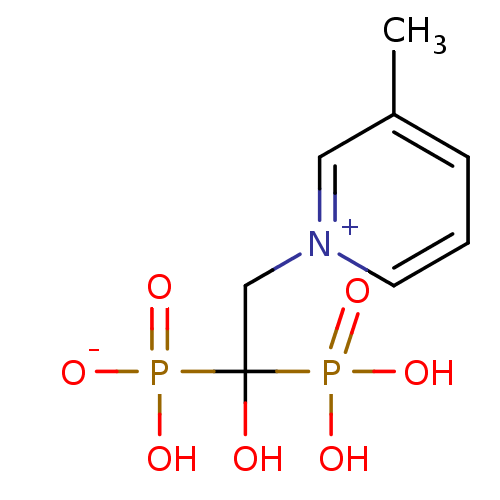 BDBM50165342 CHEMBL193356 hydrogen 1-hydroxy-2-(3-methylpyridinium-1-yl)-1-phosphonoethylphosphonate
BDBM50165342 CHEMBL193356 hydrogen 1-hydroxy-2-(3-methylpyridinium-1-yl)-1-phosphonoethylphosphonate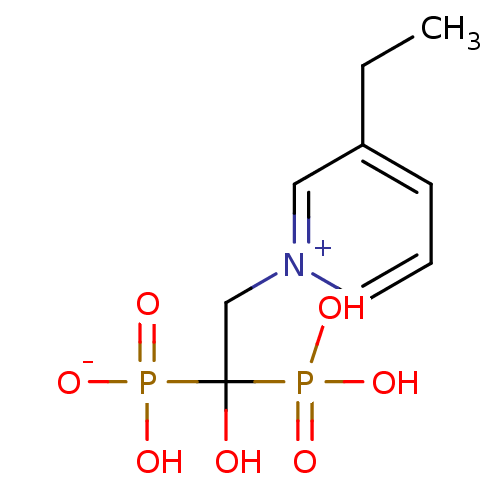 BDBM50165352 CHEMBL192043 hydrogen 2-(3-ethylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate
BDBM50165352 CHEMBL192043 hydrogen 2-(3-ethylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate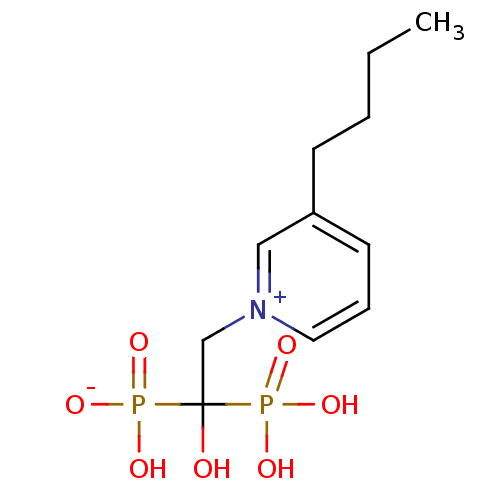 BDBM50165353 hydrogen 2-(3-butylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL373332
BDBM50165353 hydrogen 2-(3-butylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate CHEMBL373332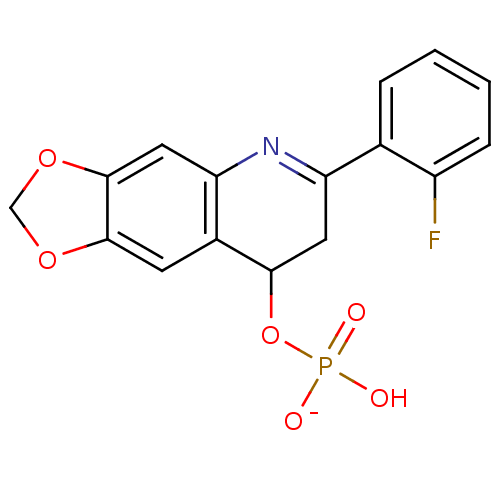 BDBM50312665 Sodium 2-(2-Fluorophenyl)-6,7-methylenedioxyquinolin-4-yl hydrogen phosphate CHEMBL1088572
BDBM50312665 Sodium 2-(2-Fluorophenyl)-6,7-methylenedioxyquinolin-4-yl hydrogen phosphate CHEMBL1088572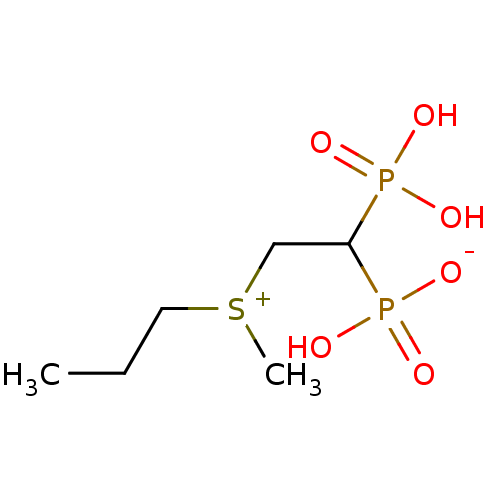 CHEMBL235059 bisphosphonate, 50 BDBM25303 hydrogen {2-[methyl(propyl)sulfanylium]-1-phosphonoethyl}phosphonate
CHEMBL235059 bisphosphonate, 50 BDBM25303 hydrogen {2-[methyl(propyl)sulfanylium]-1-phosphonoethyl}phosphonate CHEMBL235690 hydrogen {2-[methyl(octyl)sulfanylium]-1-phosphonoethyl}phosphonate BDBM25287 bisphosphonate, 37
CHEMBL235690 hydrogen {2-[methyl(octyl)sulfanylium]-1-phosphonoethyl}phosphonate BDBM25287 bisphosphonate, 37 CHEMBL238046 BDBM25272 hydrogen {2-[dodecyl(methyl)sulfanylium]-1-phosphonoethyl}phosphonate bisphosphonate, 25
CHEMBL238046 BDBM25272 hydrogen {2-[dodecyl(methyl)sulfanylium]-1-phosphonoethyl}phosphonate bisphosphonate, 25 bisphosphonate, 43 BDBM25294 CHEMBL392884 hydrogen {2-[methyl(pentyl)sulfanylium]-1-phosphonoethyl}phosphonate
bisphosphonate, 43 BDBM25294 CHEMBL392884 hydrogen {2-[methyl(pentyl)sulfanylium]-1-phosphonoethyl}phosphonate bisphosphonate, 52 hydrogen {2-[(10-carboxydecyl)(methyl)sulfanylium]-1-phosphonoethyl}phosphonate BDBM25305
bisphosphonate, 52 hydrogen {2-[(10-carboxydecyl)(methyl)sulfanylium]-1-phosphonoethyl}phosphonate BDBM25305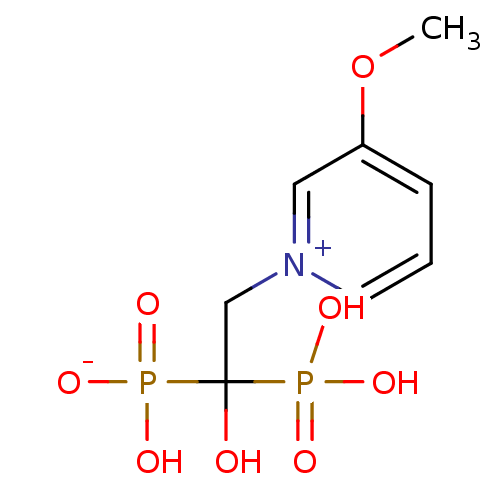 hydrogen 1-hydroxy-2-(3-methoxypyridinium-1-yl)-1-phosphonoethylphosphonate BDBM50165349 CHEMBL363145
hydrogen 1-hydroxy-2-(3-methoxypyridinium-1-yl)-1-phosphonoethylphosphonate BDBM50165349 CHEMBL363145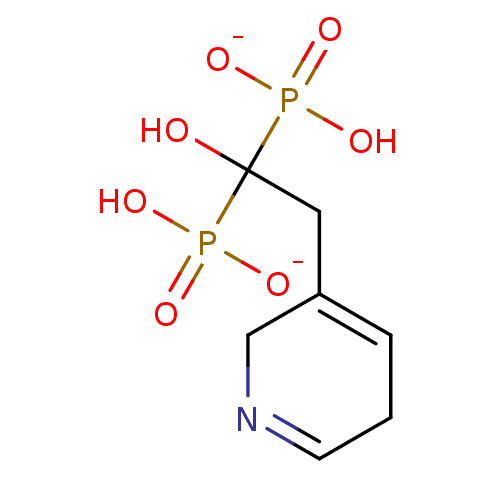 hydrogen 2-(1,2-dihydropyridin-3-yl)-1-hydroxy-1-(hydroxyphosphinato)ethylphosphonate BDBM50150875
hydrogen 2-(1,2-dihydropyridin-3-yl)-1-hydroxy-1-(hydroxyphosphinato)ethylphosphonate BDBM50150875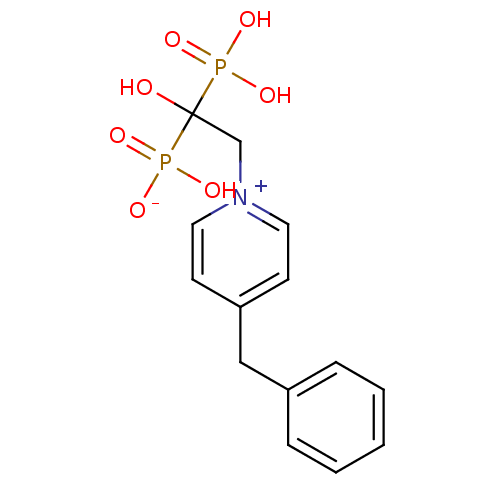 hydrogen 2-(4-benzylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate BDBM50165338 CHEMBL190258
hydrogen 2-(4-benzylpyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate BDBM50165338 CHEMBL190258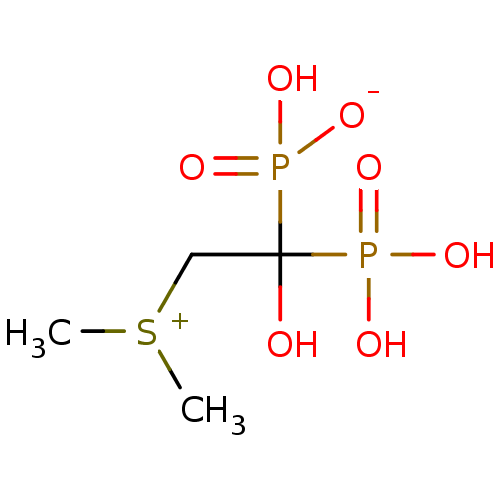 hydrogen [2-(dimethylsulfanylium)-1-hydroxy-1-phosphonoethyl]phosphonate bisphosphonate, 49 CHEMBL277580 BDBM25302
hydrogen [2-(dimethylsulfanylium)-1-hydroxy-1-phosphonoethyl]phosphonate bisphosphonate, 49 CHEMBL277580 BDBM25302 hydrogen {2-[methyl(tetradecyl)sulfanylium]-1-phosphonoethyl}phosphonate CHEMBL237808 bisphosphonate, 28 BDBM25275
hydrogen {2-[methyl(tetradecyl)sulfanylium]-1-phosphonoethyl}phosphonate CHEMBL237808 bisphosphonate, 28 BDBM25275 (R)-Fluoro-phenyl-methanephosphonic acid anion BDBM50129059 hydrogen [(R)-fluoro(phenyl)methyl]phosphonate
(R)-Fluoro-phenyl-methanephosphonic acid anion BDBM50129059 hydrogen [(R)-fluoro(phenyl)methyl]phosphonate BDBM235706 trans-(1R(S),6R(S))-6-Hydroxycyclohex-3-enyl hydrogen sulfate (4)
BDBM235706 trans-(1R(S),6R(S))-6-Hydroxycyclohex-3-enyl hydrogen sulfate (4) BDBM25296 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-phenylpyridin-1-ium bisphosphonate, 45
BDBM25296 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-phenylpyridin-1-ium bisphosphonate, 45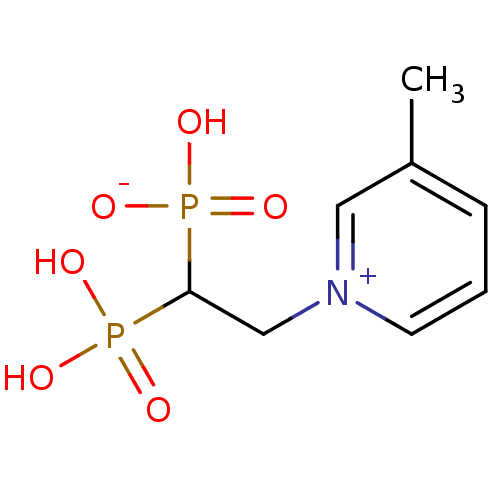 BDBM25301 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-methylpyridin-1-ium bisphosphonate, 48
BDBM25301 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-methylpyridin-1-ium bisphosphonate, 48 Blausaeure formonitrile CHEMBL183419 hydrogen cyanide hydridonitridocarbonhydrogen(nitridocarbonate)methanenitrile Cyanwasserstoff BDBM50152968 [CHN] hydrocyanic acid
Blausaeure formonitrile CHEMBL183419 hydrogen cyanide hydridonitridocarbonhydrogen(nitridocarbonate)methanenitrile Cyanwasserstoff BDBM50152968 [CHN] hydrocyanic acid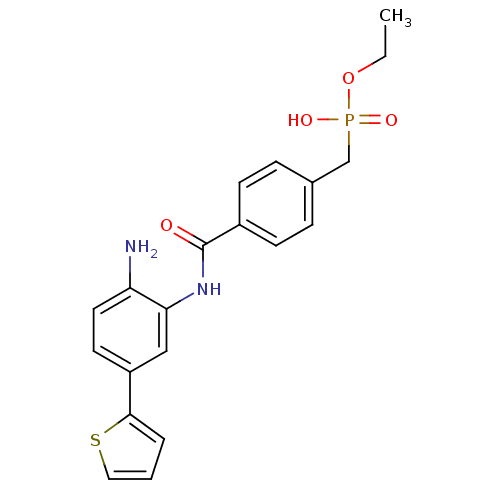 CHEMBL513014 ethyl hydrogen 4-(2-amino-5-(thiophen-2-yl)phenylcarbamoyl)benzylphosphonate BDBM50278306
CHEMBL513014 ethyl hydrogen 4-(2-amino-5-(thiophen-2-yl)phenylcarbamoyl)benzylphosphonate BDBM50278306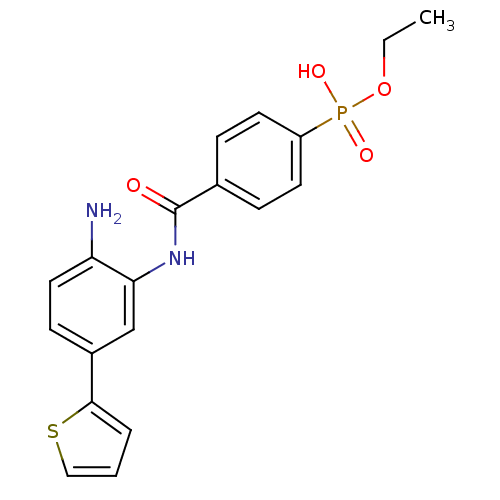 CHEMBL513255 ethyl hydrogen 4-(2-amino-5-(thiophen-2-yl)phenylcarbamoyl)phenylphosphonate BDBM50278250
CHEMBL513255 ethyl hydrogen 4-(2-amino-5-(thiophen-2-yl)phenylcarbamoyl)phenylphosphonate BDBM50278250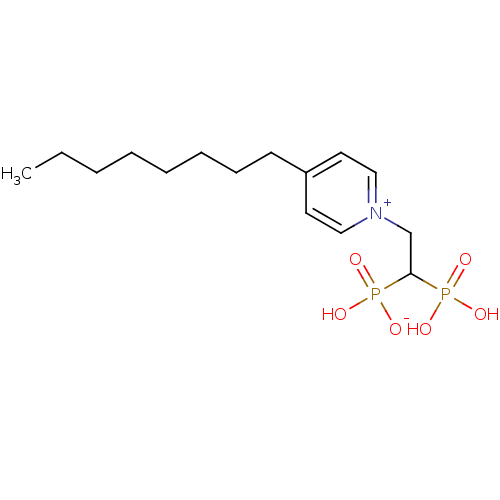 bisphosphonate, 20 BDBM25264 1-(2-hydrogen phosphonato-2-phosphonoethyl)-4-octylpyridin-1-ium
bisphosphonate, 20 BDBM25264 1-(2-hydrogen phosphonato-2-phosphonoethyl)-4-octylpyridin-1-ium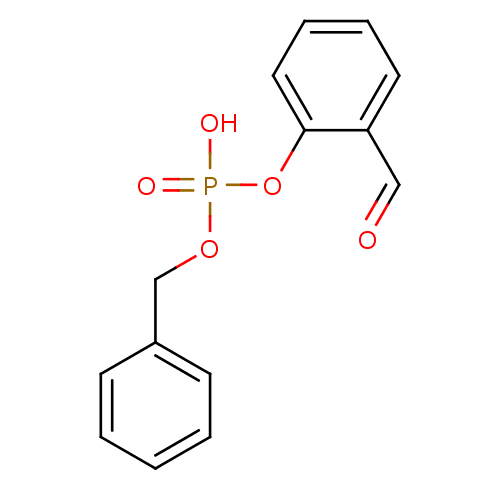 (benzyloxy)(2-formylphenoxy)phosphinic acid PASBN BDBM14688 benzyl 2-formylphenyl hydrogen phosphate Fragment 18
(benzyloxy)(2-formylphenoxy)phosphinic acid PASBN BDBM14688 benzyl 2-formylphenyl hydrogen phosphate Fragment 18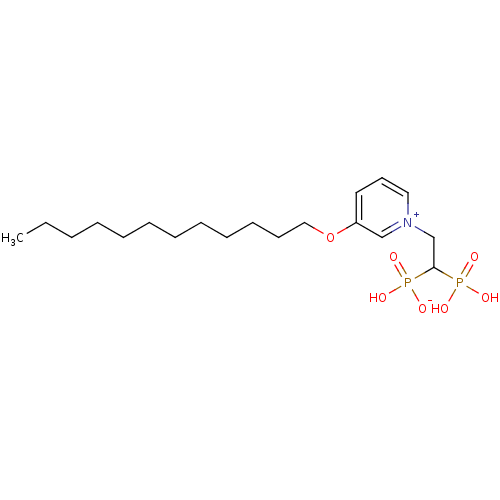 3-(dodecyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25286 bisphosphonate, 36
3-(dodecyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25286 bisphosphonate, 36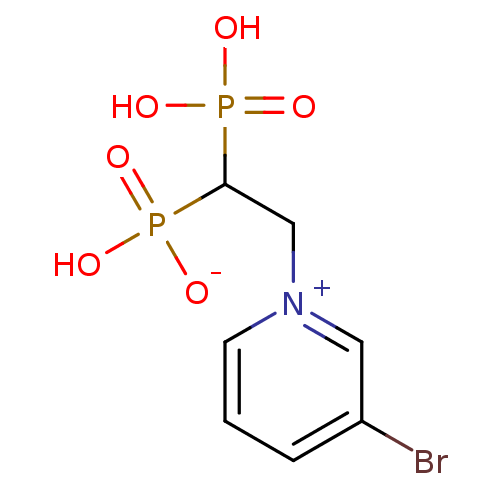 3-bromo-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25307 bisphosphonate, 56
3-bromo-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25307 bisphosphonate, 56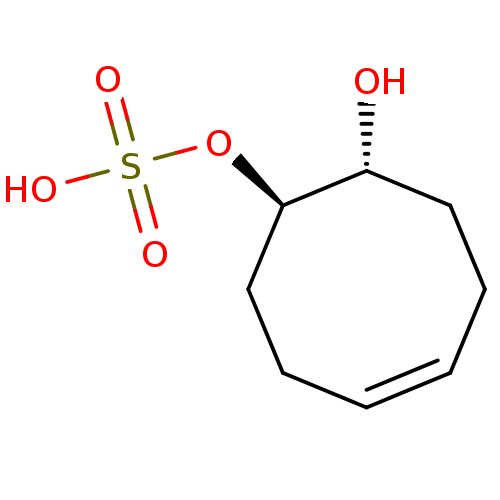 BDBM235708 trans-(1R(S),8R(S),Z)-8-Hydroxycyclooct-4-enyl hydrogen sulfate (6)
BDBM235708 trans-(1R(S),8R(S),Z)-8-Hydroxycyclooct-4-enyl hydrogen sulfate (6)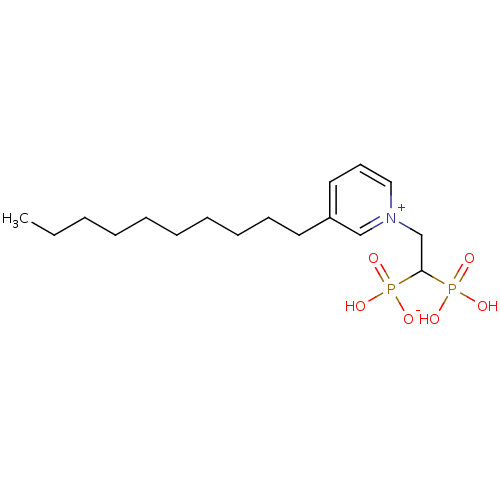 BDBM25261 3-decyl-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 17
BDBM25261 3-decyl-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 17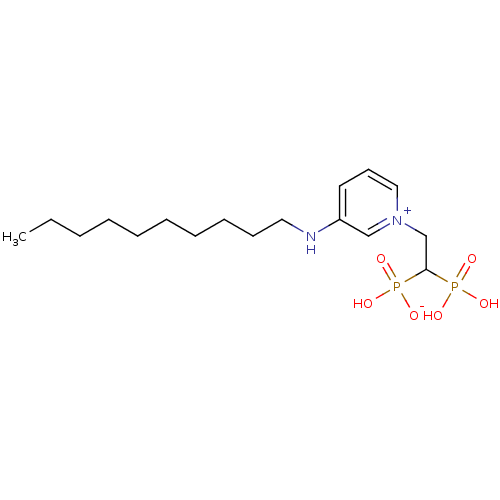 BDBM25273 3-(decylamino)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 26
BDBM25273 3-(decylamino)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 26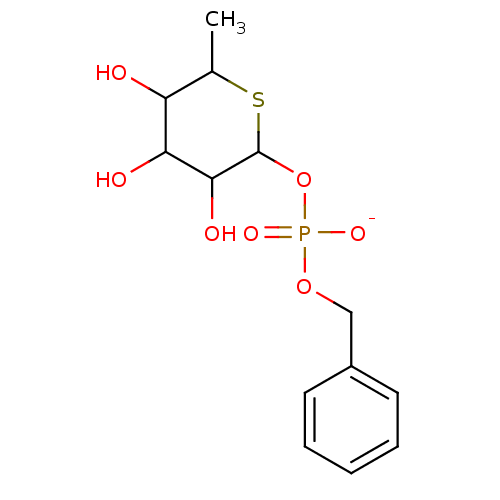 BDBM50287679 CHEMBL61555 Sodium; benzyl 3,4,5-trihydroxy-6-methyltetrahydro-2H-thiopyran-2-yl hydrogen phosphate
BDBM50287679 CHEMBL61555 Sodium; benzyl 3,4,5-trihydroxy-6-methyltetrahydro-2H-thiopyran-2-yl hydrogen phosphate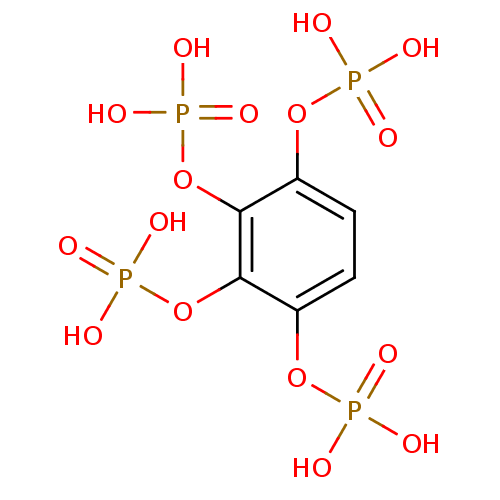 BDBM50304360 BENZENE-1,2,3,4-TETRAYL TETRAKIS[DIHYDROGEN (PHOSPHATE)] benzene-1,2,3,4-tetrayl tetrakis(hydrogen phosphate) CHEMBL595349
BDBM50304360 BENZENE-1,2,3,4-TETRAYL TETRAKIS[DIHYDROGEN (PHOSPHATE)] benzene-1,2,3,4-tetrayl tetrakis(hydrogen phosphate) CHEMBL595349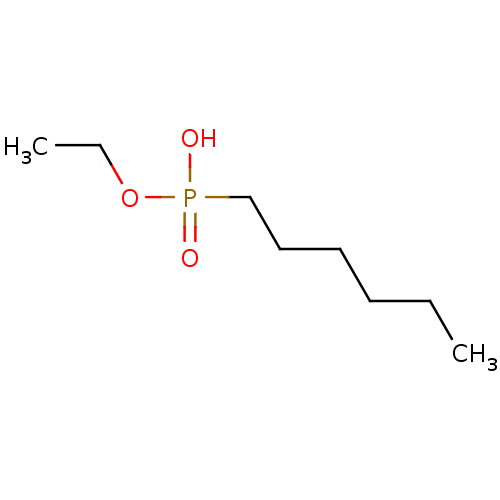 CHEMBL119521 Hexyl-phosphonic acid monoethyl ester N-HEXYLPHOSPHONATE ETHYL ESTER BDBM50148593 ethyl hydrogen hexylphosphonate
CHEMBL119521 Hexyl-phosphonic acid monoethyl ester N-HEXYLPHOSPHONATE ETHYL ESTER BDBM50148593 ethyl hydrogen hexylphosphonate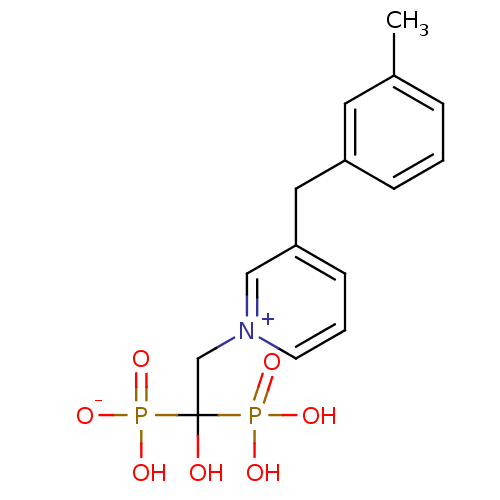 CHEMBL192938 BDBM50165350 hydrogen 1-hydroxy-2-[3-(3-methylbenzyl)pyridinium-1-yl]-1-phosphonoethylphosphonate
CHEMBL192938 BDBM50165350 hydrogen 1-hydroxy-2-[3-(3-methylbenzyl)pyridinium-1-yl]-1-phosphonoethylphosphonate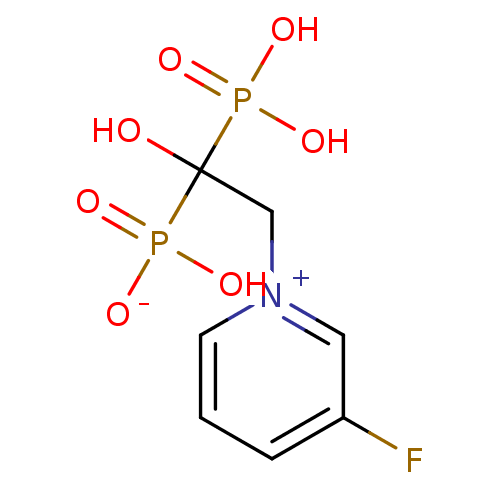 CHEMBL193722 BPH-461 hydrogen 2-(3-fluoropyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate BDBM50165340
CHEMBL193722 BPH-461 hydrogen 2-(3-fluoropyridinium-1-yl)-1-hydroxy-1-phosphonoethylphosphonate BDBM50165340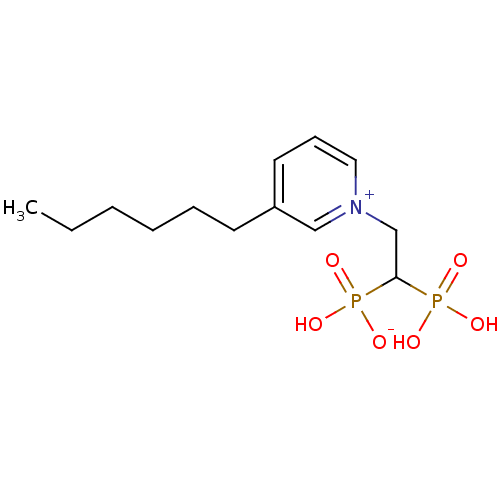 bisphosphonate, 42 BDBM25293 3-hexyl-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium
bisphosphonate, 42 BDBM25293 3-hexyl-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium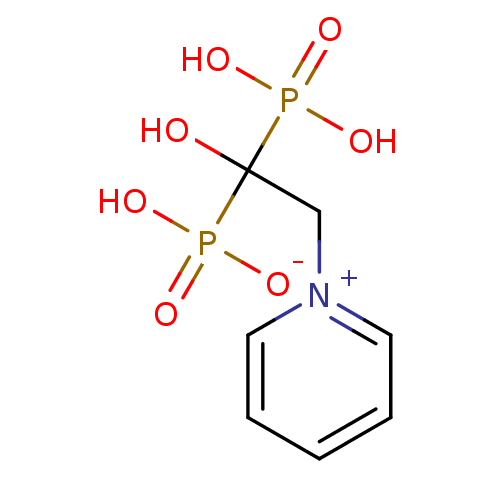 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)pyridin-1-ium CHEMBL191634 BDBM25310 bisphosphonate, 59
1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)pyridin-1-ium CHEMBL191634 BDBM25310 bisphosphonate, 59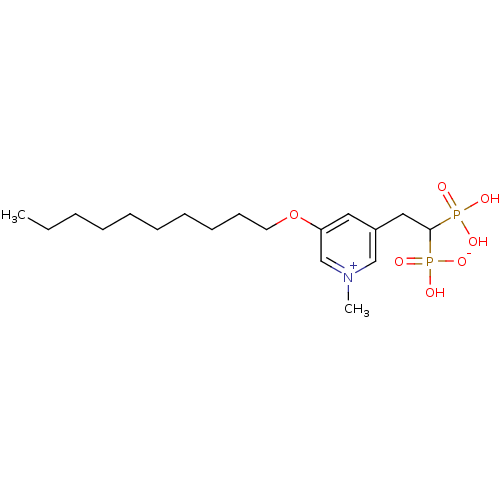 3-(decyloxy)-5-(2-hydrogen phosphonato-2-phosphonoethyl)-1-methylpyridin-1-ium BDBM25258 bisphosphonate, 14
3-(decyloxy)-5-(2-hydrogen phosphonato-2-phosphonoethyl)-1-methylpyridin-1-ium BDBM25258 bisphosphonate, 14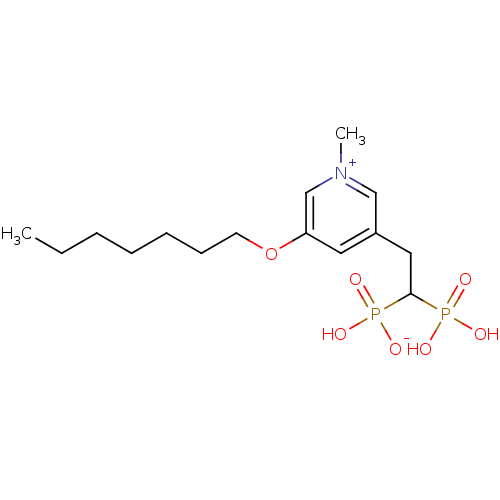 3-(heptyloxy)-5-(2-hydrogen phosphonato-2-phosphonoethyl)-1-methylpyridin-1-ium bisphosphonate, 30 BDBM25278
3-(heptyloxy)-5-(2-hydrogen phosphonato-2-phosphonoethyl)-1-methylpyridin-1-ium bisphosphonate, 30 BDBM25278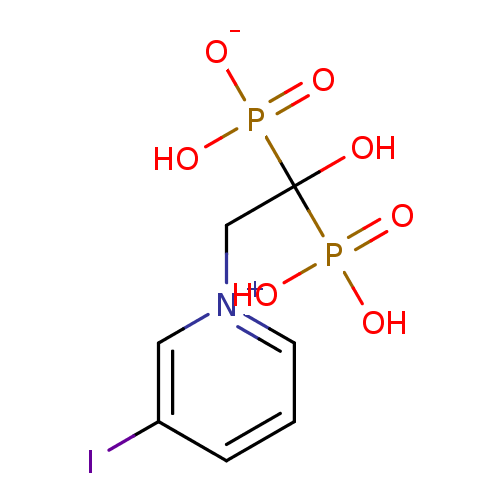 BDBM12580 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-iodopyridin-1-ium Bisphosphonate 5
BDBM12580 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-iodopyridin-1-ium Bisphosphonate 5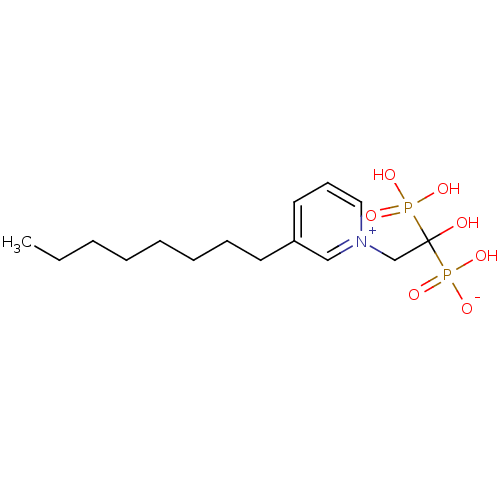 BDBM25274 bisphosphonate, 27 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-octylpyridin-1-ium
BDBM25274 bisphosphonate, 27 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-octylpyridin-1-ium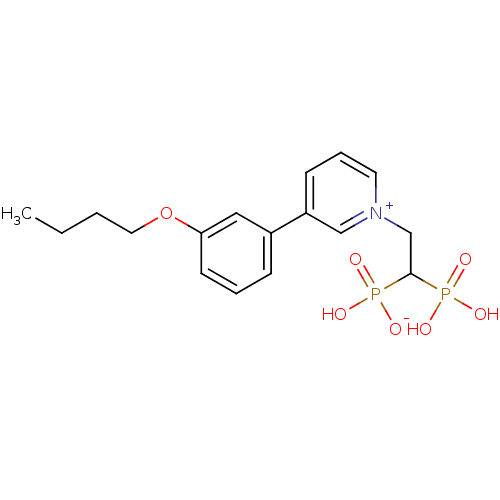 BDBM25283 3-(3-butoxyphenyl)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 35
BDBM25283 3-(3-butoxyphenyl)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 35 PAS219 (cyclohexylmethoxy)(2-formylphenoxy)phosphinic acid BDBM14685 CHEMBL24811 Fragment 15 cyclohexylmethyl 2-formylphenyl hydrogen phosphate
PAS219 (cyclohexylmethoxy)(2-formylphenoxy)phosphinic acid BDBM14685 CHEMBL24811 Fragment 15 cyclohexylmethyl 2-formylphenyl hydrogen phosphate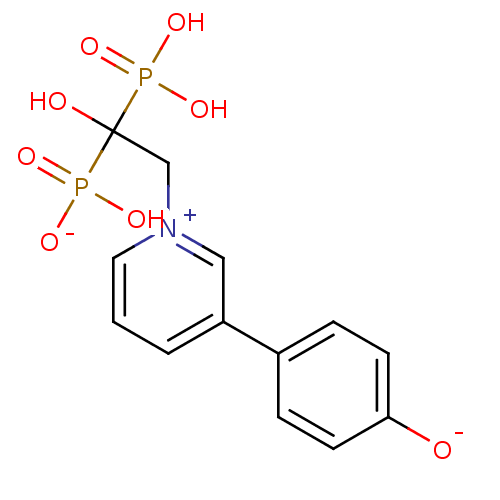 sodium hydrogen 1-hydroxy-2-[3-(4-oxidophenyl)pyridinium-1-yl]-1-phosphonoethylphosphonate CHEMBL193619 BDBM50165351
sodium hydrogen 1-hydroxy-2-[3-(4-oxidophenyl)pyridinium-1-yl]-1-phosphonoethylphosphonate CHEMBL193619 BDBM50165351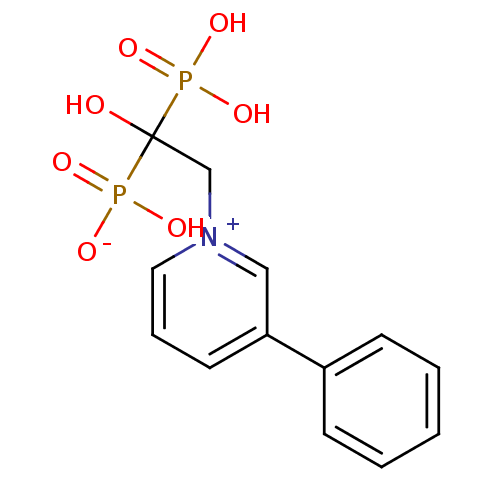 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-phenylpyridin-1-ium BDBM25299 bisphosphonate, 46 CHEMBL192342
1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-phenylpyridin-1-ium BDBM25299 bisphosphonate, 46 CHEMBL192342 BDBM25259 Bisphpshonate-715 3-(decyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 15
BDBM25259 Bisphpshonate-715 3-(decyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 15 BDBM25262 bisphosphonate, 18 3-bromo-5-(decyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium
BDBM25262 bisphosphonate, 18 3-bromo-5-(decyloxy)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25271 3-[(3,7-dimethyloctyl)oxy]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 24
BDBM25271 3-[(3,7-dimethyloctyl)oxy]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium bisphosphonate, 24 BDBM25282 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(octane-1-sulfonamido)pyridin-1-ium bisphosphonate, 34
BDBM25282 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(octane-1-sulfonamido)pyridin-1-ium bisphosphonate, 34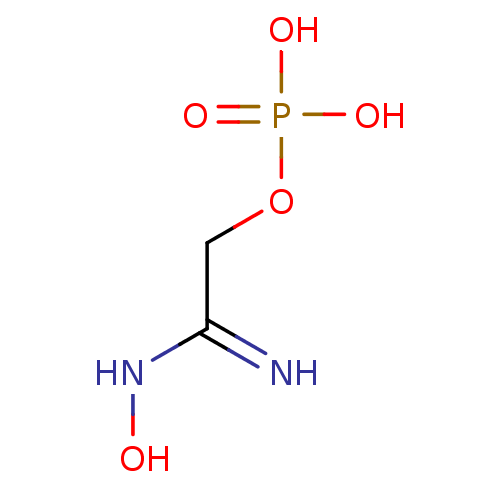 CHEMBL195976 Phosphoric acid mono-(N-hydroxycarbamimidoylmethyl) ester BDBM50167775 (Z)-2-amino-2-(hydroxyimino)ethyl hydrogen phosphate
CHEMBL195976 Phosphoric acid mono-(N-hydroxycarbamimidoylmethyl) ester BDBM50167775 (Z)-2-amino-2-(hydroxyimino)ethyl hydrogen phosphate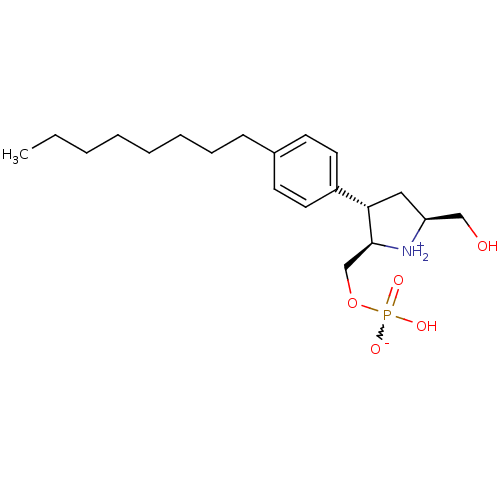 (2R,3S,5S)-2-[(hydrogen phosphonatooxy)methyl]-5-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium BDBM50197422 CHEMBL247901
(2R,3S,5S)-2-[(hydrogen phosphonatooxy)methyl]-5-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium BDBM50197422 CHEMBL247901 (2S,3R,5R)-2-[(hydrogen phosphonatooxy)methyl]-5-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium CHEMBL247698 BDBM50197421
(2S,3R,5R)-2-[(hydrogen phosphonatooxy)methyl]-5-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium CHEMBL247698 BDBM50197421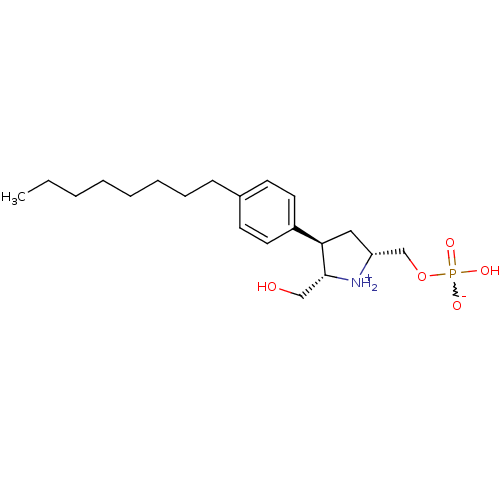 (2S,3R,5R)-5-[(hydrogen phosphonatooxy)methyl]-2-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium CHEMBL393055 BDBM50197420
(2S,3R,5R)-5-[(hydrogen phosphonatooxy)methyl]-2-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium CHEMBL393055 BDBM50197420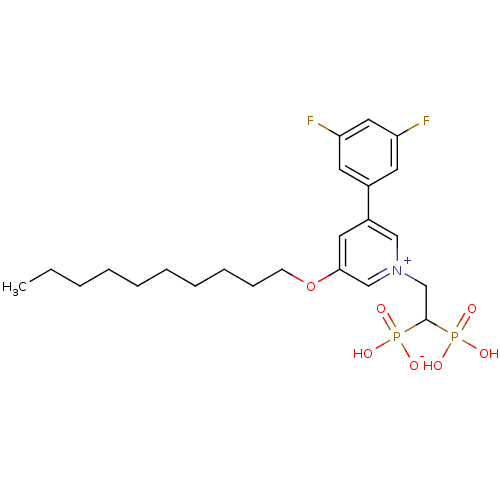 3-(decyloxy)-5-(3,5-difluorophenyl)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25267 bisphosphonate, 13
3-(decyloxy)-5-(3,5-difluorophenyl)-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium BDBM25267 bisphosphonate, 13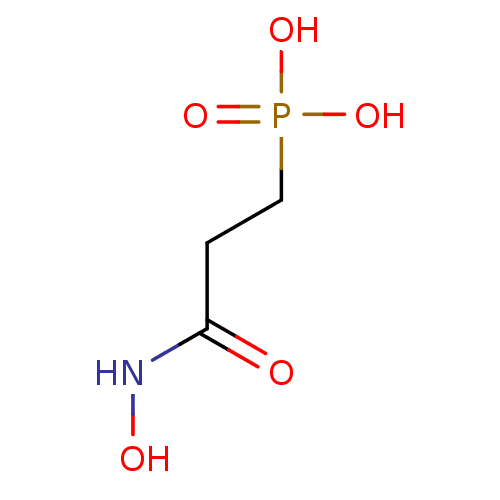 3-(hydroxyamino)-3-oxopropylphosphonic acid BDBM50167776 hydrogen 3-(hydroxyamino)-3-oxopropylphosphonate (2-Hydroxycarbamoyl-ethyl)-phosphonic acid CHEMBL196442
3-(hydroxyamino)-3-oxopropylphosphonic acid BDBM50167776 hydrogen 3-(hydroxyamino)-3-oxopropylphosphonate (2-Hydroxycarbamoyl-ethyl)-phosphonic acid CHEMBL196442 CHEMBL247699 (2R,3S,5S)-5-[(hydrogen phosphonatooxy)methyl]-2-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium BDBM50197424
CHEMBL247699 (2R,3S,5S)-5-[(hydrogen phosphonatooxy)methyl]-2-(hydroxymethyl)-3-(4-octylphenyl)pyrrolidin-1-ium BDBM50197424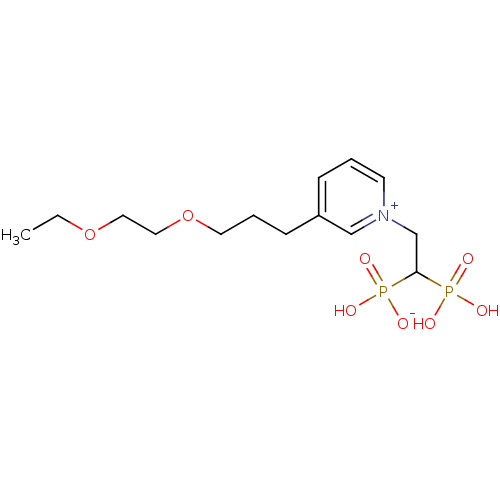 bisphosphonate, 54 BDBM25306 3-[3-(2-ethoxyethoxy)propyl]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium
bisphosphonate, 54 BDBM25306 3-[3-(2-ethoxyethoxy)propyl]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium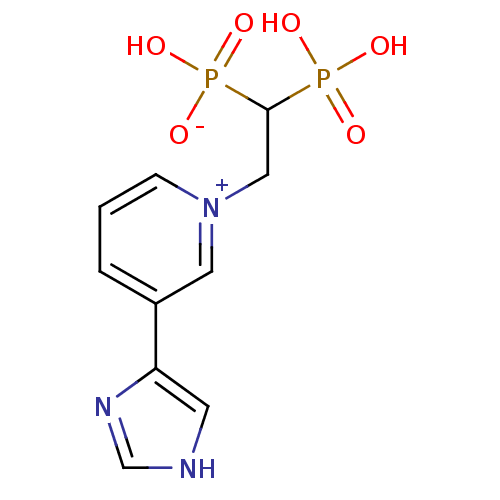 bisphosphonate, 63 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(1H-imidazol-5-yl)pyridin-1-ium BDBM25312
bisphosphonate, 63 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(1H-imidazol-5-yl)pyridin-1-ium BDBM25312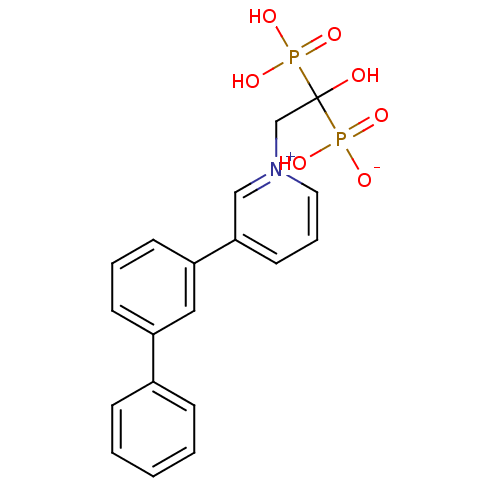 1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-(3-phenylphenyl)pyridin-1-ium CHEMBL192017 bisphosphonate, 7 BDBM25297
1-(2-hydrogen phosphonato-2-hydroxy-2-phosphonoethyl)-3-(3-phenylphenyl)pyridin-1-ium CHEMBL192017 bisphosphonate, 7 BDBM25297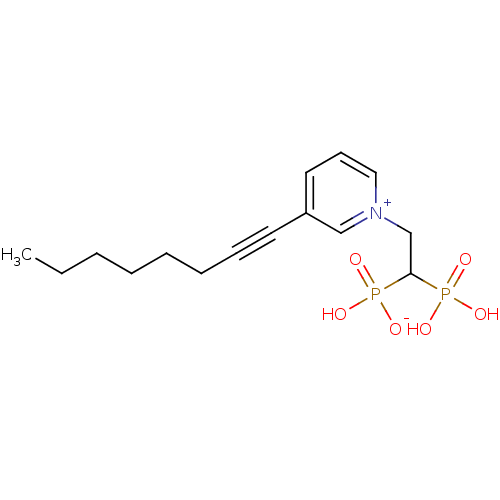 1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(oct-1-yn-1-yl)pyridin-1-ium bisphosphonate, 29 BDBM25276
1-(2-hydrogen phosphonato-2-phosphonoethyl)-3-(oct-1-yn-1-yl)pyridin-1-ium bisphosphonate, 29 BDBM25276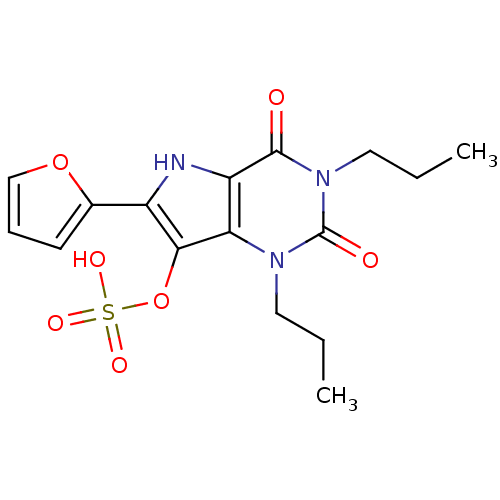 BDBM50273310 CHEMBL508090 6-(2-Furyl)-2,4dioxo-1,3-dipropyl-2,3,4,5-tetrahydro-1H-pyrrolo[3,2-d]pyrimidin-7-yl hydrogen sulfate
BDBM50273310 CHEMBL508090 6-(2-Furyl)-2,4dioxo-1,3-dipropyl-2,3,4,5-tetrahydro-1H-pyrrolo[3,2-d]pyrimidin-7-yl hydrogen sulfate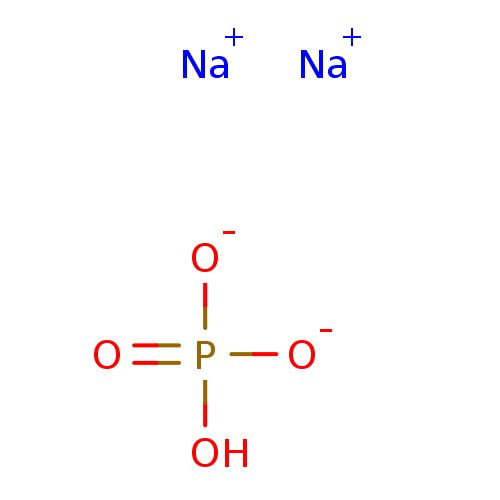 disodium monohydrogen phosphate Na2HPO4 disodium hydrogen phosphate Disodium phosphate disodium acid orthophosphate BDBM50080995 disodium hydrogenphosphate Dibasic sodium phosphate CHEMBL1060
disodium monohydrogen phosphate Na2HPO4 disodium hydrogen phosphate Disodium phosphate disodium acid orthophosphate BDBM50080995 disodium hydrogenphosphate Dibasic sodium phosphate CHEMBL1060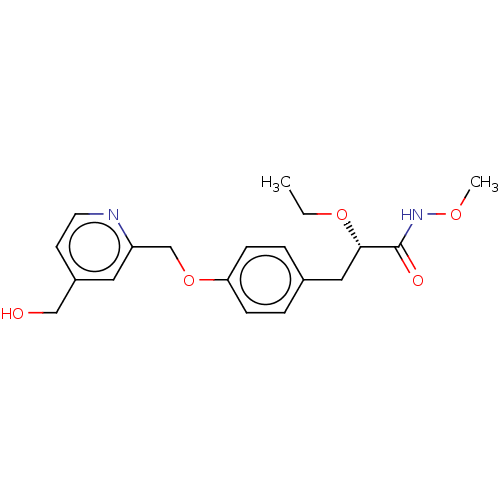 (2s)-2-ethoxy-n-hydroxy-3-[4-(pyridin-2-ylmethoxy)phenyl]propanamide hydrogen chloride (compound no. 1) US9562012, 5 BDBM228135
(2s)-2-ethoxy-n-hydroxy-3-[4-(pyridin-2-ylmethoxy)phenyl]propanamide hydrogen chloride (compound no. 1) US9562012, 5 BDBM228135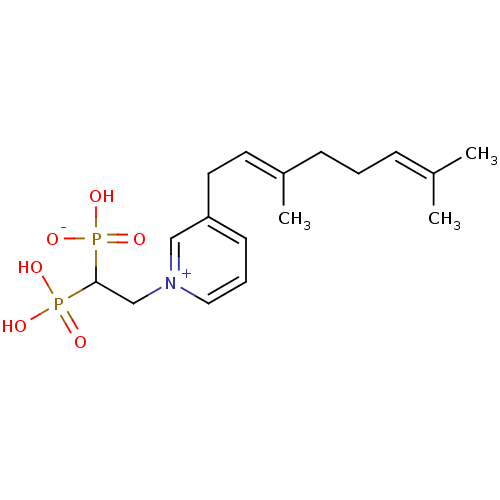 BDBM25295 bisphosphonate, 44 3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium
BDBM25295 bisphosphonate, 44 3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium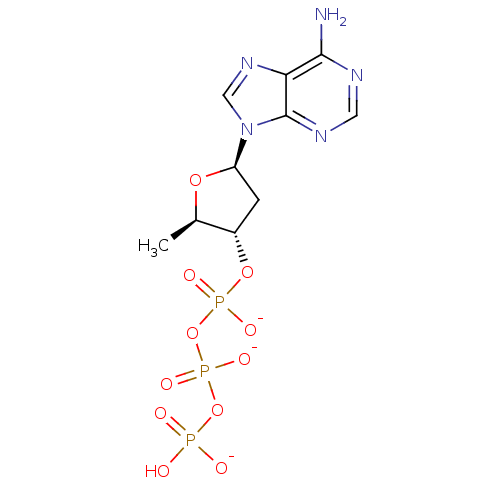 {[(2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-methyloxolan-3-yl phosphonato]oxy}(hydrogen phosphonatooxy)phosphinate BDBM50262191
{[(2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-methyloxolan-3-yl phosphonato]oxy}(hydrogen phosphonatooxy)phosphinate BDBM50262191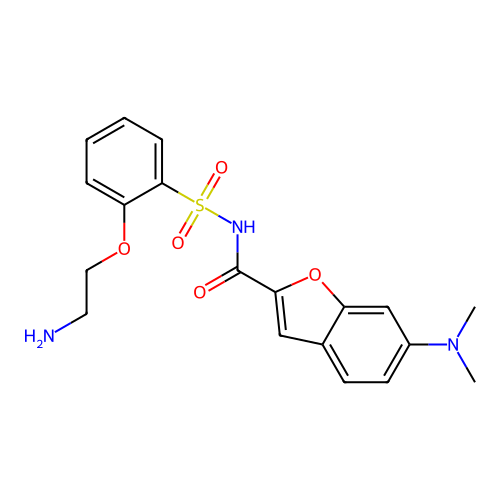 N-[2-(2-Aminoethoxy)benzene-1-sulfonyl]-6-(dimethylamino)-1-benzofuran-2-carboxamide hydrogen chloride (1/1) BDBM757529 US12357603, Example 167
N-[2-(2-Aminoethoxy)benzene-1-sulfonyl]-6-(dimethylamino)-1-benzofuran-2-carboxamide hydrogen chloride (1/1) BDBM757529 US12357603, Example 167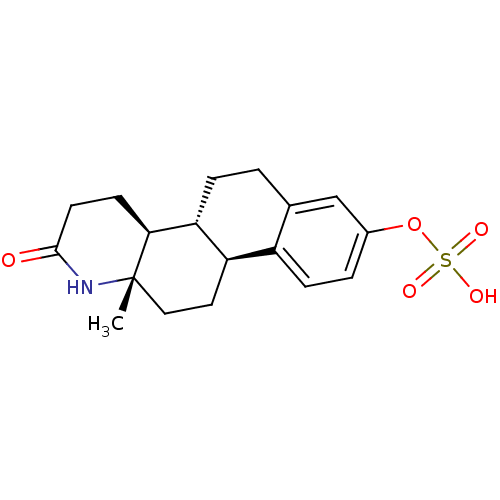 (4aS,4bR,10bS,12aS)-12a-methyl-2-oxo-1,2,3,4,4a,4b,5,6,10b,11,12,12a-dodecahydronaphtho[2,1-f]quinolin-8-yl hydrogen sulfate CHEMBL1642920 BDBM50334791
(4aS,4bR,10bS,12aS)-12a-methyl-2-oxo-1,2,3,4,4a,4b,5,6,10b,11,12,12a-dodecahydronaphtho[2,1-f]quinolin-8-yl hydrogen sulfate CHEMBL1642920 BDBM50334791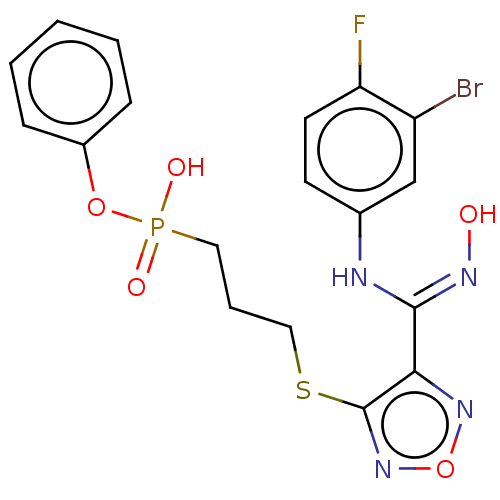 BDBM429929 US10538497, Example 118 phenyl hydrogen [3-({4-[N-(3- bromo-4-fluorophenyl)-N'- hydroxycarbamimidoyl]-1,2,5- oxadiazol-3- yl}sulfanyl)propyl]phosphonate
BDBM429929 US10538497, Example 118 phenyl hydrogen [3-({4-[N-(3- bromo-4-fluorophenyl)-N'- hydroxycarbamimidoyl]-1,2,5- oxadiazol-3- yl}sulfanyl)propyl]phosphonate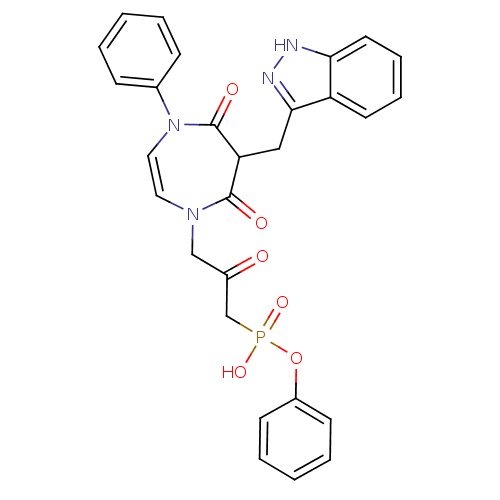 BDBM50308328 CHEMBL598208 (+/-)-Phenyl hydrogen{[6-(1H-indazol-3-ylmethyl)-5,7-dioxo-4-phenyl-4,5,6,7-tetrahydro-1H-1,4-diazepin-1-yl]methyl}phosphonate
BDBM50308328 CHEMBL598208 (+/-)-Phenyl hydrogen{[6-(1H-indazol-3-ylmethyl)-5,7-dioxo-4-phenyl-4,5,6,7-tetrahydro-1H-1,4-diazepin-1-yl]methyl}phosphonate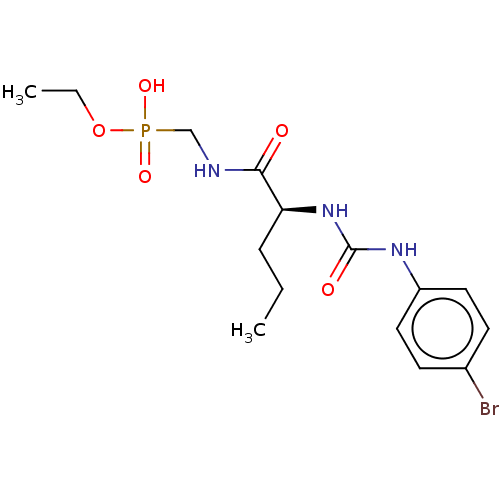 US10799518, Compound 123 US10434112, Compound 123 BDBM350661 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino} pentanoyl]amino}methyl) phosphonate US10208071, Compound 123
US10799518, Compound 123 US10434112, Compound 123 BDBM350661 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino} pentanoyl]amino}methyl) phosphonate US10208071, Compound 123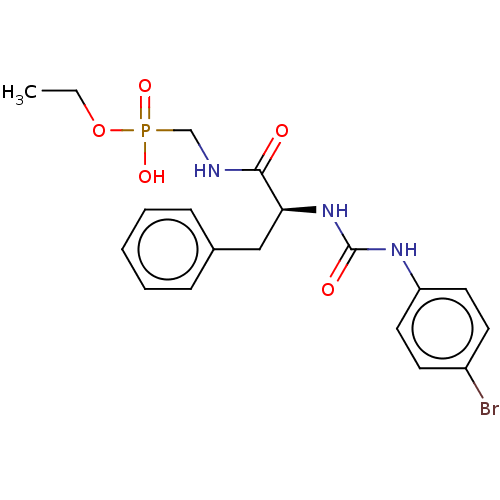 BDBM350664 US10208071, Compound 126 US10434112, Compound 126 US10799518, Compound 126 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino}- 3-phenylpropanoyl]amino} methyl)phosphonate
BDBM350664 US10208071, Compound 126 US10434112, Compound 126 US10799518, Compound 126 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino}- 3-phenylpropanoyl]amino} methyl)phosphonate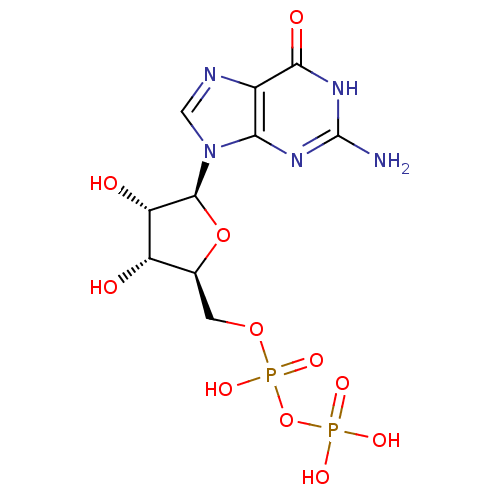 BDBM50300554 ((2S,3R,4S,5S)-5-(2-amino-6-oxo-1H-purin-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen diphosphate CHEMBL574200
BDBM50300554 ((2S,3R,4S,5S)-5-(2-amino-6-oxo-1H-purin-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen diphosphate CHEMBL574200 NaHCO3 sodium hydrogen carbonate bicarbonate of soda SODIUM BICARBONATE BDBM50155533 carbonic acid monosodium salt sodium hydrogencarbonate CHEMBL1353 sodium acid carbonate baking soda Natriumhydrogenkarbonat
NaHCO3 sodium hydrogen carbonate bicarbonate of soda SODIUM BICARBONATE BDBM50155533 carbonic acid monosodium salt sodium hydrogencarbonate CHEMBL1353 sodium acid carbonate baking soda Natriumhydrogenkarbonat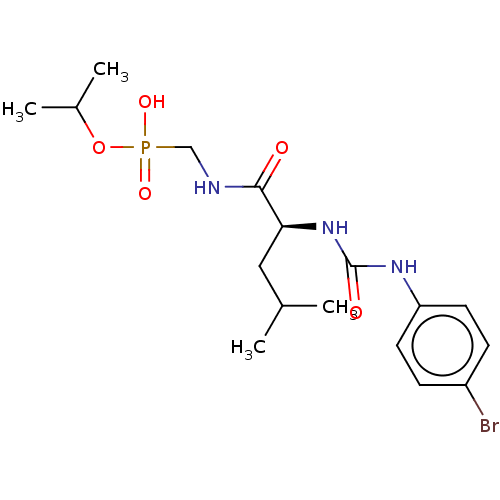 US10208071, Compound 129 BDBM350667 US10799518, Compound 129 US10434112, Compound 129 propan-2-yl hydrogen {[(2- {[(4- bromophenyl)carbamoyl]amino} pentanoyl)amino]methyl} phosphonate
US10208071, Compound 129 BDBM350667 US10799518, Compound 129 US10434112, Compound 129 propan-2-yl hydrogen {[(2- {[(4- bromophenyl)carbamoyl]amino} pentanoyl)amino]methyl} phosphonate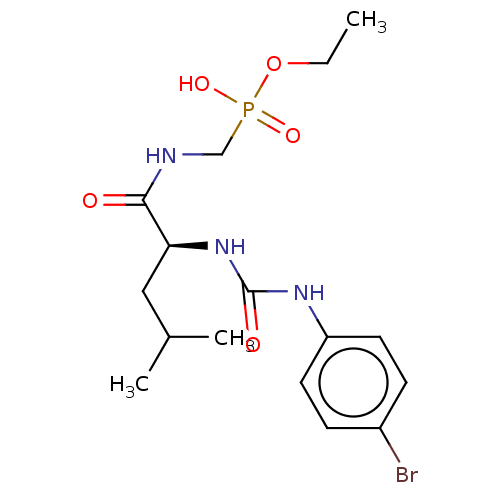 US10799518, Compound 105 US10434112, Compound 105 BDBM350643 US10208071, Compound 105 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino}- 4-methylpentanoyl]amino} methyl)phosphonate
US10799518, Compound 105 US10434112, Compound 105 BDBM350643 US10208071, Compound 105 ethyl hydrogen ({[(2S)-2-{[(4- bromophenyl)carbamoyl]amino}- 4-methylpentanoyl]amino} methyl)phosphonate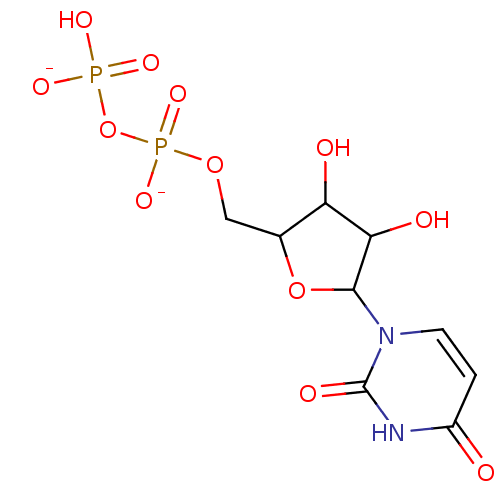 hydrogen ({[5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphonato}oxy)phosphonate BDBM50096295 Uridine dinucleoside 5'-polyphosphate analogue Uridine
hydrogen ({[5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphonato}oxy)phosphonate BDBM50096295 Uridine dinucleoside 5'-polyphosphate analogue Uridine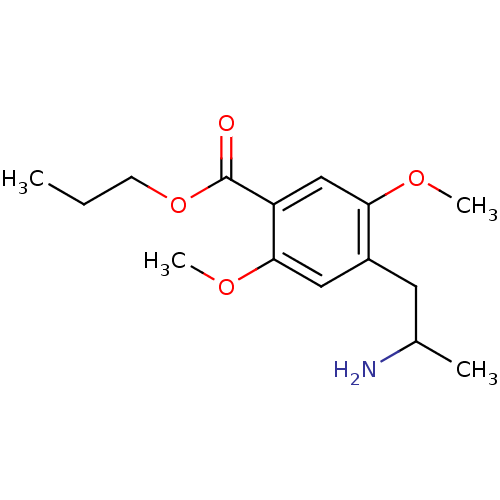 4-(2-Amino-propyl)-2,5-dimethoxy-benzoic acid propyl ester 4-(2-Amino-propyl)-2,5-dimethoxy-benzoic acid propyl ester : (Hydrogen oxalate) BDBM50005263 CHEMBL273251
4-(2-Amino-propyl)-2,5-dimethoxy-benzoic acid propyl ester 4-(2-Amino-propyl)-2,5-dimethoxy-benzoic acid propyl ester : (Hydrogen oxalate) BDBM50005263 CHEMBL273251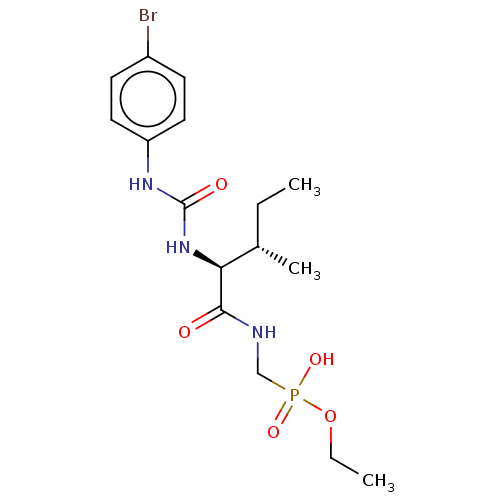 BDBM350653 US10208071, Compound 115 US10434112, Compound 115 US10799518, Compound 115 ethyl hydrogen ({[(2S,3S)-2- {[(4- bromophenyl)carbamoyl]amino}- 3-methylpentanoyl]amino} methyl)phosphonate
BDBM350653 US10208071, Compound 115 US10434112, Compound 115 US10799518, Compound 115 ethyl hydrogen ({[(2S,3S)-2- {[(4- bromophenyl)carbamoyl]amino}- 3-methylpentanoyl]amino} methyl)phosphonate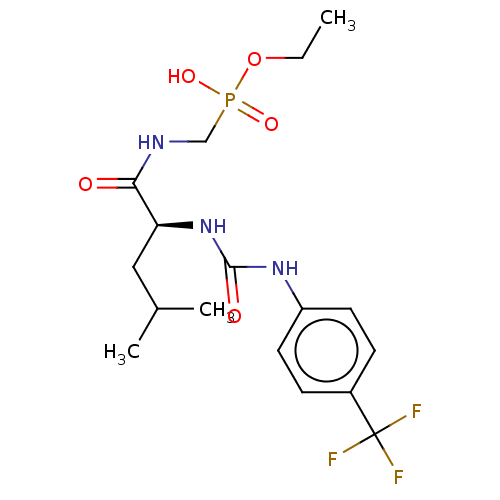 BDBM350673 US10434112, Compound 135 US10799518, Compound 135 US10208071, Compound 135 ethyl hydrogen ({[(2S)-4- methyl-2-({[4- (trifluoromethyl)phenyl]carbamoyl} amino)pentanoyl]amino} methyl)phosphonate
BDBM350673 US10434112, Compound 135 US10799518, Compound 135 US10208071, Compound 135 ethyl hydrogen ({[(2S)-4- methyl-2-({[4- (trifluoromethyl)phenyl]carbamoyl} amino)pentanoyl]amino} methyl)phosphonate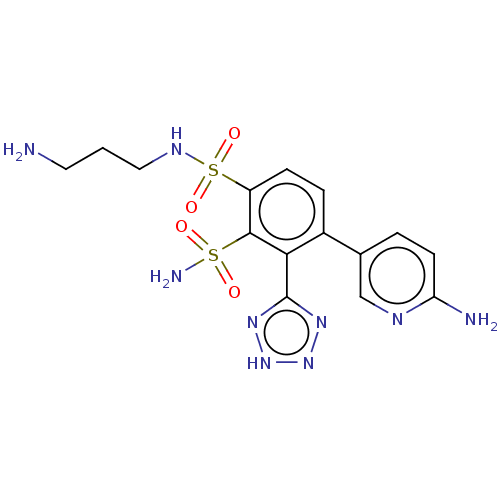 US10544130, Example 496 US10221163, Example 496 BDBM361339 N1-(3-aminopropyl)-4-(6- aminopyridin-3-yl)-3-(2H- tetrazol-5-yl)benzene-1,2- disulfonamide hydrogen chloride
US10544130, Example 496 US10221163, Example 496 BDBM361339 N1-(3-aminopropyl)-4-(6- aminopyridin-3-yl)-3-(2H- tetrazol-5-yl)benzene-1,2- disulfonamide hydrogen chloride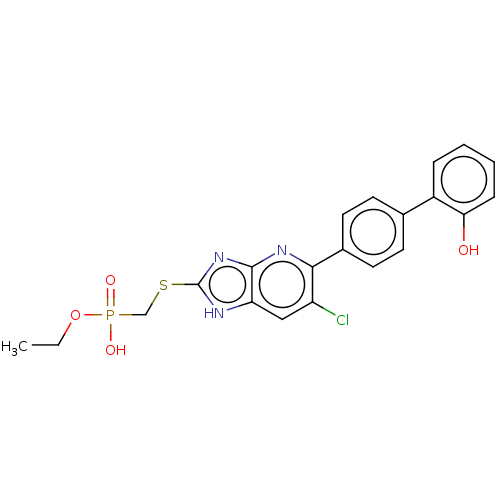 ethyl hydrogen (((6-chloro-5-(2'-hydroxy- [1,1'-biphenyl]-4-yl)-1H-imidazo[4,5- b]pyridin-2-yl)thio)methyl)phosphonate BDBM543472 US11279702, Compound 3
ethyl hydrogen (((6-chloro-5-(2'-hydroxy- [1,1'-biphenyl]-4-yl)-1H-imidazo[4,5- b]pyridin-2-yl)thio)methyl)phosphonate BDBM543472 US11279702, Compound 3 [5-[5-fluoranyl-2,4-bis(oxidanylidene)pyrimidin-1-yl]-3-oxidanyl-oxolan-2-yl]methyl [5-[4-(octadecylamino)-2-oxidanylidene-pyrimidin-1-yl]-3,4-bis(oxidanyl)oxolan-2-yl]methyl hydrogen phosphate cid_392613 SMR001565974 [3,4-dihydroxy-5-[2-keto-4-(stearylamino)pyrimidin-1-yl]tetrahydrofuran-2-yl]methyl [5-(5-fluoro-2,4-diketo-pyrimidin-1-yl)-3-hydroxy-tetrahydrofuran-2-yl]methyl hydrogen phosphate MLS002702412 [3,4-dihydroxy-5-[4-(octadecylamino)-2-oxopyrimidin-1-yl]oxolan-2-yl]methyl [5-(5-fluoro-2,4-dioxopyrimidin-1-yl)-3-hydroxyoxolan-2-yl]methyl hydrogen phosphate [3,4-dihydroxy-5-[4-(octadecylamino)-2-oxo-1-pyrimidinyl]-2-oxolanyl]methyl [5-(5-fluoro-2,4-dioxo-1-pyrimidinyl)-3-hydroxy-2-oxolanyl]methyl hydrogen phosphate BDBM80994
[5-[5-fluoranyl-2,4-bis(oxidanylidene)pyrimidin-1-yl]-3-oxidanyl-oxolan-2-yl]methyl [5-[4-(octadecylamino)-2-oxidanylidene-pyrimidin-1-yl]-3,4-bis(oxidanyl)oxolan-2-yl]methyl hydrogen phosphate cid_392613 SMR001565974 [3,4-dihydroxy-5-[2-keto-4-(stearylamino)pyrimidin-1-yl]tetrahydrofuran-2-yl]methyl [5-(5-fluoro-2,4-diketo-pyrimidin-1-yl)-3-hydroxy-tetrahydrofuran-2-yl]methyl hydrogen phosphate MLS002702412 [3,4-dihydroxy-5-[4-(octadecylamino)-2-oxopyrimidin-1-yl]oxolan-2-yl]methyl [5-(5-fluoro-2,4-dioxopyrimidin-1-yl)-3-hydroxyoxolan-2-yl]methyl hydrogen phosphate [3,4-dihydroxy-5-[4-(octadecylamino)-2-oxo-1-pyrimidinyl]-2-oxolanyl]methyl [5-(5-fluoro-2,4-dioxo-1-pyrimidinyl)-3-hydroxy-2-oxolanyl]methyl hydrogen phosphate BDBM80994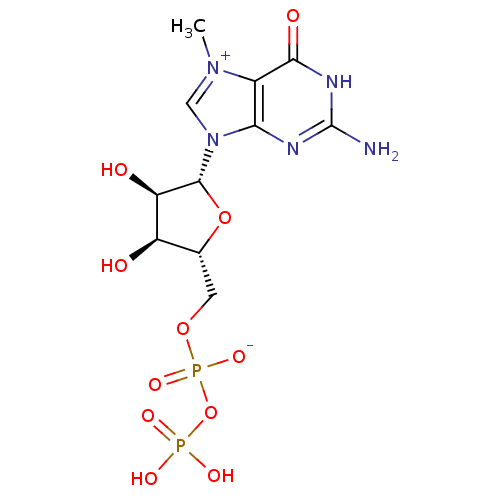 ((2R,3S,4R,5R)-5-(2-amino-7-methyl-6-oxo-1H-purin-1-ium-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen diphosphate CHEMBL1094974 BDBM50316303
((2R,3S,4R,5R)-5-(2-amino-7-methyl-6-oxo-1H-purin-1-ium-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen diphosphate CHEMBL1094974 BDBM50316303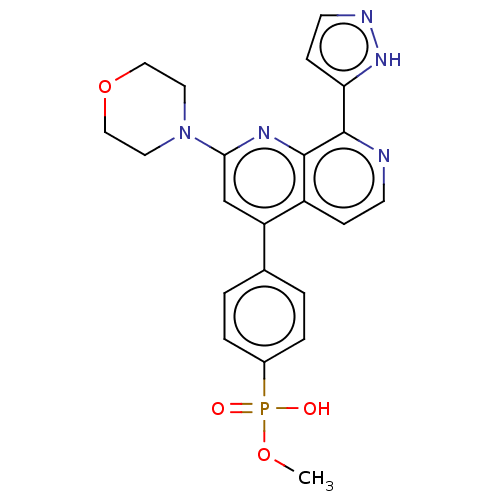 BDBM268149 US9549932, 181 US10772893, Example 181 US11529356, Example 181 Methyl hydrogen {4-[2-(morpholin-4-yl)-8-(1H-pyrazol-5-yl)-1,7-naphthyridin-4-yl]phenyl}phosphonate
BDBM268149 US9549932, 181 US10772893, Example 181 US11529356, Example 181 Methyl hydrogen {4-[2-(morpholin-4-yl)-8-(1H-pyrazol-5-yl)-1,7-naphthyridin-4-yl]phenyl}phosphonate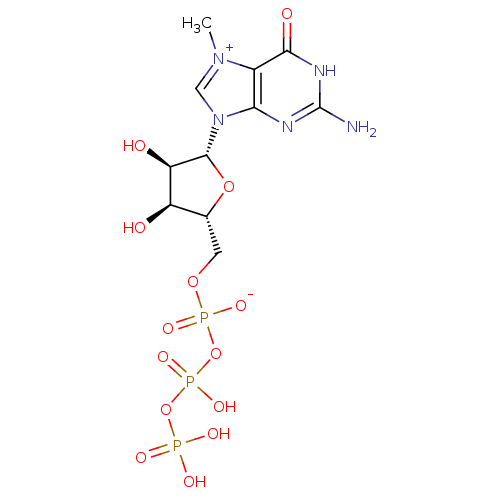 CHEMBL1094973 ((2R,3S,4R,5R)-5-(2-amino-7-methyl-6-oxo-1H-purin-1-ium-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen triphosphate BDBM50316302
CHEMBL1094973 ((2R,3S,4R,5R)-5-(2-amino-7-methyl-6-oxo-1H-purin-1-ium-9(6H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl hydrogen triphosphate BDBM50316302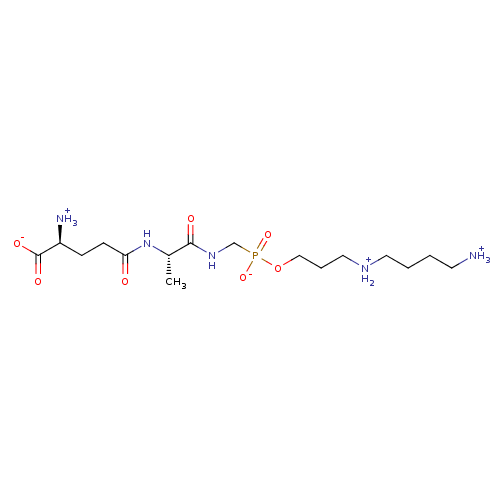 2-Amino-4-[1-({[3-(4-amino-butylamino)-propoxy]-hydroxy-phosphorylmethyl}-carbamoyl)-ethylcarbamoyl]-butyric acid BDBM50060962 Hydrogen O-[3-[N-(4-Aminobutyl)amino]propyl][[(glutamylalanyl) amino]methyl]phosphonate
2-Amino-4-[1-({[3-(4-amino-butylamino)-propoxy]-hydroxy-phosphorylmethyl}-carbamoyl)-ethylcarbamoyl]-butyric acid BDBM50060962 Hydrogen O-[3-[N-(4-Aminobutyl)amino]propyl][[(glutamylalanyl) amino]methyl]phosphonate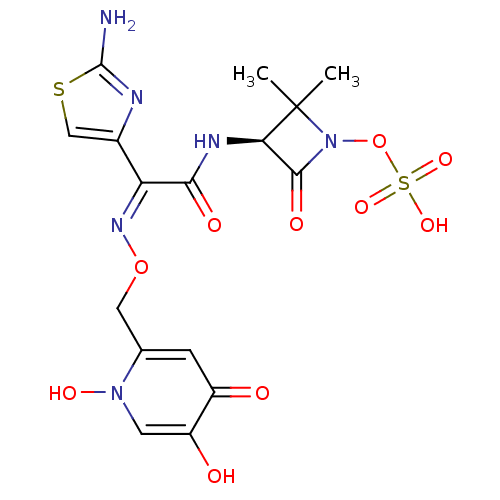 CHEMBL1256967 BDBM50327179 (S)-3-(2-(2-aminothiazol-4-yl)-2-((1,5-dihydroxy-4-oxo-1,4-dihydropyridin-2-yl)methoxyimino)acetamido)-2,2-dimethyl-4-oxoazetidin-1-yl hydrogen sulfate
CHEMBL1256967 BDBM50327179 (S)-3-(2-(2-aminothiazol-4-yl)-2-((1,5-dihydroxy-4-oxo-1,4-dihydropyridin-2-yl)methoxyimino)acetamido)-2,2-dimethyl-4-oxoazetidin-1-yl hydrogen sulfate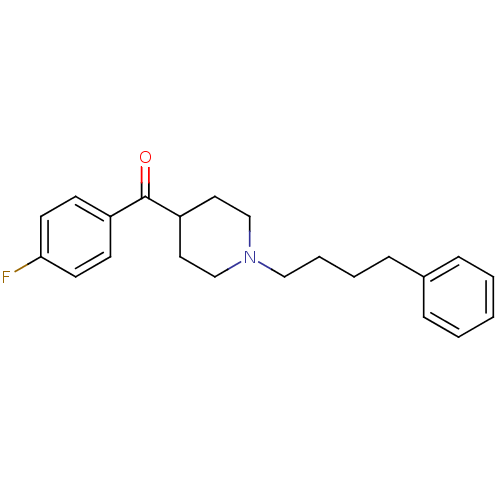 CHEMBL19259 CHEMBL1788135 BDBM50004337 (4-Fluoro-phenyl)-[1-(4-phenyl-butyl)-piperidin-4-yl]-methanone (4-Fluoro-phenyl)-[1-(4-phenyl-butyl)-piperidin-4-yl]-methanone; hydrogen oxalate salt
CHEMBL19259 CHEMBL1788135 BDBM50004337 (4-Fluoro-phenyl)-[1-(4-phenyl-butyl)-piperidin-4-yl]-methanone (4-Fluoro-phenyl)-[1-(4-phenyl-butyl)-piperidin-4-yl]-methanone; hydrogen oxalate salt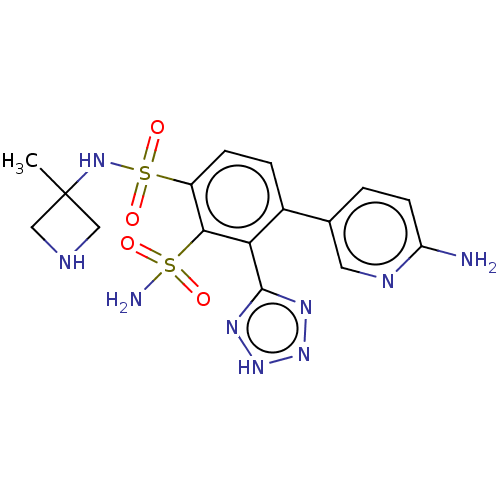 US10544130, Example 497 US10221163, Example 497 4-(6-aminopyridin-3-yl)- 1-N-(3-methylazetidin-3- yl)-3-(2H-1,2,3,4-tetrazol- 5-yl)benzene-1,2- disulfonamide hydrogen BDBM361340
US10544130, Example 497 US10221163, Example 497 4-(6-aminopyridin-3-yl)- 1-N-(3-methylazetidin-3- yl)-3-(2H-1,2,3,4-tetrazol- 5-yl)benzene-1,2- disulfonamide hydrogen BDBM361340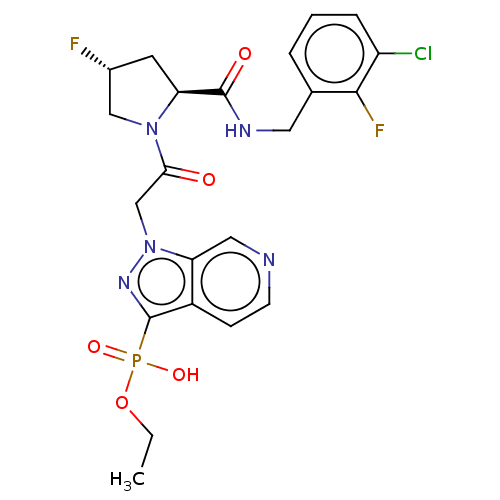 ethyl hydrogen 1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)- 4-fluoropyrrolidin-1- yl)-2-oxoethyl)-1H- pyrazolo[3,4-e]pyridin- 3-ylphosphonate BDBM340257 US9758537, Compound 60
ethyl hydrogen 1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)- 4-fluoropyrrolidin-1- yl)-2-oxoethyl)-1H- pyrazolo[3,4-e]pyridin- 3-ylphosphonate BDBM340257 US9758537, Compound 60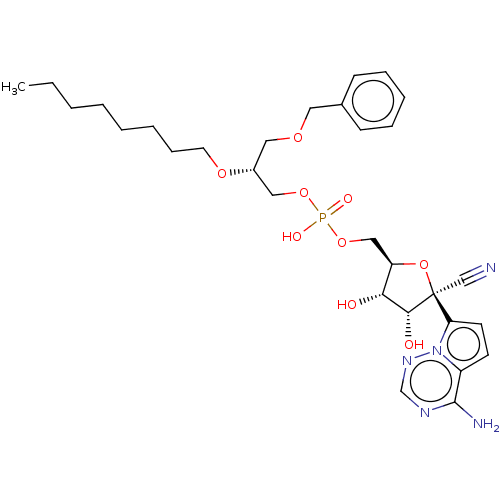 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (2-(octyloxy)ethyl) hydrogen phosphate (26) WO2022081973, Example 26 BDBM534222
((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (2-(octyloxy)ethyl) hydrogen phosphate (26) WO2022081973, Example 26 BDBM534222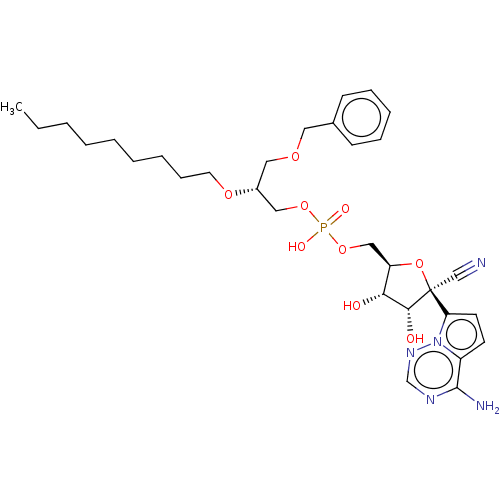 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (3-(nonyloxy)propyl) hydrogen phosphate (25) BDBM534221 WO2022081973, Example 25
((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (3-(nonyloxy)propyl) hydrogen phosphate (25) BDBM534221 WO2022081973, Example 25 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl (2-(octadecyloxy)ethyl) hydrogen phosphate (21) BDBM534217 WO2022081973, Example 21
((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl (2-(octadecyloxy)ethyl) hydrogen phosphate (21) BDBM534217 WO2022081973, Example 21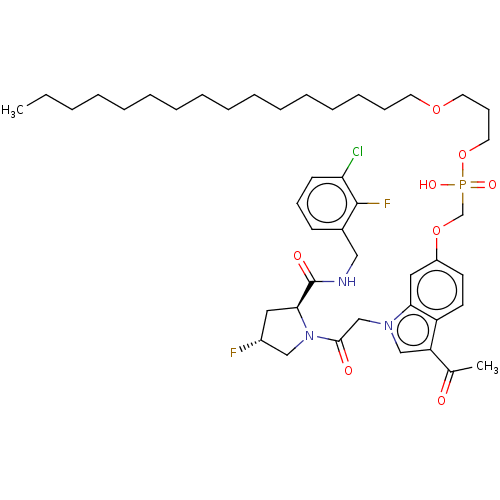 BDBM335530 US9732104, Compound 20 3-(hexadecyloxy)propyl hydrogen (3-acetyl-1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)- 4-fluoropyrrolidin-1-yl)- 2-oxoethyl)-1H-indol-6- yloxy)methylphosphonate
BDBM335530 US9732104, Compound 20 3-(hexadecyloxy)propyl hydrogen (3-acetyl-1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)- 4-fluoropyrrolidin-1-yl)- 2-oxoethyl)-1H-indol-6- yloxy)methylphosphonate US10550140, Compound 20 BDBM431954 3- (hexadecyloxy)propyl hydrogen (3-acetyl-1- (2-((2S,4R)-2-(3- chloro-2- fluorobenzylcarbamoyl)- 4- fluoropyrrolidin-1- yl)-2-oxoethyl)-1H- indol-6- yloxy) methylphosphonate
US10550140, Compound 20 BDBM431954 3- (hexadecyloxy)propyl hydrogen (3-acetyl-1- (2-((2S,4R)-2-(3- chloro-2- fluorobenzylcarbamoyl)- 4- fluoropyrrolidin-1- yl)-2-oxoethyl)-1H- indol-6- yloxy) methylphosphonate US11124518, Example 21 2-Allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-(isoindolin-5-ylamino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518547
US11124518, Example 21 2-Allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-(isoindolin-5-ylamino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518547 WO2022081973, Example 19 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (3-(hexadecylthio)propyl) hydrogen phosphate (19) BDBM534215
WO2022081973, Example 19 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (3-(hexadecylthio)propyl) hydrogen phosphate (19) BDBM534215 WO2022081973, Example 27 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (2-(undecyloxy)ethyl) hydrogen phosphate (27) BDBM534223
WO2022081973, Example 27 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl (2-(undecyloxy)ethyl) hydrogen phosphate (27) BDBM534223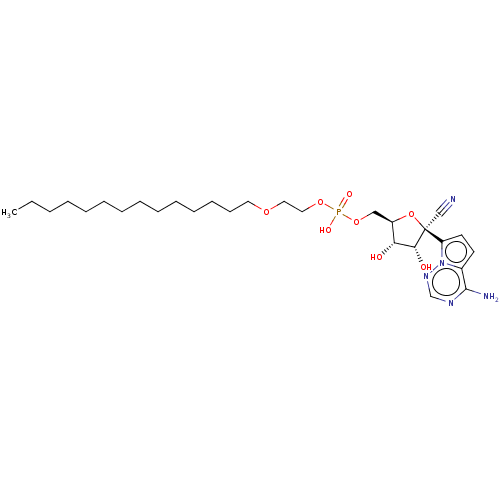 WO2022081973, Example 59 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-tetradecoxyethyl hydrogen phosphate (59) BDBM534255
WO2022081973, Example 59 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-tetradecoxyethyl hydrogen phosphate (59) BDBM534255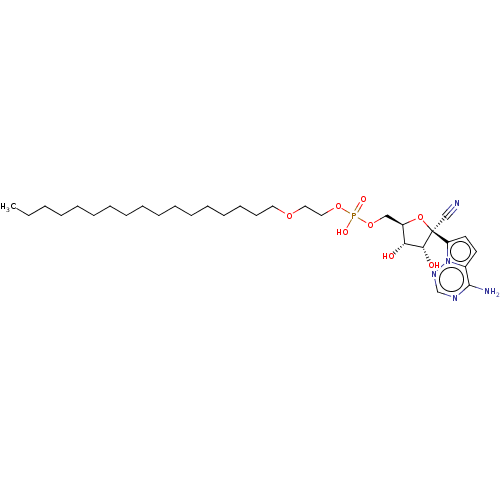 WO2022081973, Example 60 BDBM534256 [(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-heptadecoxyethyl hydrogen phosphate (60)
WO2022081973, Example 60 BDBM534256 [(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-heptadecoxyethyl hydrogen phosphate (60)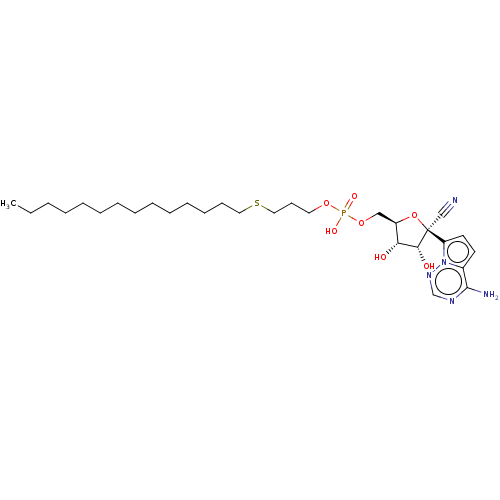 WO2022081973, Example 61 [(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-tetradecylsulfanylpropyl hydrogen phosphate (61) BDBM534257
WO2022081973, Example 61 [(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-tetradecylsulfanylpropyl hydrogen phosphate (61) BDBM534257 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-hexadecoxyethyl hydrogen phosphate (66) BDBM534262 WO2022081973, Example 66
[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 2-hexadecoxyethyl hydrogen phosphate (66) BDBM534262 WO2022081973, Example 66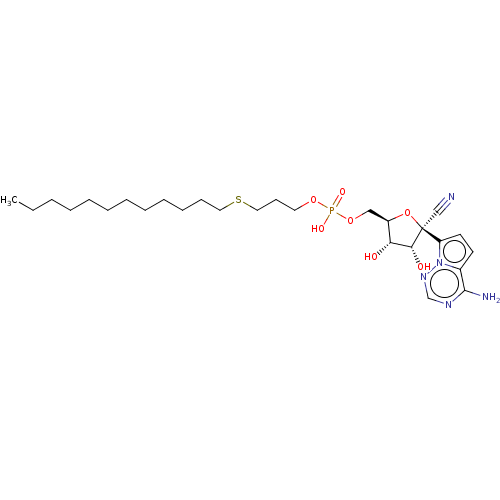 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-dodecylsulfanylpropyl hydrogen phosphate (63) BDBM534259 WO2022081973, Example 63
[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-dodecylsulfanylpropyl hydrogen phosphate (63) BDBM534259 WO2022081973, Example 63 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-hexadecylsulfanylpropyl hydrogen phosphate (65) BDBM534261 WO2022081973, Example 65
[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-hexadecylsulfanylpropyl hydrogen phosphate (65) BDBM534261 WO2022081973, Example 65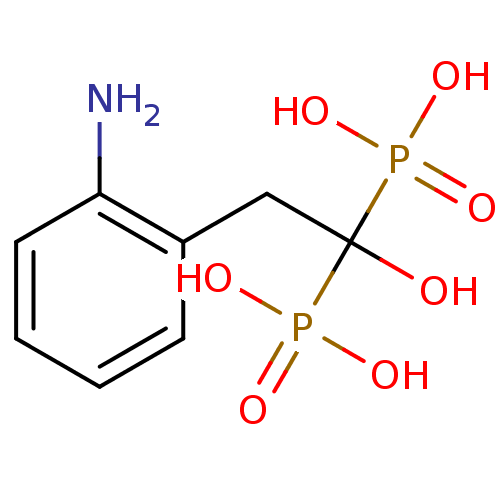 [2-(2-amino-phenyl)-1-hydroxy-1-phosphono-ethyl]-phosphonic acid CHEMBL414849 2-(2-aminophenyl)-1-hydroxyethane-1,1-diyldiphosphonic acid BDBM50173792 hydrogen [2-(2-azaniumylphenyl)-1-hydroxy-1-phosphonatoethyl]phosphonate
[2-(2-amino-phenyl)-1-hydroxy-1-phosphono-ethyl]-phosphonic acid CHEMBL414849 2-(2-aminophenyl)-1-hydroxyethane-1,1-diyldiphosphonic acid BDBM50173792 hydrogen [2-(2-azaniumylphenyl)-1-hydroxy-1-phosphonatoethyl]phosphonate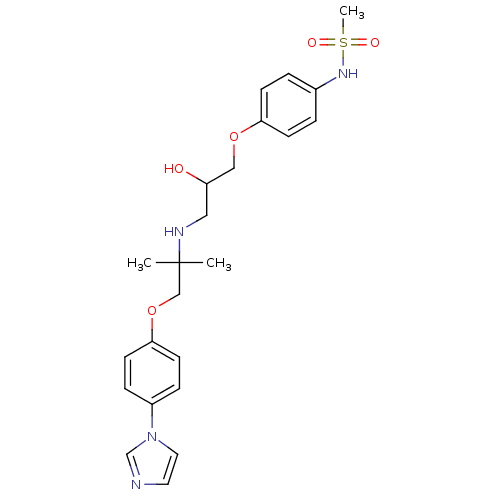 BDBM50015190 N-(4-{2-Hydroxy-3-[2-(4-imidazol-1-yl-phenoxy)-1,1-dimethyl-ethylamino]-propoxy}-phenyl)-methanesulfonamidecompound with 2mol of HCl and 1mol of water CHEMBL536800
BDBM50015190 N-(4-{2-Hydroxy-3-[2-(4-imidazol-1-yl-phenoxy)-1,1-dimethyl-ethylamino]-propoxy}-phenyl)-methanesulfonamidecompound with 2mol of HCl and 1mol of water CHEMBL536800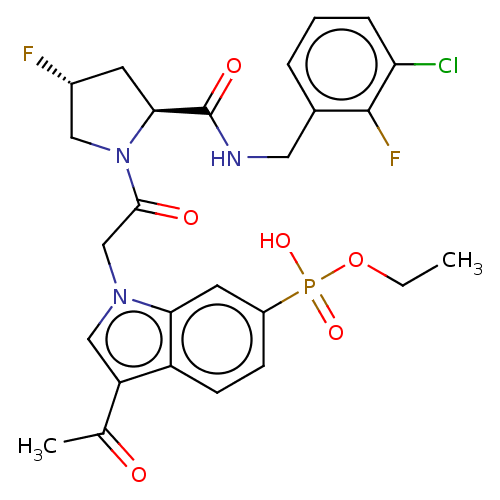 BDBM330226 ethyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate US9663543, Compound 2 US10301336, Comp No. 2
BDBM330226 ethyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate US9663543, Compound 2 US10301336, Comp No. 2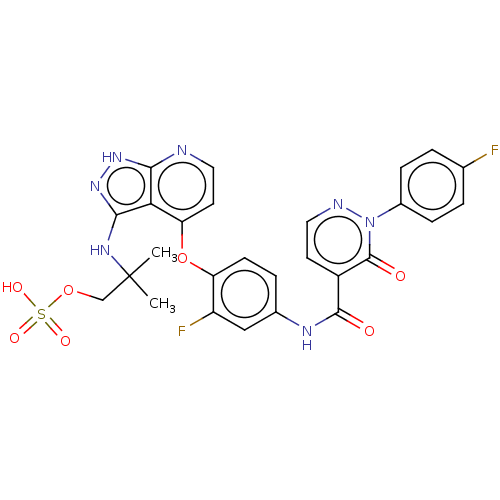 BDBM517113 2-((4-(2-fluoro-4-(2-(4-fluorophenyl)-3-oxo-2,3-dihydropyridazine-4-carboxamido)phenoxy)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)-2-methylpropyl hydrogen sulfate US11104676, Example 183
BDBM517113 2-((4-(2-fluoro-4-(2-(4-fluorophenyl)-3-oxo-2,3-dihydropyridazine-4-carboxamido)phenoxy)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)-2-methylpropyl hydrogen sulfate US11104676, Example 183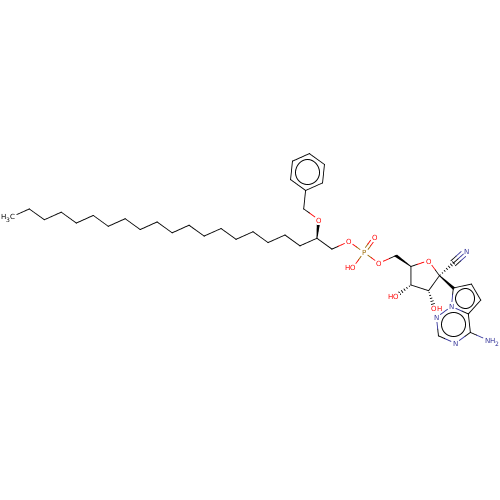 WO2022081973, Example 72 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyloxy)henicosyl) hydrogen phosphate (72) BDBM534269
WO2022081973, Example 72 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyloxy)henicosyl) hydrogen phosphate (72) BDBM534269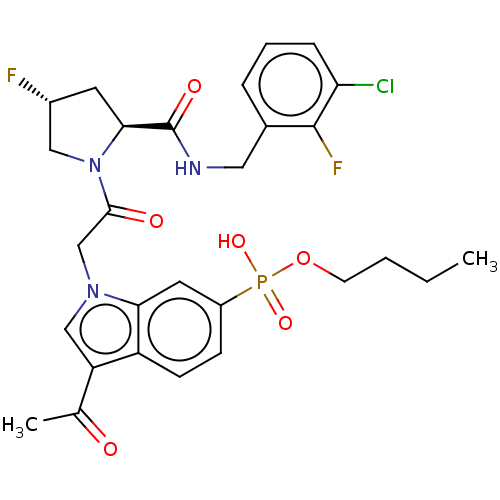 butyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate BDBM330245 US10301336, Comp No. 21 US9663543, Compound 21
butyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate BDBM330245 US10301336, Comp No. 21 US9663543, Compound 21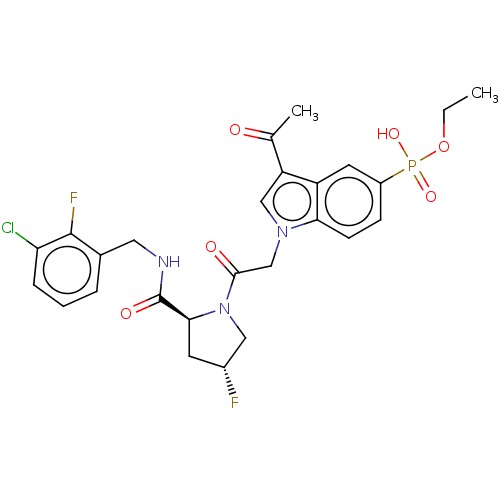 ethyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-5- ylphosphonate US10301336, Comp No. 9 BDBM330233 US9663543, Compound 9
ethyl hydrogen 3-acetyl-1- (2-((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-5- ylphosphonate US10301336, Comp No. 9 BDBM330233 US9663543, Compound 9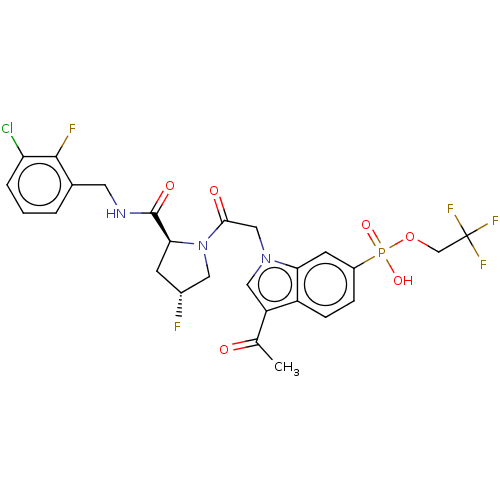 2,2,2-trifluoroethyl hydrogen 3-acetyl-1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate BDBM330253 US10301336, Comp No. 29 US9663543, Compound 29
2,2,2-trifluoroethyl hydrogen 3-acetyl-1-(2- ((2S,4R)-2-(3-chloro-2- fluorobenzylcarbamoyl)-4- fluoropyrrolidin-1-yl)-2- oxoethyl)-1H-indol-6- ylphosphonate BDBM330253 US10301336, Comp No. 29 US9663543, Compound 29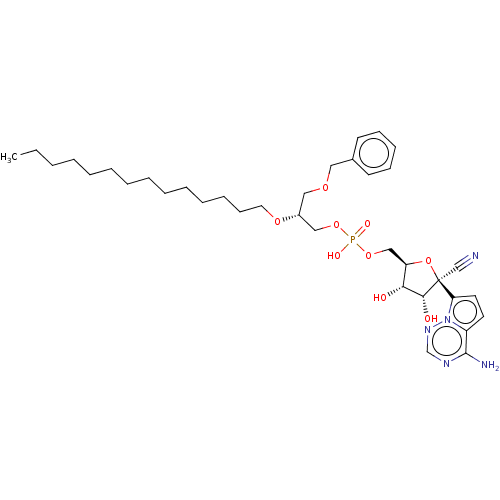 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(tetradecyloxy)propyl) hydrogen phosphate (24) BDBM534220 WO2022081973, Example 24
((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(tetradecyloxy)propyl) hydrogen phosphate (24) BDBM534220 WO2022081973, Example 24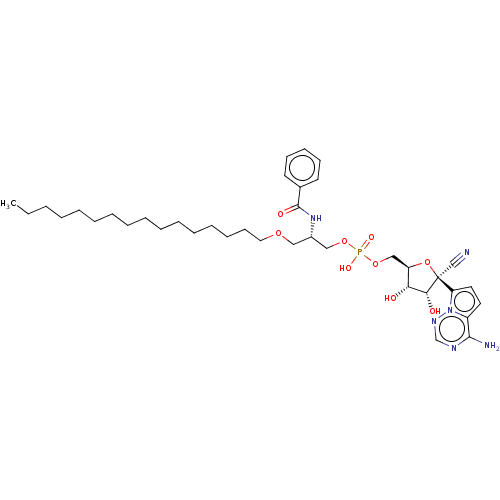 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-benzamido-3-(octadecyloxy)propyl) hydrogen phosphate (30) BDBM534226 WO2022081973, Example 30
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-benzamido-3-(octadecyloxy)propyl) hydrogen phosphate (30) BDBM534226 WO2022081973, Example 30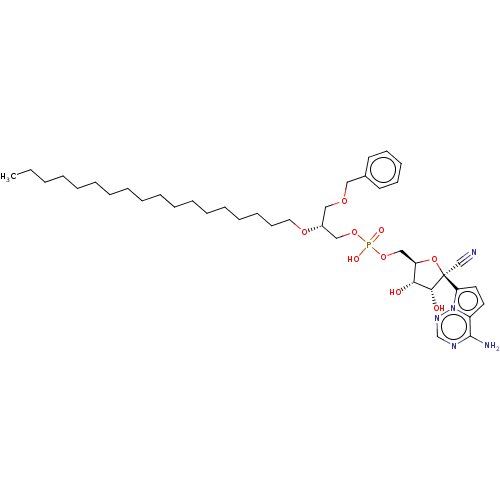 BDBM534218 WO2022081973, Example 22 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(octadecyloxy)propyl) hydrogen phosphate (22)
BDBM534218 WO2022081973, Example 22 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(octadecyloxy)propyl) hydrogen phosphate (22)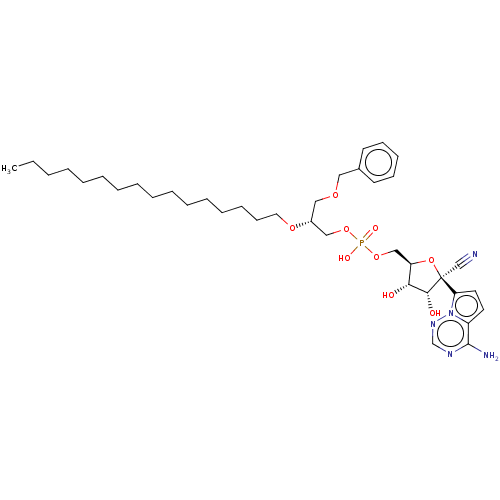 BDBM534219 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(hexadecyloxy)propyl) hydrogen phosphate (23) WO2022081973, Example 23
BDBM534219 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(benzyloxy)-2-(hexadecyloxy)propyl) hydrogen phosphate (23) WO2022081973, Example 23 US20230357239, Example 26 1-{4-[2-methyl-4-({(1R)-1-[2-methyl-3-(trifluoromethyl)phenyl]ethyl}amino)pyrido[2,3-d]pyrimidin-6-yl]-3,6-dihydropyridin-1(2H)-yl}ethan-1-one hydrogen BDBM634058
US20230357239, Example 26 1-{4-[2-methyl-4-({(1R)-1-[2-methyl-3-(trifluoromethyl)phenyl]ethyl}amino)pyrido[2,3-d]pyrimidin-6-yl]-3,6-dihydropyridin-1(2H)-yl}ethan-1-one hydrogen BDBM634058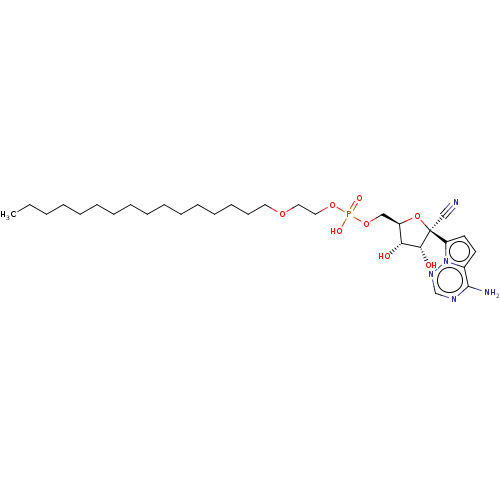 WO2022081973, Example 66 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl (2-(hexadecyloxy)ethyl) hydrogen phosphate (20) BDBM534216 WO2022081973, Example 20
WO2022081973, Example 66 ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl (2-(hexadecyloxy)ethyl) hydrogen phosphate (20) BDBM534216 WO2022081973, Example 20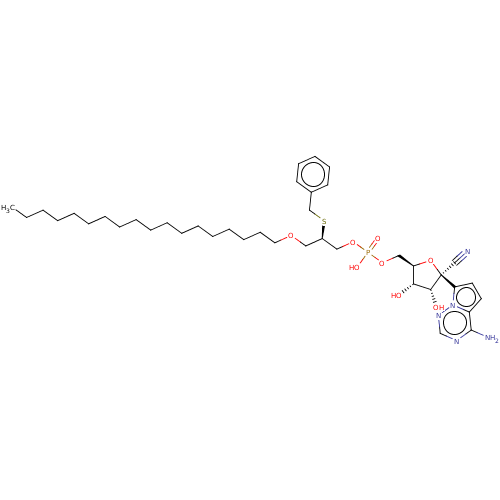 WO2022081973, Example 7 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzylthio)-3-(octadecyloxy)propyl) hydrogen phosphate: (7) BDBM534203
WO2022081973, Example 7 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzylthio)-3-(octadecyloxy)propyl) hydrogen phosphate: (7) BDBM534203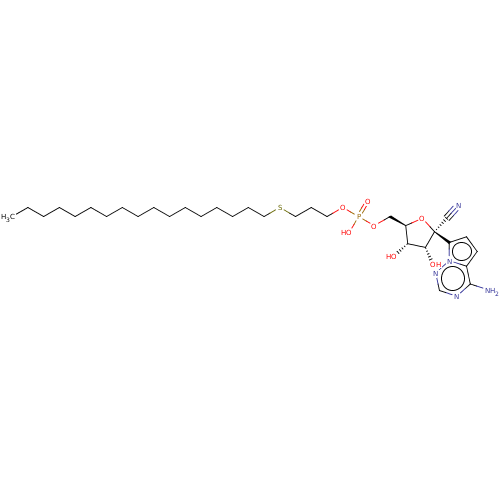 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-heptadecylsulfanylpropyl hydrogen phosphate (62) BDBM534258 WO2022081973, Example 62 WO2022081973, Example 65
[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl 3-heptadecylsulfanylpropyl hydrogen phosphate (62) BDBM534258 WO2022081973, Example 62 WO2022081973, Example 65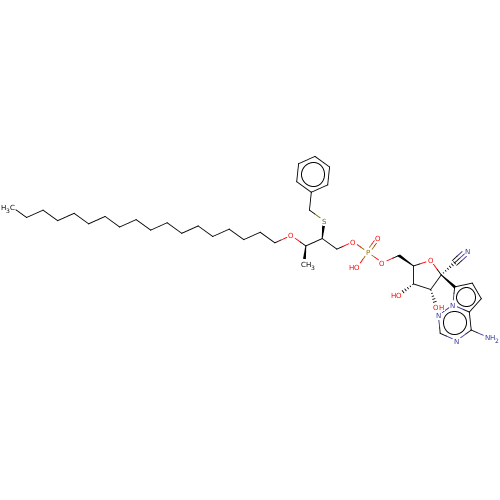 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((2S,3R)-2-(benzyloxy)-3-(octadecyloxy)butyl) hydrogen phosphate (8) BDBM534204 WO2022081973, Example 8
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((2S,3R)-2-(benzyloxy)-3-(octadecyloxy)butyl) hydrogen phosphate (8) BDBM534204 WO2022081973, Example 8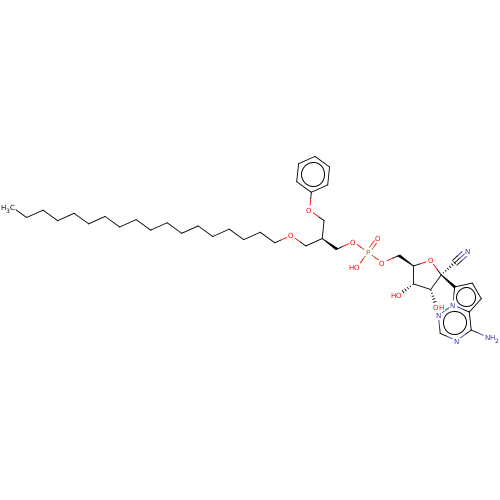 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-(octadecyloxy)-3-phenoxypropan-2-yl) hydrogen phosphate (18) WO2022081973, Example 18 BDBM534214
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-(octadecyloxy)-3-phenoxypropan-2-yl) hydrogen phosphate (18) WO2022081973, Example 18 BDBM534214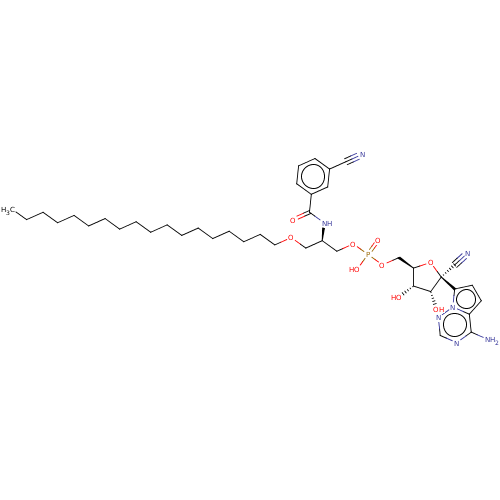 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(3-cyanobenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (57) BDBM534253 WO2022081973, Example 57
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(3-cyanobenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (57) BDBM534253 WO2022081973, Example 57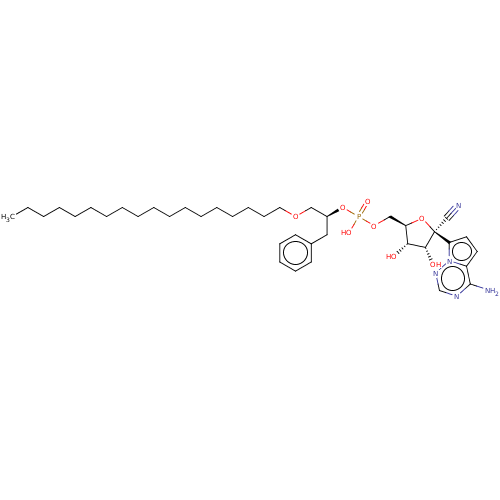 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-1-(octadecyloxy)-3-phenylpropan-2-yl) hydrogen phosphate (40) BDBM534236 WO2022081973, Example 40
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-1-(octadecyloxy)-3-phenylpropan-2-yl) hydrogen phosphate (40) BDBM534236 WO2022081973, Example 40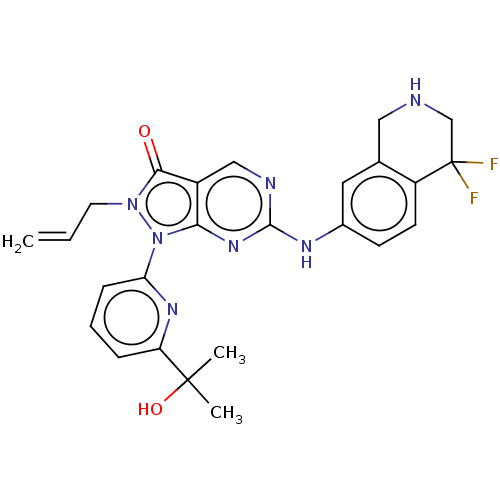 2-Allyl-6-((4,4-difluoro-1,2,3,4-tetrahydroisoquinolin-7-yl)amino)-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518554 US11124518, Example 28
2-Allyl-6-((4,4-difluoro-1,2,3,4-tetrahydroisoquinolin-7-yl)amino)-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518554 US11124518, Example 28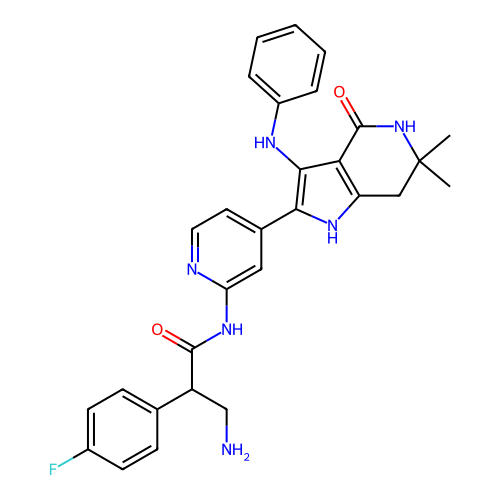 3-amino-N-[4-(3-anilino-6,6-dimethyl-4-oxo-4,5,6,7-tetrahydro-1H-pyrrolo[3,2-c]pyridin-2-yl)pyridin-2-yl]-2-(4-fluorophenyl)propanamide-hydrogen chloride salt (Racemate) BDBM721181 US12227501, Example 139
3-amino-N-[4-(3-anilino-6,6-dimethyl-4-oxo-4,5,6,7-tetrahydro-1H-pyrrolo[3,2-c]pyridin-2-yl)pyridin-2-yl]-2-(4-fluorophenyl)propanamide-hydrogen chloride salt (Racemate) BDBM721181 US12227501, Example 139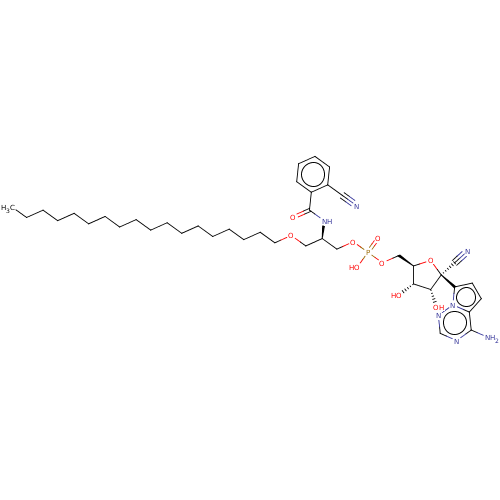 BDBM534254 WO2022081973, Example 58 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(2-cyanobenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (58)
BDBM534254 WO2022081973, Example 58 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(2-cyanobenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (58)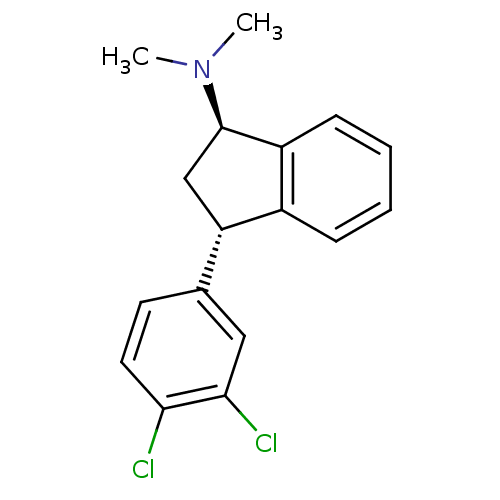 CHEMBL356750 [3-(3,4-Dichloro-phenyl)-indan-1-yl]-ethyl-amine;chloride [3-(3,4-Dichloro-phenyl)-indan-1-yl]-dimethyl-amine;hydrogen maleate tert-Butyl-[3-(3,4-dichloro-phenyl)-indan-1-yl]-amine;chloride BDBM50095611
CHEMBL356750 [3-(3,4-Dichloro-phenyl)-indan-1-yl]-ethyl-amine;chloride [3-(3,4-Dichloro-phenyl)-indan-1-yl]-dimethyl-amine;hydrogen maleate tert-Butyl-[3-(3,4-dichloro-phenyl)-indan-1-yl]-amine;chloride BDBM50095611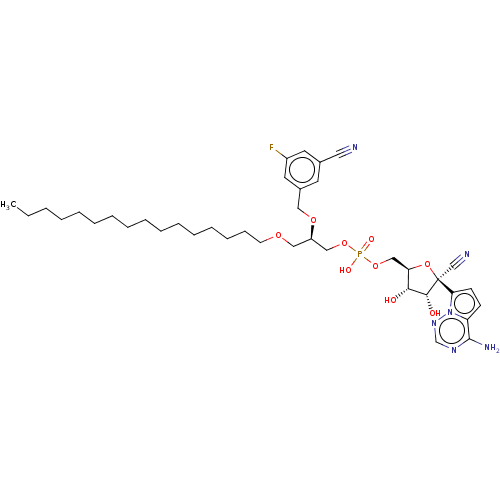 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)henicosyl) hydrogen phosphate (35) BDBM534231 WO2022081973, Example 35
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)henicosyl) hydrogen phosphate (35) BDBM534231 WO2022081973, Example 35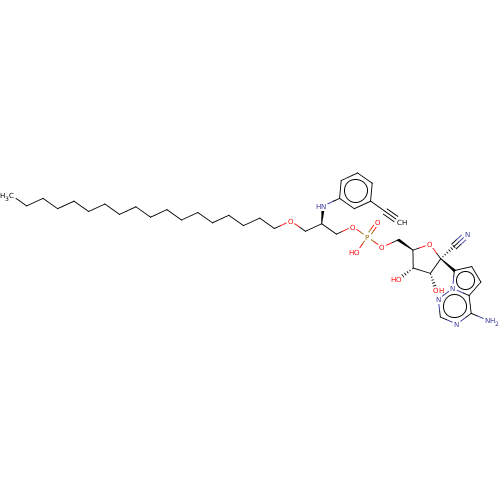 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyanophenyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (16) WO2022081973, Example 16 BDBM534212
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyanophenyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (16) WO2022081973, Example 16 BDBM534212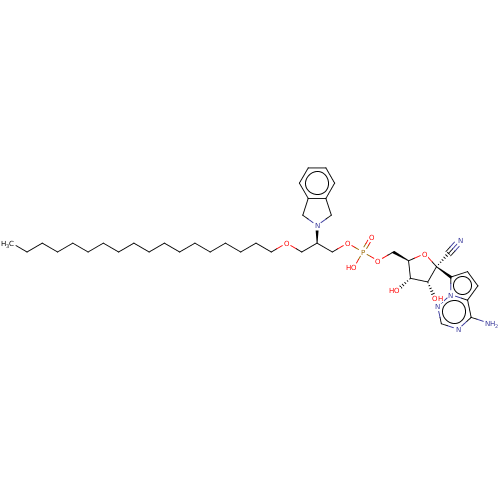 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(isoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (1) WO2022081973, Example 1 BDBM534197
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(isoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (1) WO2022081973, Example 1 BDBM534197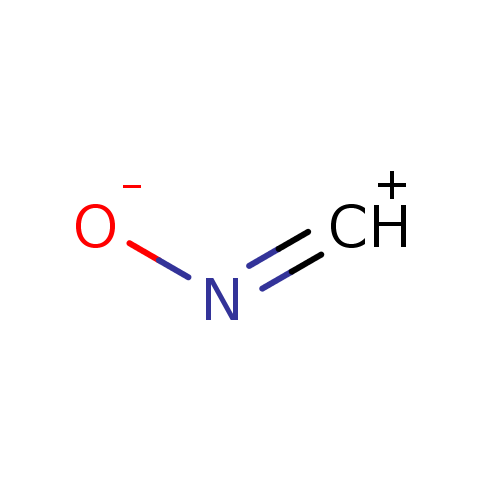 (methylidyneammoniumyl)oxidanide H-C#NO formonitrile oxideformonitrile-N-oxidehydrido(nitrosyl-kappaN)carbonhydrido(oxidonitrato-N)carbon [C(H)NO] hydrogen cyanide N-oxide fulminic acid methylidyne(oxo)-lambda(5)-azane BDBM50152965 [CH(NO)] CHEMBL185198 HCNO Knallsaeure
(methylidyneammoniumyl)oxidanide H-C#NO formonitrile oxideformonitrile-N-oxidehydrido(nitrosyl-kappaN)carbonhydrido(oxidonitrato-N)carbon [C(H)NO] hydrogen cyanide N-oxide fulminic acid methylidyne(oxo)-lambda(5)-azane BDBM50152965 [CH(NO)] CHEMBL185198 HCNO Knallsaeure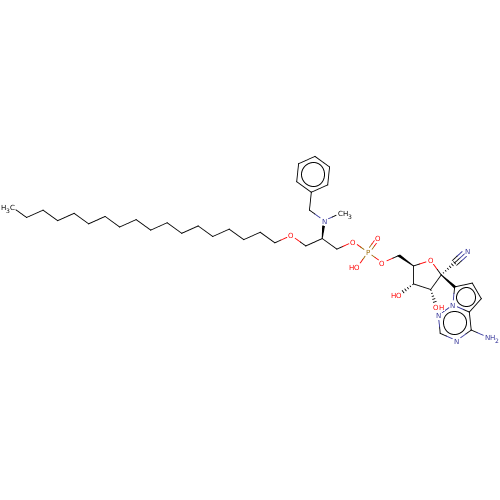 BDBM534198 WO2022081973, Example 2 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyl(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (2)
BDBM534198 WO2022081973, Example 2 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyl(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (2)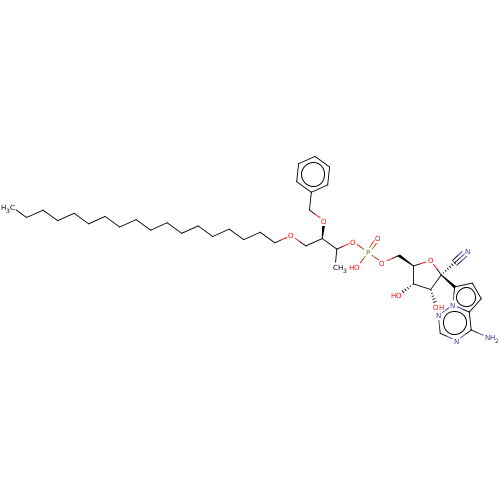 BDBM534208 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((3R)-3-(benzyloxy)-4-(octadecyloxy)butan-2-yl) hydrogen phosphate (12) WO2022081973, Example 12
BDBM534208 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((3R)-3-(benzyloxy)-4-(octadecyloxy)butan-2-yl) hydrogen phosphate (12) WO2022081973, Example 12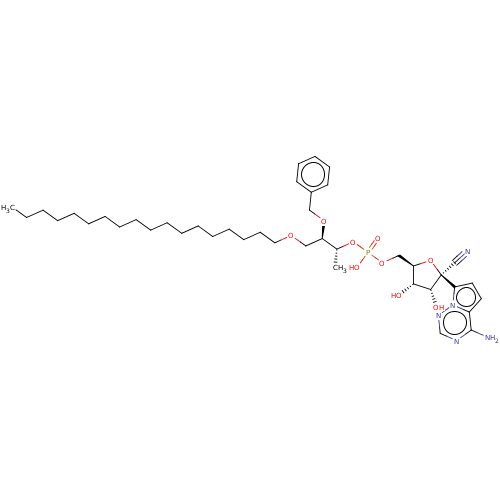 BDBM534209 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((3R)-3-(benzyloxy)-4-(octadecyloxy)butan-2-yl) hydrogen phosphate (13) WO2022081973, Example 13
BDBM534209 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((3R)-3-(benzyloxy)-4-(octadecyloxy)butan-2-yl) hydrogen phosphate (13) WO2022081973, Example 13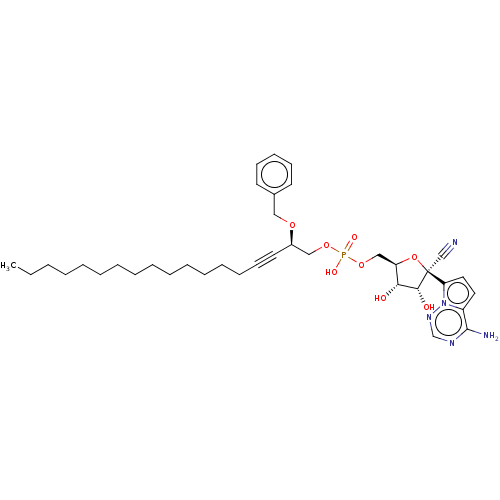 BDBM534270 WO2022081973, Example 73 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyloxy)nonadec-4-yn-1-yl) hydrogen phosphate (73)
BDBM534270 WO2022081973, Example 73 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(benzyloxy)nonadec-4-yn-1-yl) hydrogen phosphate (73)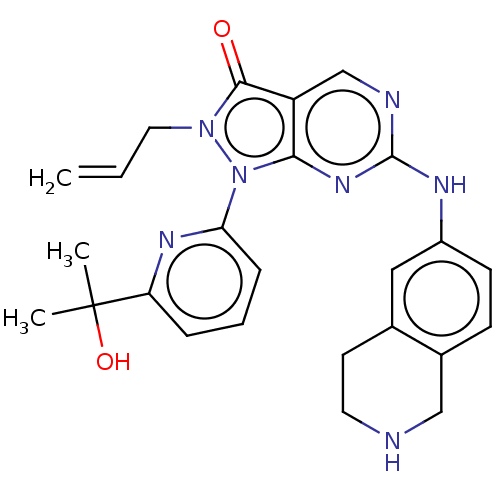 US11124518, Example 15 2-Allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-((1,2,3,4-tetrahydroisoquinolin-6-yl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518539 US11299493, Compound 1.8
US11124518, Example 15 2-Allyl-1-(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-((1,2,3,4-tetrahydroisoquinolin-6-yl)amino)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one hydrogen chloride BDBM518539 US11299493, Compound 1.8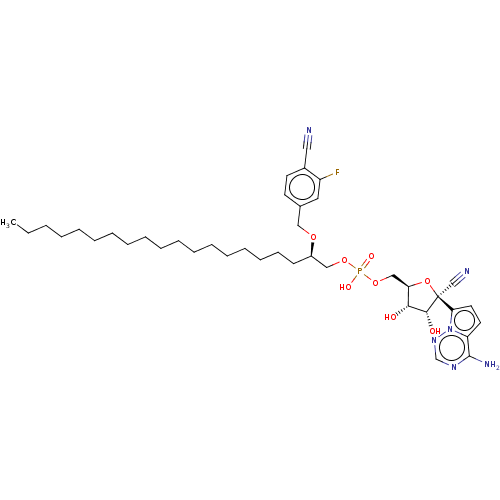 WO2022081973, Example 71 BDBM534268 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3-fluorobenzyl)oxy)henicosyl) hydrogen phosphate (71)
WO2022081973, Example 71 BDBM534268 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3-fluorobenzyl)oxy)henicosyl) hydrogen phosphate (71)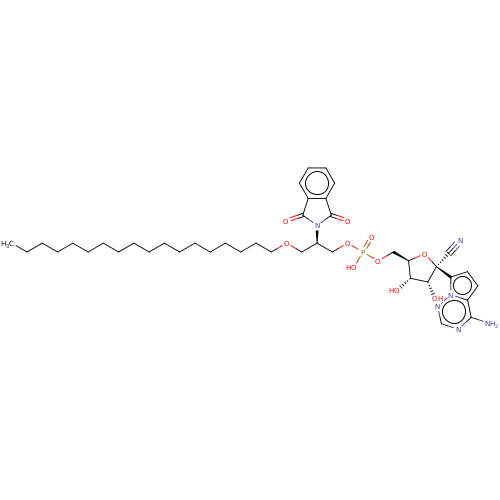 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(1,3-dioxoisoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (15) WO2022081973, Example 15 BDBM534211
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(1,3-dioxoisoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (15) WO2022081973, Example 15 BDBM534211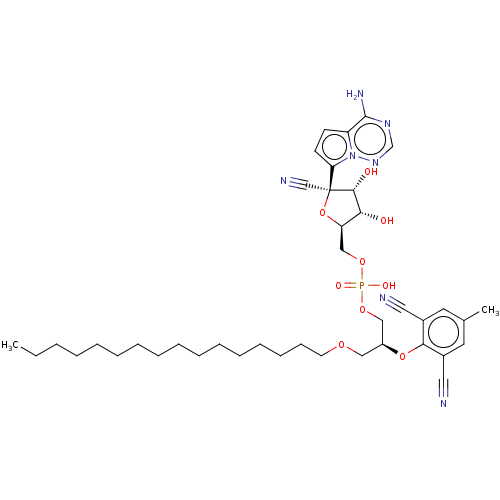 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(2,6-dicyano-4-methylphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (29) WO2022081973, Example 29 BDBM534225
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(2,6-dicyano-4-methylphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (29) WO2022081973, Example 29 BDBM534225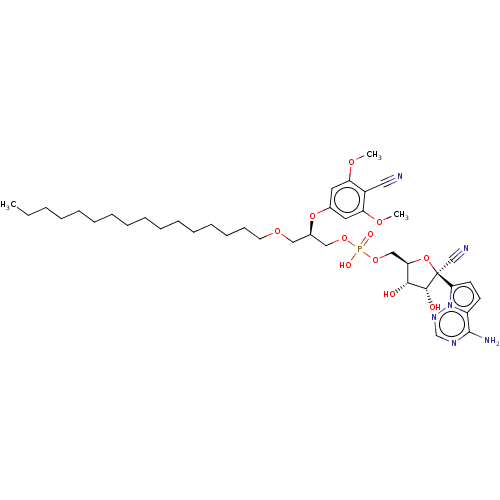 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-3,5-dimethoxyphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (34) BDBM534230 WO2022081973, Example 34
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-3,5-dimethoxyphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (34) BDBM534230 WO2022081973, Example 34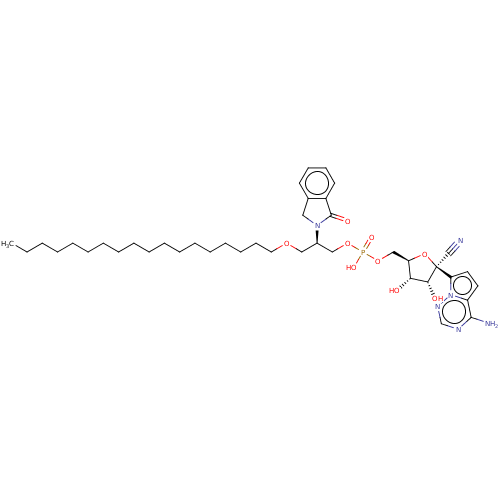 BDBM534207 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(octadecyloxy)-2-(1-oxoisoindolin-2-yl)propyl) hydrogen phosphate (11) WO2022081973, Example 11
BDBM534207 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-3-(octadecyloxy)-2-(1-oxoisoindolin-2-yl)propyl) hydrogen phosphate (11) WO2022081973, Example 11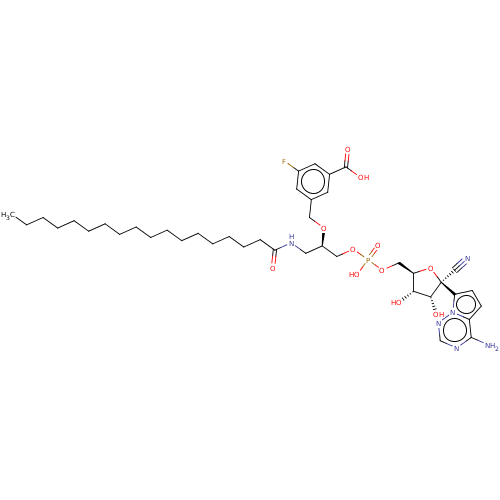 BDBM534241 WO2022081973, Example 45 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)-3-stearamidopropyl) hydrogen phosphate (45)
BDBM534241 WO2022081973, Example 45 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)-3-stearamidopropyl) hydrogen phosphate (45)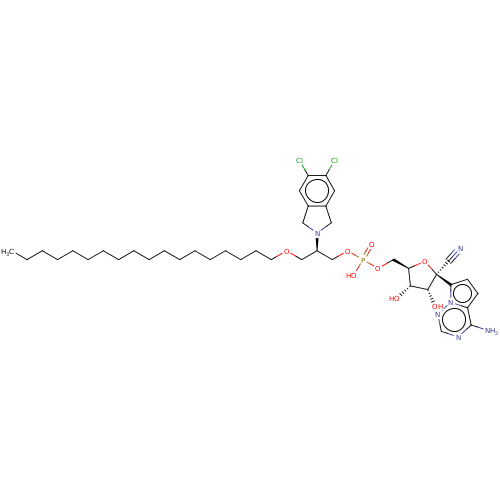 WO2022081973, Example 9 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(5,6-dichloroisoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (9) BDBM534205
WO2022081973, Example 9 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(5,6-dichloroisoindolin-2-yl)-3-(octadecyloxy)propyl) hydrogen phosphate (9) BDBM534205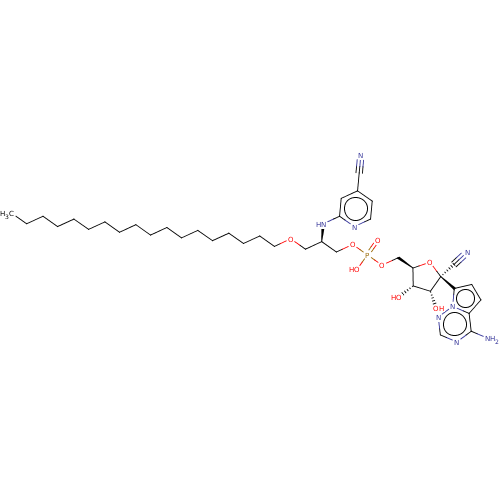 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((5-cyanopyridin-2-yl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (42) BDBM534238 WO2022081973, Example 42
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((5-cyanopyridin-2-yl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (42) BDBM534238 WO2022081973, Example 42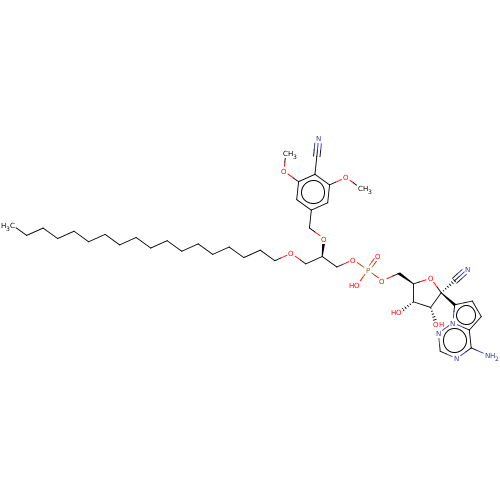 BDBM534251 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3,5-dimethoxybenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (55) WO2022081973, Example 55
BDBM534251 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3,5-dimethoxybenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (55) WO2022081973, Example 55 BDBM534263 WO2022081973, Example 67 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) O-hydrogen (R)-phosphorothioate (67)
BDBM534263 WO2022081973, Example 67 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) O-hydrogen (R)-phosphorothioate (67)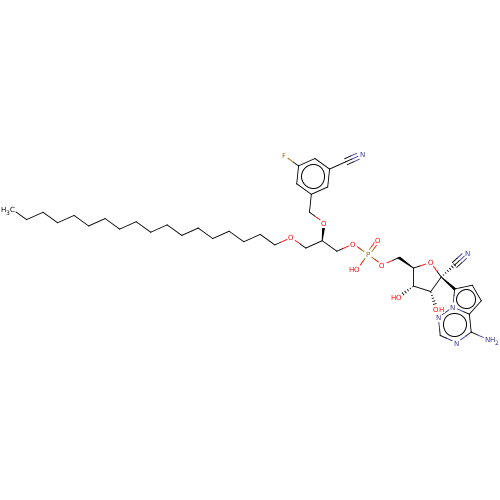 BDBM534265 WO2022081973, Example 68 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) O-hydrogen (S)-phosphorothioate (68)
BDBM534265 WO2022081973, Example 68 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) O-hydrogen (S)-phosphorothioate (68)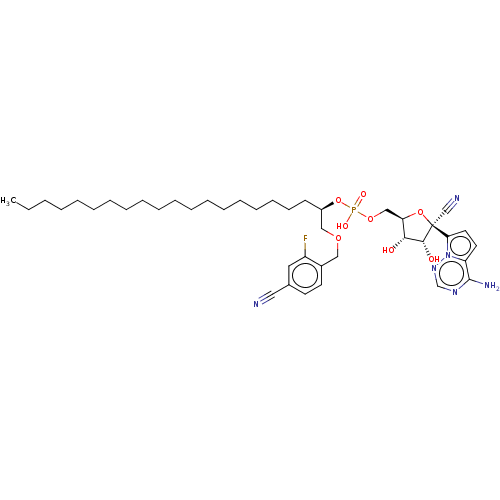 BDBM534266 WO2022081973, Example 69 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((4-cyano-2-fluorobenzyl)oxy)henicosan-2-yl) hydrogen phosphate (69)
BDBM534266 WO2022081973, Example 69 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((4-cyano-2-fluorobenzyl)oxy)henicosan-2-yl) hydrogen phosphate (69) BDBM634045 1-{6-[2-methyl-4-({(1R)-1-[2-methyl-3-(trifluoromethyl)phenyl]ethyl}amino)pyrido[2,3-d]pyrimidin-6-yl]-2,6-diazaspiro[3.3]heptan-2-yl}ethan-1-one-hydrogen chloride (1/1) US20230357239, Example 13
BDBM634045 1-{6-[2-methyl-4-({(1R)-1-[2-methyl-3-(trifluoromethyl)phenyl]ethyl}amino)pyrido[2,3-d]pyrimidin-6-yl]-2,6-diazaspiro[3.3]heptan-2-yl}ethan-1-one-hydrogen chloride (1/1) US20230357239, Example 13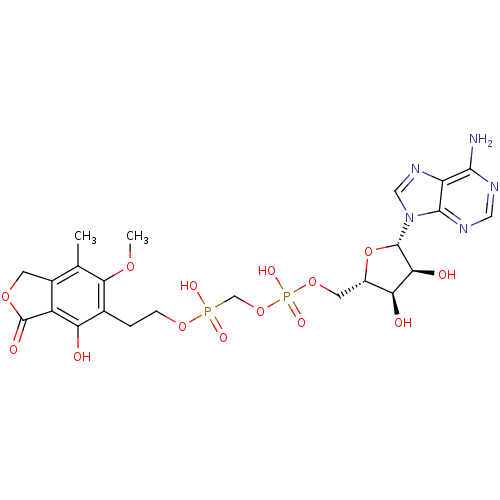 CHEMBL1683742 ((2S,3R,4S,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl(hydroxy(2-(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)ethoxy)phosphoryl)methyl hydrogen phosphate BDBM50338540
CHEMBL1683742 ((2S,3R,4S,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl(hydroxy(2-(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)ethoxy)phosphoryl)methyl hydrogen phosphate BDBM50338540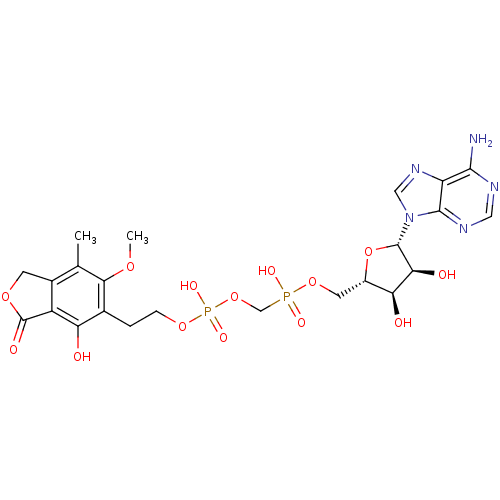 CHEMBL1683743 ((((2S,3R,4S,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(hydroxy)phosphoryl)methyl 2-(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)ethyl hydrogen phosphate BDBM50338541
CHEMBL1683743 ((((2S,3R,4S,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(hydroxy)phosphoryl)methyl 2-(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)ethyl hydrogen phosphate BDBM50338541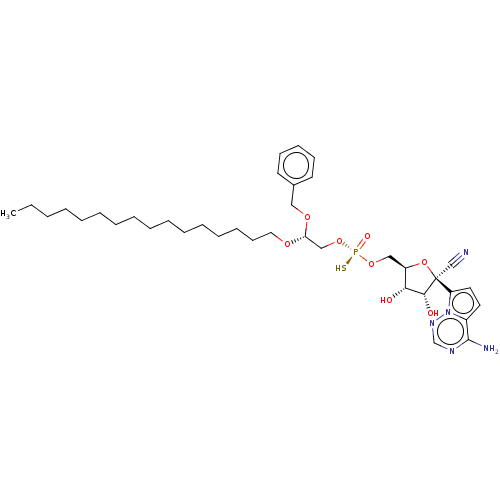 O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) S- hydrogen (R)-phosphorothioate (28) WO2022081973, Example 28 BDBM534224
O-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methyl) O-((R)-2-(benzyloxy)-3-(octadecyloxy)propyl) S- hydrogen (R)-phosphorothioate (28) WO2022081973, Example 28 BDBM534224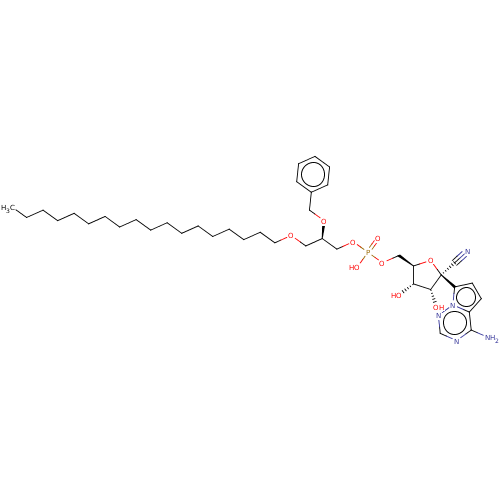 WO2022081973, Example 67 WO2022081973, Example 5 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((2S,3S)-2-(benzyloxy)-3-(octadecyloxy)butyl) hydrogen phosphate (5) BDBM534201
WO2022081973, Example 67 WO2022081973, Example 5 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((2S,3S)-2-(benzyloxy)-3-(octadecyloxy)butyl) hydrogen phosphate (5) BDBM534201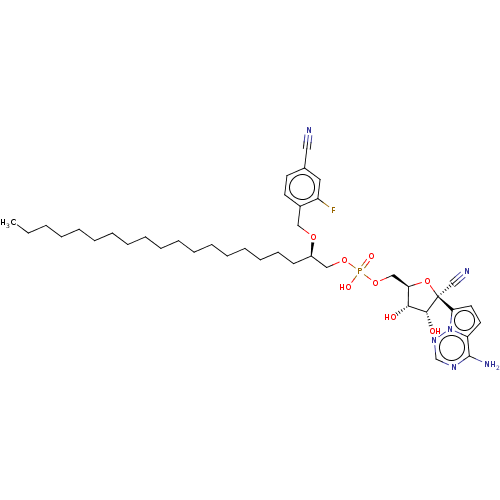 WO2022081973, Example 70 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((4-cyano-2-fluorobenzyl)oxy)henicosan-2-yl) hydrogen phosphate (70) BDBM534267
WO2022081973, Example 70 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((4-cyano-2-fluorobenzyl)oxy)henicosan-2-yl) hydrogen phosphate (70) BDBM534267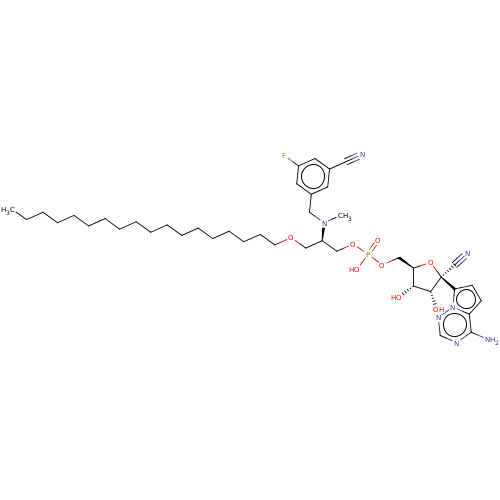 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (6) BDBM534202 WO2022081973, Example 6
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (6) BDBM534202 WO2022081973, Example 6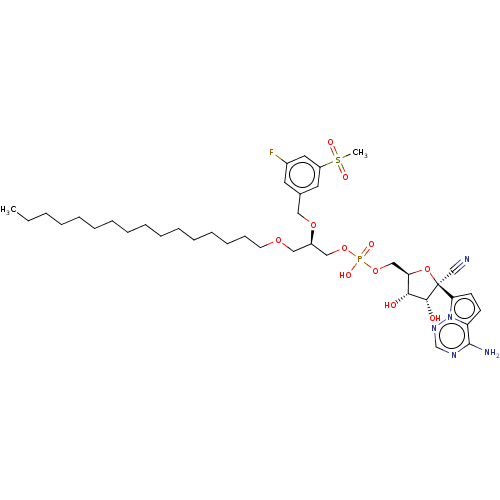 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-fluoro-5-(methylsulfonyl)benzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (32) BDBM534228 WO2022081973, Example 32
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-fluoro-5-(methylsulfonyl)benzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (32) BDBM534228 WO2022081973, Example 32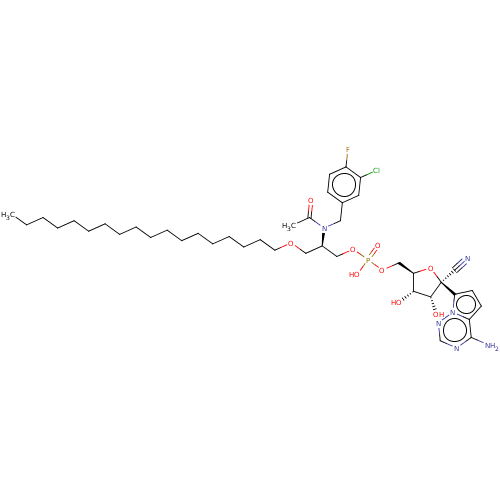 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(N-(3-chloro-4-fluorobenzyl)acetamido)-3-(octadecyloxy)propyl) hydrogen phosphate (10) BDBM534206 WO2022081973, Example 10
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(N-(3-chloro-4-fluorobenzyl)acetamido)-3-(octadecyloxy)propyl) hydrogen phosphate (10) BDBM534206 WO2022081973, Example 10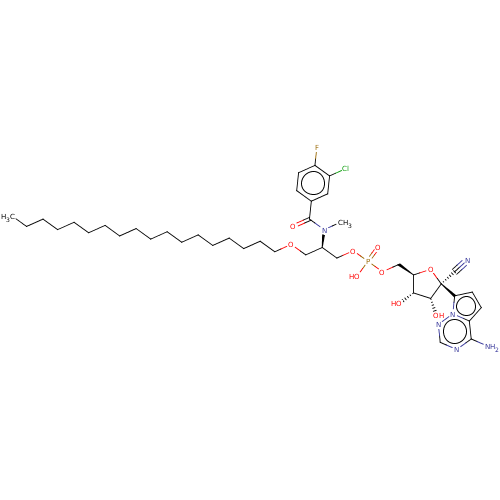 BDBM534200 WO2022081973, Example 4 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(3-chloro-4-fluoro-N-methylbenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (4)
BDBM534200 WO2022081973, Example 4 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(3-chloro-4-fluoro-N-methylbenzamido)-3-(octadecyloxy)propyl) hydrogen phosphate (4)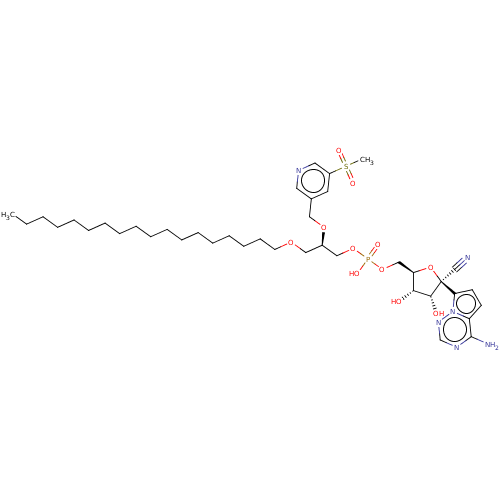 BDBM534239 WO2022081973, Example 43 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((5-(methylsulfonyl)pyridin-3-yl)methoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (43)
BDBM534239 WO2022081973, Example 43 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((5-(methylsulfonyl)pyridin-3-yl)methoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (43)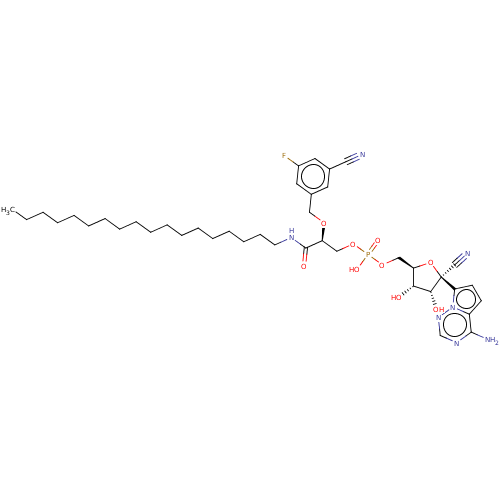 BDBM534243 WO2022081973, Example 47 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-2-((3-cyano-5-fluorobenzyl)oxy)-3-(octadecylamino)-3-oxopropyl) hydrogen phosphate (47)
BDBM534243 WO2022081973, Example 47 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-2-((3-cyano-5-fluorobenzyl)oxy)-3-(octadecylamino)-3-oxopropyl) hydrogen phosphate (47)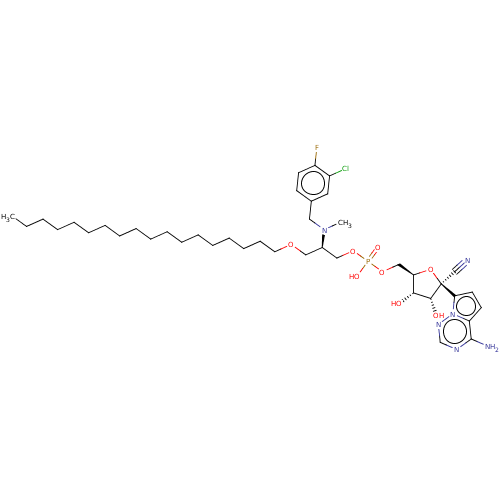 WO2022081973, Example 3 BDBM534199 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-chloro-4-fluorobenzyl)(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (3)
WO2022081973, Example 3 BDBM534199 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-chloro-4-fluorobenzyl)(methyl)amino)-3-(octadecyloxy)propyl) hydrogen phosphate (3)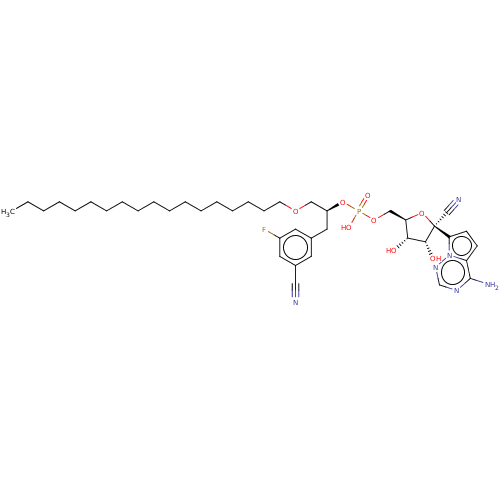 WO2022081973, Example 39 BDBM534235 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-(3-cyano-5-fluorophenoxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (39)
WO2022081973, Example 39 BDBM534235 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-(3-cyano-5-fluorophenoxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (39) WO2022081973, Example 48 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-2-fluoro-3-methoxyphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (48) BDBM534244
WO2022081973, Example 48 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-2-fluoro-3-methoxyphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (48) BDBM534244 WO2022081973, Example 49 BDBM534245 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-2-fluoro-5-methylphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (49)
WO2022081973, Example 49 BDBM534245 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-(4-cyano-2-fluoro-5-methylphenoxy)-3-(octadecyloxy)propyl) hydrogen phosphate (49) ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((5-cyanopyridin-2-yl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (41) BDBM534237 WO2022081973, Example 41
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((5-cyanopyridin-2-yl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (41) BDBM534237 WO2022081973, Example 41 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluoro-4-methylbenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (53) BDBM534249 WO2022081973, Example 53
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluoro-4-methylbenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (53) BDBM534249 WO2022081973, Example 53 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluoro-4-propylbenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (51) BDBM534247 WO2022081973, Example 51
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluoro-4-propylbenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (51) BDBM534247 WO2022081973, Example 51 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3-fluorobenzyl)oxy)nonadec-4-yn-1-yl) hydrogen phosphate (75) BDBM534272 WO2022081973, Example 75
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-cyano-3-fluorobenzyl)oxy)nonadec-4-yn-1-yl) hydrogen phosphate (75) BDBM534272 WO2022081973, Example 75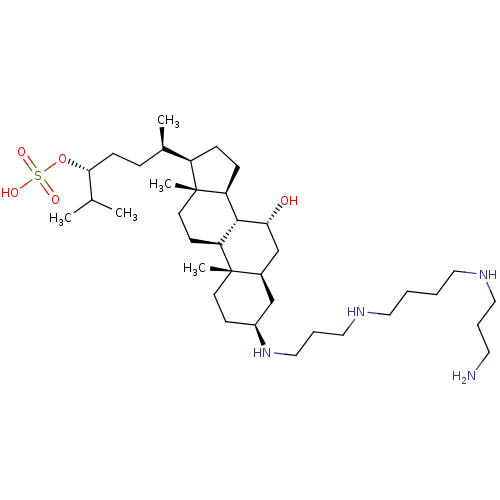 (3R,6R)-6-((3S,5R,7R,8R,9S,10S,13R,14S,17R)-3-(3-(4-(3-aminopropylamino)butylamino)propylamino)-7-hydroxy-10,13-dimethyl-hexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-methylheptan-3-yl hydrogen sulfate CHEMBL508583 BDBM50333649 trodusquemine
(3R,6R)-6-((3S,5R,7R,8R,9S,10S,13R,14S,17R)-3-(3-(4-(3-aminopropylamino)butylamino)propylamino)-7-hydroxy-10,13-dimethyl-hexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-methylheptan-3-yl hydrogen sulfate CHEMBL508583 BDBM50333649 trodusquemine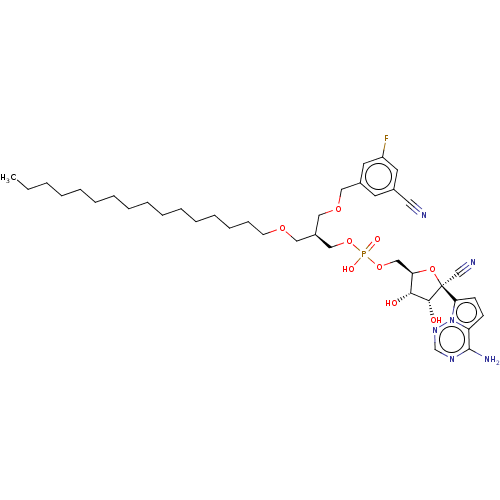 BDBM534232 WO2022081973, Example 36 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((3-cyano-5-fluorobenzyl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (36)
BDBM534232 WO2022081973, Example 36 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((3-cyano-5-fluorobenzyl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (36) BDBM534242 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((6-cyanopyridazin-3-yl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (46) WO2022081973, Example 46
BDBM534242 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((6-cyanopyridazin-3-yl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (46) WO2022081973, Example 46 BDBM534250 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-chloro-3-cyano-5-methoxybenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (54) WO2022081973, Example 54
BDBM534250 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-chloro-3-cyano-5-methoxybenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (54) WO2022081973, Example 54 BDBM534260 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl [(2R)-2-[(3-chloro-2,4-difluoro-phenyl)methoxy]-3-octadecoxy-propyl] hydrogen phosphate (64) WO2022081973, Example 64
BDBM534260 [(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl [(2R)-2-[(3-chloro-2,4-difluoro-phenyl)methoxy]-3-octadecoxy-propyl] hydrogen phosphate (64) WO2022081973, Example 64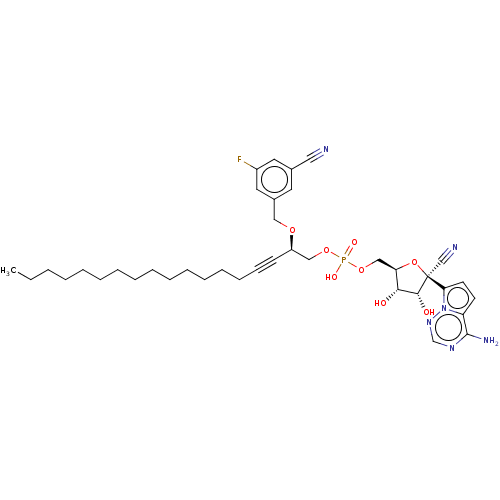 BDBM534271 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)nonadec-4-yn-1-yl) hydrogen phosphate (74) WO2022081973, Example 74
BDBM534271 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((3-cyano-5-fluorobenzyl)oxy)nonadec-4-yn-1-yl) hydrogen phosphate (74) WO2022081973, Example 74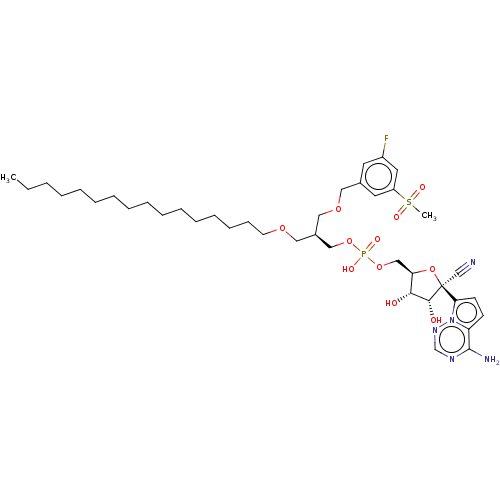 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((3-fluoro-5-(methylsulfonyl)benzyl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (33) BDBM534229 WO2022081973, Example 33
((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-1-((3-fluoro-5-(methylsulfonyl)benzyl)oxy)-3-(octadecyloxy)propan-2-yl) hydrogen phosphate (33) BDBM534229 WO2022081973, Example 33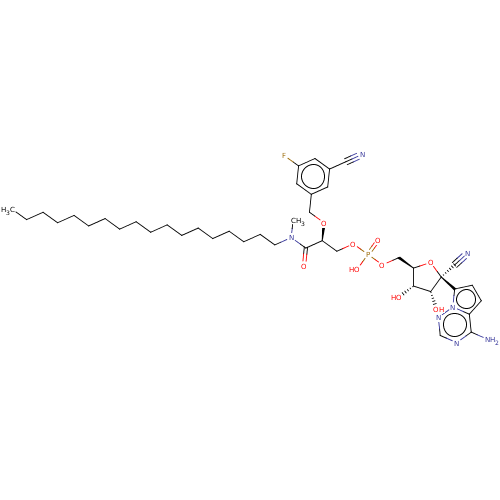 BDBM534252 WO2022081973, Example 56 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-2-((3-cyano-5-fluorobenzyl)oxy)-3-(methyl(octadecyl)amino)-3-oxopropyl) hydrogen phosphate (56)
BDBM534252 WO2022081973, Example 56 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((S)-2-((3-cyano-5-fluorobenzyl)oxy)-3-(methyl(octadecyl)amino)-3-oxopropyl) hydrogen phosphate (56) erythro-Phenyl-2-piperidyl-carbinol,(-) trans-N-Methylphenylcyclopropylamine 3,4-DNH Benzine Cc-34,(+/-) benzene Benzol phenyl hydride CHEMBL277500 Pyrobenzol 5-Hydroxy Tryptamine Mineral naphtha Coal naphtha Bicarburet of hydrogen cyclohexatriene Benzen benzole [6]annulene trans-N, N-Dimethylphenylcyclopropylamine Phene Pyrobenzole BDBM50167939
erythro-Phenyl-2-piperidyl-carbinol,(-) trans-N-Methylphenylcyclopropylamine 3,4-DNH Benzine Cc-34,(+/-) benzene Benzol phenyl hydride CHEMBL277500 Pyrobenzol 5-Hydroxy Tryptamine Mineral naphtha Coal naphtha Bicarburet of hydrogen cyclohexatriene Benzen benzole [6]annulene trans-N, N-Dimethylphenylcyclopropylamine Phene Pyrobenzole BDBM50167939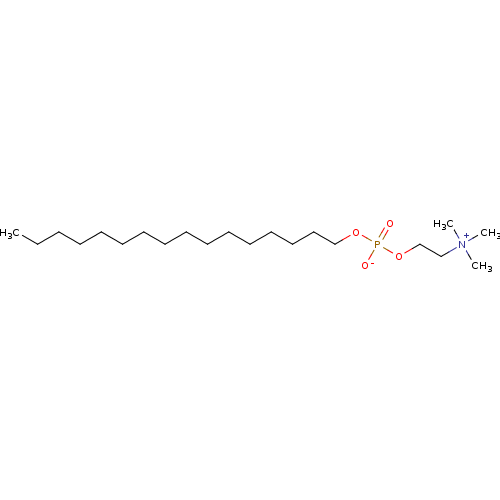 hexadecyl 2-(trimethylammonio)ethyl phosphate [2-(Hexadecyloxy-hydroxy-phosphoryloxy)-ethyl]-trimethyl-ammonium hexadecylphosphocholine hexadecyl 2-(trimethyl-lambda~5~-azanyl)ethyl hydrogen phosphate hexadecylphosphocholine, miltefosine MILTEFOSINE CHEMBL125 hexadecyloxy-2-trimethylammonioethylphosphorate 2-(((Hexadecyloxy)hydroxyphosphinyl)oxy)-N,N,N-trimethylethanaminium hydroxide BDBM50034220 n-hexadecylphosphocholine
hexadecyl 2-(trimethylammonio)ethyl phosphate [2-(Hexadecyloxy-hydroxy-phosphoryloxy)-ethyl]-trimethyl-ammonium hexadecylphosphocholine hexadecyl 2-(trimethyl-lambda~5~-azanyl)ethyl hydrogen phosphate hexadecylphosphocholine, miltefosine MILTEFOSINE CHEMBL125 hexadecyloxy-2-trimethylammonioethylphosphorate 2-(((Hexadecyloxy)hydroxyphosphinyl)oxy)-N,N,N-trimethylethanaminium hydroxide BDBM50034220 n-hexadecylphosphocholine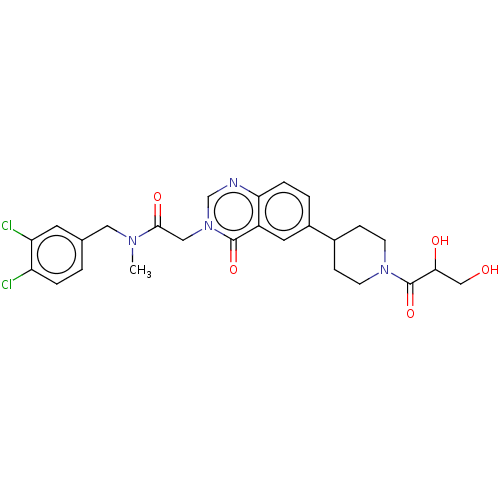 N-[(3,4- Dichlorophenyl)methyl]-N- methyl-2-(4-oxo-6-piperidin-4- ylquinazolin-3-yl)acetamide (Example 165, step A) and 2,3- dihydroxypropanoic acid (40% in water) ([CAS RN 600-19-1]) BDBM450593 US10676446, Example 177
N-[(3,4- Dichlorophenyl)methyl]-N- methyl-2-(4-oxo-6-piperidin-4- ylquinazolin-3-yl)acetamide (Example 165, step A) and 2,3- dihydroxypropanoic acid (40% in water) ([CAS RN 600-19-1]) BDBM450593 US10676446, Example 177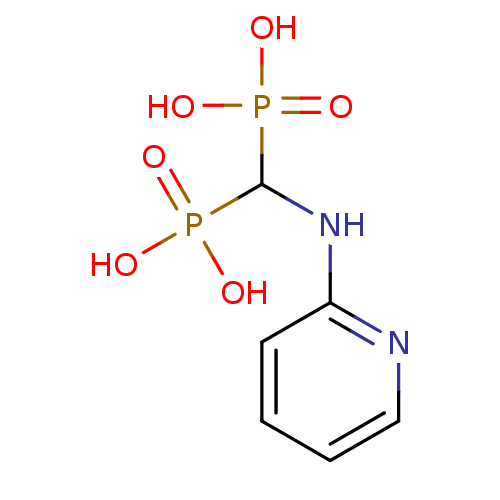 2-[(Bis-phosphono-methyl)-amino]-pyridinium CHEMBL291736 2-{[hydrogen phosphonato(phosphonato)methyl]amino}pyridin-1-ium [Phosphono-(pyridin-2-ylamino)-methyl]-phosphonic acid BDBM50115104 (pyridin-2-ylamino)methylenediphosphonic acid [Phosphono-(pyridin-2-ylamino)-methyl]-phosphonic acid(NE11807) [(2-pyridinylamino)methylene]-1,1-bisphosphonate
2-[(Bis-phosphono-methyl)-amino]-pyridinium CHEMBL291736 2-{[hydrogen phosphonato(phosphonato)methyl]amino}pyridin-1-ium [Phosphono-(pyridin-2-ylamino)-methyl]-phosphonic acid BDBM50115104 (pyridin-2-ylamino)methylenediphosphonic acid [Phosphono-(pyridin-2-ylamino)-methyl]-phosphonic acid(NE11807) [(2-pyridinylamino)methylene]-1,1-bisphosphonate WO2022081973, Example 50 BDBM534246 WO2022081973, Example 54 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-chloro-3-cyano-5-fluorobenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (50)
WO2022081973, Example 50 BDBM534246 WO2022081973, Example 54 ((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl ((R)-2-((4-chloro-3-cyano-5-fluorobenzyl)oxy)-3-(octadecyloxy)propyl) hydrogen phosphate (50)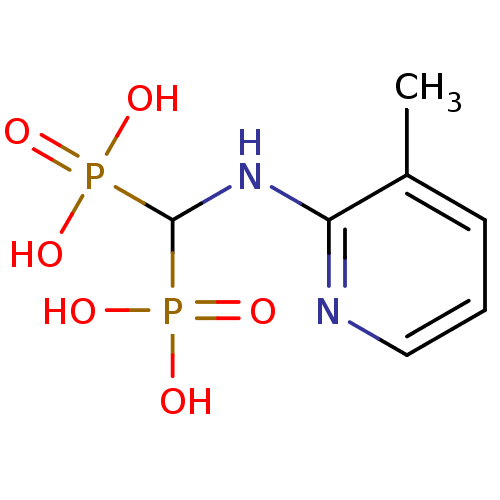 BDBM50098390 2-[(Bis-phosphono-methyl)-amino]-3-methyl-pyridinium [(3-Methyl-pyridin-2-ylamino)-phosphono-methyl]-phosphonic acid(NE97200) (3-methylpyridin-2-ylamino)methylenediphosphonic acid CHEMBL55140 [(3-methyl-pyridin-2-ylamino)-phosphono-methyl]-phosphonic acid 2-{[hydrogen phosphonato(phosphonato)methyl]amino}-3-methylpyridin-1-ium
BDBM50098390 2-[(Bis-phosphono-methyl)-amino]-3-methyl-pyridinium [(3-Methyl-pyridin-2-ylamino)-phosphono-methyl]-phosphonic acid(NE97200) (3-methylpyridin-2-ylamino)methylenediphosphonic acid CHEMBL55140 [(3-methyl-pyridin-2-ylamino)-phosphono-methyl]-phosphonic acid 2-{[hydrogen phosphonato(phosphonato)methyl]amino}-3-methylpyridin-1-ium {4-[5-bromo-3-(sulfooxy)-1H-indol-2-yl]-13-chloro-3-thiatricyclo[7.4.0.0^{2,6}]trideca-1(9),2(6),4,7,10,12-hexaen-5-yl}oxidanesulfonic acid 5-bromo-2-[9-chloro-3-(sulfooxy)naphtho[1,2-b]thiophen-2-yl]-1H-indol-3-yl hydrogen sulfate SALOR2 BDBM22584
{4-[5-bromo-3-(sulfooxy)-1H-indol-2-yl]-13-chloro-3-thiatricyclo[7.4.0.0^{2,6}]trideca-1(9),2(6),4,7,10,12-hexaen-5-yl}oxidanesulfonic acid 5-bromo-2-[9-chloro-3-(sulfooxy)naphtho[1,2-b]thiophen-2-yl]-1H-indol-3-yl hydrogen sulfate SALOR2 BDBM22584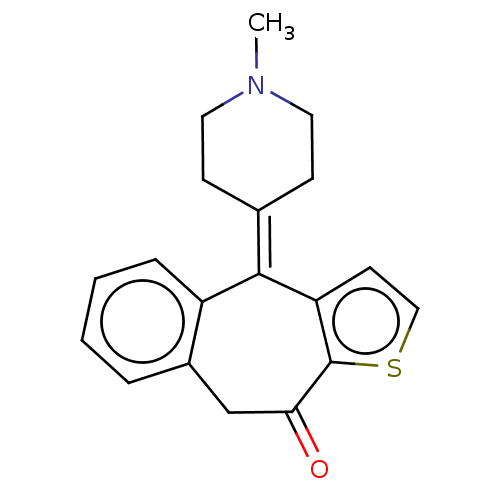 Zaditor 4-(1-Methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one(Ketotifen hydrogen malate) 4-(1-methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one CHEMBL534 BDBM50002087 KETOTIFEN 4-(1-Methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one(Ketotifen)
Zaditor 4-(1-Methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one(Ketotifen hydrogen malate) 4-(1-methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one CHEMBL534 BDBM50002087 KETOTIFEN 4-(1-Methyl-piperidin-4-ylidene)-4,9-dihydro-1-thia-benzo[f]azulen-10-one(Ketotifen)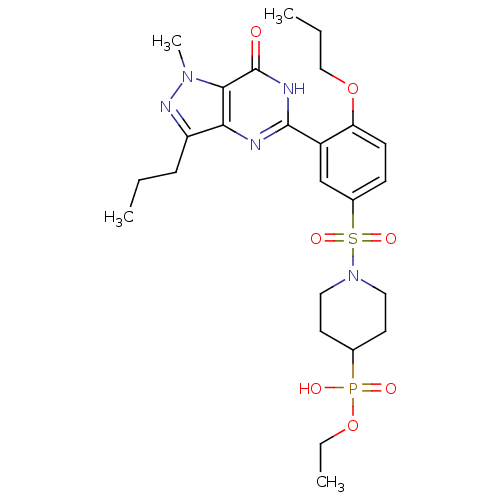 {1-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperidin-4-yl}-phosphonic acid monoethyl ester CHEMBL245315 ethyl hydrogen 1-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperidin-4-ylphosphonate BDBM50241302
{1-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperidin-4-yl}-phosphonic acid monoethyl ester CHEMBL245315 ethyl hydrogen 1-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperidin-4-ylphosphonate BDBM50241302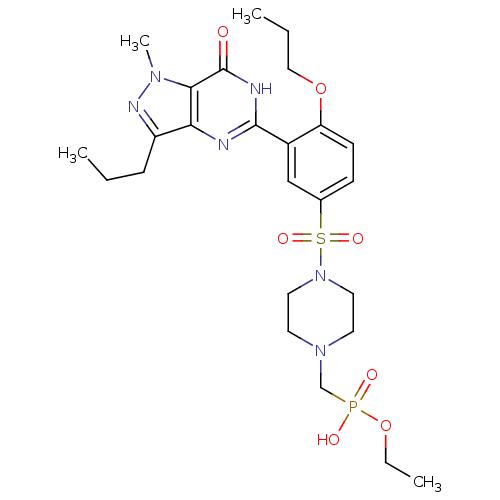 ethyl hydrogen (4-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperazin-1-yl)methylphosphonate {4-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperazin-1-ylmethyl}-phosphonic acid monoethyl ester BDBM50241300 CHEMBL245517
ethyl hydrogen (4-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperazin-1-yl)methylphosphonate {4-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperazin-1-ylmethyl}-phosphonic acid monoethyl ester BDBM50241300 CHEMBL245517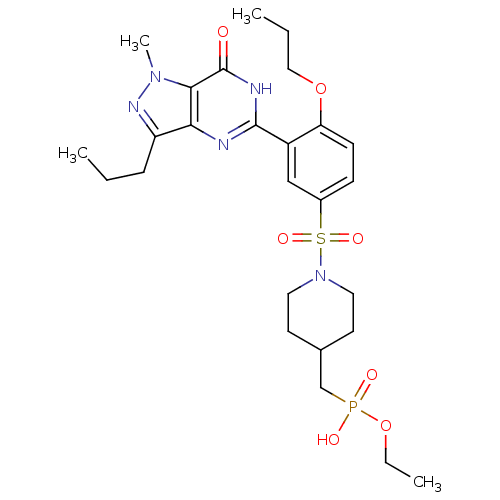 {1-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperidin-4-ylmethyl}-phosphonic acid monoethyl ester ethyl hydrogen (1-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperidin-4-yl)methylphosphonate CHEMBL245317 BDBM50241303
{1-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperidin-4-ylmethyl}-phosphonic acid monoethyl ester ethyl hydrogen (1-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperidin-4-yl)methylphosphonate CHEMBL245317 BDBM50241303 Foscarnet Foscavir trisodium dioxidophosphinecarboxylate oxide (phosphonoformate)Trisodium phosphonoformate BDBM50027536 trisodium phosphonoformate CHEMBL754 FOSCARNET SODIUM EHB-776 phosphonoformate trisodium salt trisodium dioxidophosphinecarboxylate oxide with 6 molecules of water(foscarnet) Antiviral agent : Inhibitor of reverse transcriptase of Human T-cell lymphotropic, virus type III, useful in the treatment of HSV-1 infections
Foscarnet Foscavir trisodium dioxidophosphinecarboxylate oxide (phosphonoformate)Trisodium phosphonoformate BDBM50027536 trisodium phosphonoformate CHEMBL754 FOSCARNET SODIUM EHB-776 phosphonoformate trisodium salt trisodium dioxidophosphinecarboxylate oxide with 6 molecules of water(foscarnet) Antiviral agent : Inhibitor of reverse transcriptase of Human T-cell lymphotropic, virus type III, useful in the treatment of HSV-1 infections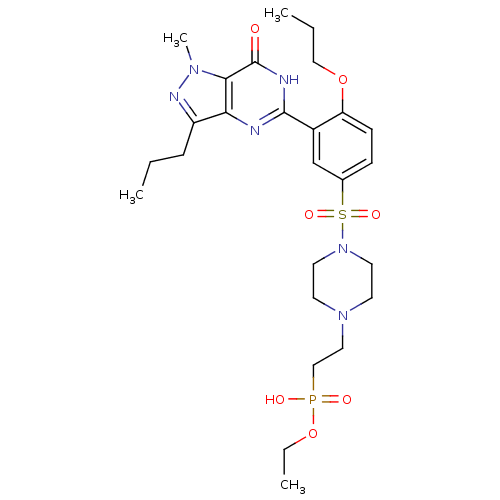 CHEMBL541468 ethyl hydrogen 2-(4-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperazin-1-yl)ethylphosphonate (2-{4-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperazin-1-yl}-ethyl)-phosphonic acid monoethyl ester BDBM50241301
CHEMBL541468 ethyl hydrogen 2-(4-(3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxyphenylsulfonyl)piperazin-1-yl)ethylphosphonate (2-{4-[3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-benzenesulfonyl]-piperazin-1-yl}-ethyl)-phosphonic acid monoethyl ester BDBM50241301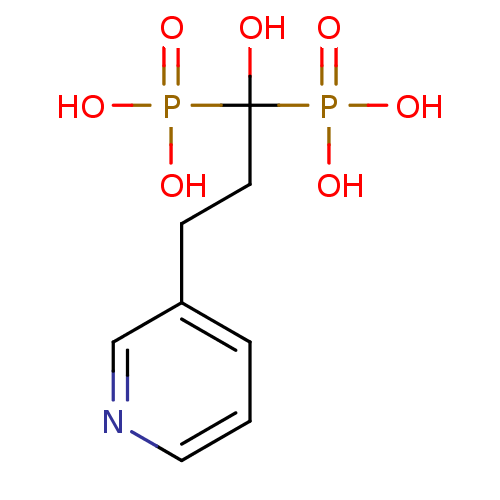 (1-Hydroxy-1-phosphono-3-pyridin-3-yl-propyl)-phosphonic acid(NE58051,homorisedronate) 3-(3-Hydroxy-3,3-bis-phosphono-propyl)-pyridinium (1-Hydroxy-1-phosphono-3-pyridin-3-yl-propyl)-phosphonic acid BDBM50098382 CHEMBL293522 1-hydroxy-3-(pyridin-3-yl)propane-1,1-diyldiphosphonic acid 3-(3-pyridyl)-1-hydroxy-propane-1,1-bisphosphonate 3-(3-hydrogen phosphonato-3-hydroxy-3-phosphonatopropyl)pyridin-1-ium
(1-Hydroxy-1-phosphono-3-pyridin-3-yl-propyl)-phosphonic acid(NE58051,homorisedronate) 3-(3-Hydroxy-3,3-bis-phosphono-propyl)-pyridinium (1-Hydroxy-1-phosphono-3-pyridin-3-yl-propyl)-phosphonic acid BDBM50098382 CHEMBL293522 1-hydroxy-3-(pyridin-3-yl)propane-1,1-diyldiphosphonic acid 3-(3-pyridyl)-1-hydroxy-propane-1,1-bisphosphonate 3-(3-hydrogen phosphonato-3-hydroxy-3-phosphonatopropyl)pyridin-1-ium BDBM50057660 CHEMBL1116 Keoxifene Hydrochloride Evista RALOXIFENE HYDROCHLORIDE 1-(2-{4-[6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophene-3-carbonyl]-phenoxy}-ethyl)-piperidinium; chloride Raloxifene LY-156758 [6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone; Hydrogen chloride [6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone; hydrochloride
BDBM50057660 CHEMBL1116 Keoxifene Hydrochloride Evista RALOXIFENE HYDROCHLORIDE 1-(2-{4-[6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophene-3-carbonyl]-phenoxy}-ethyl)-piperidinium; chloride Raloxifene LY-156758 [6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone; Hydrogen chloride [6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone; hydrochloride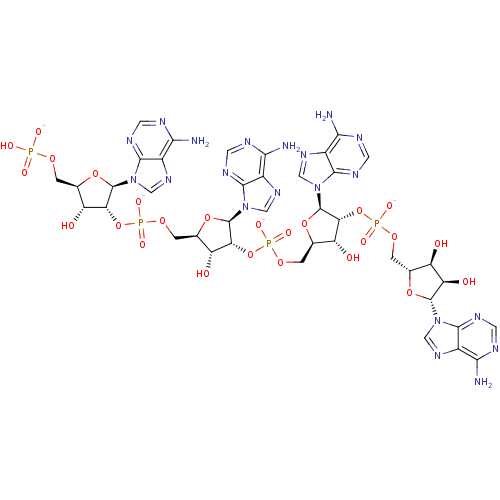 [(2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-[({[(2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}phosphinato)oxy]-3-hydroxyoxolan-2-yl]methyl (2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-({[(2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-[(hydrogen phosphonatooxy)methyl]-4-hydroxyoxolan-3-yl phosphonato]oxy}methyl)-4-hydroxyoxolan-3-yl phosphate 2''-5--oligoadenylate derivative BDBM50152834
[(2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-[({[(2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}phosphinato)oxy]-3-hydroxyoxolan-2-yl]methyl (2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-({[(2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-[(hydrogen phosphonatooxy)methyl]-4-hydroxyoxolan-3-yl phosphonato]oxy}methyl)-4-hydroxyoxolan-3-yl phosphate 2''-5--oligoadenylate derivative BDBM50152834 US10072025, Example 3-14 tert-butyl 4-[3-({4-[(2,3- dimethylmorpholin-4- yl)carbonyl]-3- methylphenyl}amino)-4- oxo-4,5-dihydro-1H- pyrazolo[4,3-c] pyridin-1- yl]-4-ethylpiperidine-1- carboxylate (Peak 1, separated by mass triggered reverse phase HPLC (ACN/water with 0.1% NH4OH modifier) then was separated by SFC using Phenomenex Lux-4 column, eluting with 40% methanol +0.25% dimethyl ethyl amine in CO2, retention time peak A: 8.2 minutes.) BDBM277380
US10072025, Example 3-14 tert-butyl 4-[3-({4-[(2,3- dimethylmorpholin-4- yl)carbonyl]-3- methylphenyl}amino)-4- oxo-4,5-dihydro-1H- pyrazolo[4,3-c] pyridin-1- yl]-4-ethylpiperidine-1- carboxylate (Peak 1, separated by mass triggered reverse phase HPLC (ACN/water with 0.1% NH4OH modifier) then was separated by SFC using Phenomenex Lux-4 column, eluting with 40% methanol +0.25% dimethyl ethyl amine in CO2, retention time peak A: 8.2 minutes.) BDBM277380 US10072025, Example 3-15 BDBM277381 tert-butyl 4-[3-({4-[(2,3- dimethylmorpholin-4- yl)carbonyl]-3- methylphenyl}amino)-4- oxo-4,5-dihydro-1H- pyrazolo[4,3-c] pyridin-1- yl]-4-ethylpiperidine-1- carboxylate (Peak 1, separated by mass triggered reverse phase HPLC (ACN/water with 0.1% NH4OH modifier) then was separated by SFC using Phenomenex Lux-4 column, eluting with 40% methanol +0.25% dimethyl ethyl amine in CO2, retention time peak B: 9.8 minutes.)
US10072025, Example 3-15 BDBM277381 tert-butyl 4-[3-({4-[(2,3- dimethylmorpholin-4- yl)carbonyl]-3- methylphenyl}amino)-4- oxo-4,5-dihydro-1H- pyrazolo[4,3-c] pyridin-1- yl]-4-ethylpiperidine-1- carboxylate (Peak 1, separated by mass triggered reverse phase HPLC (ACN/water with 0.1% NH4OH modifier) then was separated by SFC using Phenomenex Lux-4 column, eluting with 40% methanol +0.25% dimethyl ethyl amine in CO2, retention time peak B: 9.8 minutes.) US10072025, Example 3-16 BDBM277382 tert-butyl 4-[3-({4-[(2,3- dimethylmorpholin-4- yl)carbonyl]-3- methylphenyl}amino)-4- oxo-4,5-dihydro-1H- pyrazolo[4,3-c] pyridin-1- yl]-4-ethylpiperidine-1- carboxylate (Peak 2, separated by mass triggered reverse phase HPLC (ACN/water with 0.1% NH4OH modifier) then was separated by SFC using Phenomenex Lux-4 column, eluting with 40% methanol +0.25% dimethyl ethyl amine in CO2, retention time peak A: 8.4 minutes.)
US10072025, Example 3-16 BDBM277382 tert-butyl 4-[3-({4-[(2,3- dimethylmorpholin-4- yl)carbonyl]-3- methylphenyl}amino)-4- oxo-4,5-dihydro-1H- pyrazolo[4,3-c] pyridin-1- yl]-4-ethylpiperidine-1- carboxylate (Peak 2, separated by mass triggered reverse phase HPLC (ACN/water with 0.1% NH4OH modifier) then was separated by SFC using Phenomenex Lux-4 column, eluting with 40% methanol +0.25% dimethyl ethyl amine in CO2, retention time peak A: 8.4 minutes.)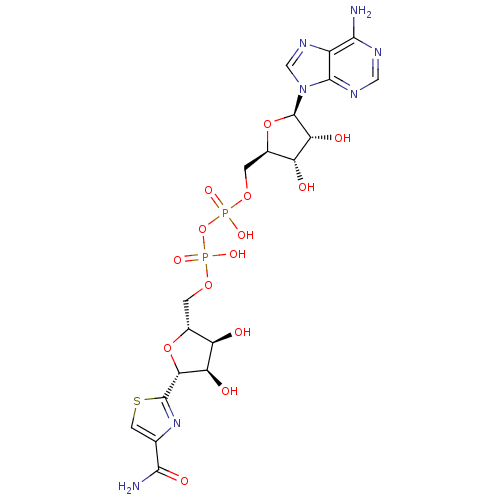 CHEMBL394276 BDBM19253 NSC 358285 [(2R,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [[(2R,3S,4R,5R)-5-(4-carbamoyl-1,3-thiazol-2-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]hydrogen phosphate tiazofurin adenine dinucleotide Tiazofurin Adenine Dinucleotide (TAD) [({[(2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)oxy]({[(2R,3S,4R,5R)-5-(4-carbamoyl-1,3-thiazol-2-yl)-3,4-dihydroxyoxolan-2-yl]methoxy})phosphinic acid
CHEMBL394276 BDBM19253 NSC 358285 [(2R,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [[(2R,3S,4R,5R)-5-(4-carbamoyl-1,3-thiazol-2-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]hydrogen phosphate tiazofurin adenine dinucleotide Tiazofurin Adenine Dinucleotide (TAD) [({[(2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)oxy]({[(2R,3S,4R,5R)-5-(4-carbamoyl-1,3-thiazol-2-yl)-3,4-dihydroxyoxolan-2-yl]methoxy})phosphinic acid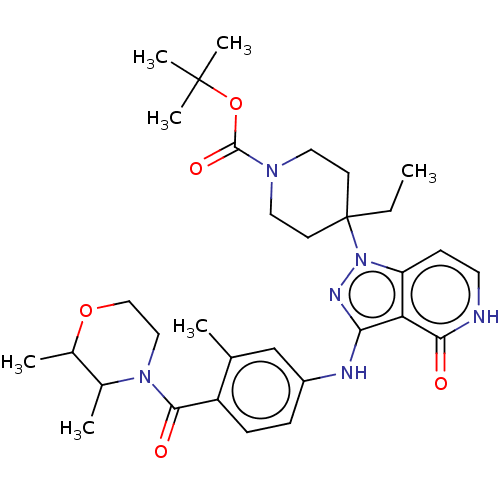 US10072025, Example 3-13 US10072025, Example 3-16 US10072025, Example 3-14 tert-butyl 4-[3-({4-[(2,3- dimethylmorpholin-4- yl)carbonyl]-3- methylphenyl}amino)-4- oxo-4,5-dihydro-1H- pyrazolo[4,3-c] pyridin-1- yl]-4-ethylpiperidine-1- carboxylate (Peak 2, separated by mass triggered reverse phase HPLC (ACN/water with 0.1% NH4OH modifier) then was separated by SFC using Phenomenex Lux-4 column, eluting with 40% methanol +0.25% dimethyl ethyl amine in CO2, retention time peak B: 10.2 minutes.) US10072025, Example 3-15 BDBM277379
US10072025, Example 3-13 US10072025, Example 3-16 US10072025, Example 3-14 tert-butyl 4-[3-({4-[(2,3- dimethylmorpholin-4- yl)carbonyl]-3- methylphenyl}amino)-4- oxo-4,5-dihydro-1H- pyrazolo[4,3-c] pyridin-1- yl]-4-ethylpiperidine-1- carboxylate (Peak 2, separated by mass triggered reverse phase HPLC (ACN/water with 0.1% NH4OH modifier) then was separated by SFC using Phenomenex Lux-4 column, eluting with 40% methanol +0.25% dimethyl ethyl amine in CO2, retention time peak B: 10.2 minutes.) US10072025, Example 3-15 BDBM277379